Hedgehog Signaling Pathway and Autophagy in Cancer

1
Department of Microbiological and Biochemical Pharmacy & The Key Laboratory of Smart Drug Delivery MOE, School of Pharmacy, Fudan University, Shanghai 201203, China
2
Department of Pharmacy, Faculty of Science, National University of Singapore, Singapore 117543, Singapore
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(8), 2279; https://doi.org/10.3390/ijms19082279
Submission received: 11 July 2018
/
Revised: 29 July 2018
/
Accepted: 31 July 2018
/
Published: 3 August 2018
(This article belongs to the Special Issue Hedgehog Signaling)
Abstract
:Hedgehog (Hh) pathway controls complex developmental processes in vertebrates. Abnormal activation of Hh pathway is responsible for tumorigenesis and maintenance of multiple cancers, and thus addressing this represents promising therapeutic opportunities. In recent years, two Hh inhibitors have been approved for basal cell carcinoma (BCC) treatment and show extraordinary clinical outcomes. Meanwhile, a series of novel agents are being developed for the treatment of several cancers, including lung cancer, leukemia, and pancreatic cancer. Unfortunately, Hh inhibition fails to show satisfactory benefits in these cancer types compared with the success stories in BCC, highlighting the need for better understanding of Hh signaling in cancer. Autophagy, a conserved biological process for cellular component elimination, plays critical roles in the initiation, progression, and drug resistance of cancer, and therefore, implied potential to be targeted. Recent evidence demonstrated that Hh signaling interplays with autophagy in multiple cancers. Importantly, modulating this crosstalk exhibited noteworthy capability to sensitize primary and drug-resistant cancer cells to Hh inhibitors, representing an emerging opportunity to reboot the efficacy of Hh inhibition in those insensitive tumors, and to tackle drug resistance challenges. This review will highlight recent advances of Hh pathway and autophagy in cancers, and focus on their crosstalk and the implied therapeutic opportunities.
1. Introduction
Hedgehog (Hh) signaling pathway regulates multiple key developmental processes, including embryogenesis and cell proliferation, cell fate determination, and tissue patterning in vertebrates [1,2]. In the adult, controlled Hh signaling activity is crucial for wound healing, tissue regeneration, and homeostasis maintenance [3,4]. However, ectopic Hh pathway activation has been documented to be responsible for tumorigenesis, progression, metastasis, and drug resistance of various cancers, including basal cell carcinoma (BCC), medulloblastoma (MB), and many other solid and hematological tumors [5]. Due to these extensive involvements in cancers, the Hh pathway has emerged as an attractive target for cancer therapy. Over the last two decades, mountains of efforts have been dedicated to the fundamental understanding of Hh pathway, as well as the development of clinical-level Hh pathway modulators. Thus far, two Hh pathway inhibitors, vismodegib and sonidegib, have been approved by the US Food and Drug Administration (FDA) for treatment of BCC in 2012 and 2015, respectively. In parallel, a series of novel small molecules are being developed under ongoing clinical trials or preclinical investigations [6].
However, remarkably, clinical benefits of targeting the Hh pathway are only limited to a few cancers, such as BCC and MB [7,8]. Results of clinical trials in other cancers indicated that Hh pathway inhibitors failed to show significant clinical benefit. For instance, a phase I study showed that 60% of BCC patients (19 out of 33) exhibited a partial or complete response to vismodegib treatment, and opposite to this encouraging outcome, none of 34 patients with other solid tumors showed partial or complete responses [9]. Moreover, clinical trials of the combination of vismodegib and gemcitabine demonstrated that the addition of vismodegib did not enhance overall response rate and extend patient survival time in metastatic pancreatic cancer, though the Hh signaling activity has been significantly decreased [10,11]. These unexcepted failures imply the complexity of Hh signaling responses and its varied roles in cancers. In addition, acquired resistance is emerging as a big challenge that compromises the clinical benefits of Hh inhibitors, and data suggested that mutations of Hh signaling components [12] and the activation of bypass signaling, such as phosphatidylinositol 3-kinase (PI3K) pathway [13], represent the potential mechanisms. Importantly, these findings indicated that combined inhibition of Hh signaling and other oncogenic pathways may be effective for improving the antitumor efficacy of Hh inhibitors and circumventing drug resistance.
Autophagy, a conservative metabolic process for degradation and recycling of damaged organelles or misfolded proteins, is responsible for maintaining cellular homeostasis and adapting to changes and stimuli imposed by the intracellular or surrounding environmental conditions [14]. Mounting evidence has demonstrated that autophagy plays critical roles in carcinogenesis, progression, and treatment resistance of various cancers, and has thus emerged as a possible anticancer target for alone or in conjunction with other targets [15,16]. Based on these advances, about 50 clinical trials have been proposed or are undergoing to evaluate the anticancer activity of combining autophagy modulators with chemotherapies or targeted drugs, according to the records on the ClinicalTrials.gov [17]. Recent studies have revealed that the Hh signaling pathway interplays with autophagy in various conditions. Some studies have attempted to elucidate the underlying molecular mechanisms. Moreover, co-manipulating Hh pathway and autophagy has been demonstrated to be able to show favorable anticancer effects in both in vitro and in vivo models. This review will briefly highlight the current clinical updates of Hh signaling and autophagy in cancer therapy, then focus on the crosstalk between Hh pathway and autophagy, and review the preclinical attempts that combined targeting Hh signaling and autophagy in the view of potential therapeutic implications for cancer therapy.
2. Hedgehog Signaling Pathway at a Glance
Attempts to decipher the transduction mechanisms of Hh pathway have never been stopped since the first disclosure of the Hh pathway in 1980 by Nüsslein-Volhard and Wieschaus [1]. Understanding of the biological fundamentals of Hh pathway has accelerated its translation to clinical therapeutics. Below sections will firstly summarize current understanding on the Hh signaling cascades, followed by a brief update of clinical advances in targeting Hh signaling, especially combined with other oncogenic pathways, for cancer therapy.
2.1. Canonical and Non-Canonical Hedgehog (Hh) Signaling Transductions
Canonical Hh signaling pathway involves multiple major components: the Hh ligand family member proteins (include three isoforms: Sonic hedgehog (SHH), Indian hedgehog (IHH) and Desert hedgehog (DHH)), the 12-transmembrane domain receptor protein Patched (PTCH) which locates in the primary cilium, the 7-transmembrane G-protein coupled receptor Smoothened (SMO), the suppressor of fused protein (SUFU) in cytoplasm, and the glioma-associated oncogene (GLI) transcription factors. In the absence of Hh ligands, PTCH represses the accumulation of SMO on the primary cilia, thus resulting in the inactivation of the downstream cascade. By contrast, the presence of Hh ligands will lead to the binding of Hh to PTCH so that PTCH unleashes SMO, which consequently triggers downstream activation of GLI transcription factors. GLI transcription factors consist of three proteins: GLI1, GLI2, and GLI3. Among them, GLI1 only has its activator form, while GLI2 and GLI3 have both activators (GLI2A and GLI3A) and repressor forms (GLI2R and GLI3R). GLI transcription factors are the nuclear executors at the end of the Hh pathway which are responsible for regulating downstream target genes.
Compared with canonical Hh signaling, the non-canonical Hh pathway, whose activation is independent of Hh ligands, is less well-studied. Current understanding of noncanonical Hh signaling suggests two distinct types: type I—SMO-independent but GLI-dependent, and type II—SMO-dependent but GLI-independent. Much evidence has demonstrated that GLI1 can be activated by other signaling pathways, such as PI3K/Protein kinase B (AKT)/mammalian target of rapamycin (mTOR) [18,19,20], transforming growth factor-β (TGFβ)/Sma- and Mad-related Protein 3 (SMAD3) [21,22], Raf serine/threonine kinase (RAF)/Meiosis-specific serine/threonine-protein kinase (MEK)/Mitogen-activated protein kinase (MAPK) [23,24], and Kirsten rat sarcoma 2 viral oncogene homolog (KRAS)/androgen receptor (AR) pathways [25], in a SMO-independent manner. These studies also indicated that such activation of type I noncanonical Hh activity is closely related to tumorigenesis, progression, and drug resistance in multiple cancers. Type II non-canonical Hh signaling activation has been observed to positively regulate Wingless-related integration site (WNT) pathway in colon cancer stem cells [26] and decrease intracellular levels of 3′,5′-cyclic adenosine monophosphate (cAMP) in airway epithelial cells [27]. Moreover, the coexistence of canonical and noncanonical Hh signaling has also been reported. For example, in multiple myeloma (MM) cells, SMO inhibitor sonidegib downregulated GLI1 and PTCH1 and led to significant cell death, indicating that the canonical Hh signaling exists and contributes to MM cell proliferation. Meanwhile, both cytosolic and nuclear GLI1 could be detected, and the nuclear localization of GLI1 could be completely abolished by a GLI1 inhibitor forskolin, suggesting the non-canonical activation of Hh signaling in MM cells [28].
2.2. Clinical Advances in the Combination of Hh Signaling Inhibition and Other Targeted Therapies
Good understanding of mechanisms and roles of interplay between Hh pathway and other oncogenic pathways opens new opportunities for rational design of combination cancer therapies. In recent years, there are at least 16 clinical trials that have been proposed to prove the combinatory concept, as shown in Table 1 (combination with chemotherapies are not included). The majority of them attempt to combine SMO inhibitors with other targeted drugs. These combined drugs include small molecular inhibitors of the Janus kinase (JAK) pathway, PI3K, mTOR, and epidermal growth factor receptor (EGFR), and macromolecular agents that target EGFR, vascular endothelial growth factor (VEGF), and programmed death-ligand 1 (PD-1), for treatment of various solid tumors. Besides, SMO inhibitors are combined with BCR-ABL modulators for chronic myeloid leukemia (CML) treatment. Only three proposals combine GLI inhibitors with others, which is easy to understand because there are no GLI targeted agents that have been approved for clinical use, so far. In fact, the arsenic trioxide (ATO) used in these combinations has been approved by US FDA for acute promyelocytic leukemia (APL) treatment. Recent studies revealed its potent inhibitory effect on GLI, and therefore, it has been suggested as a promising Hh signaling inhibitor [29,30]. In addition to ATO, there is another repositioning drug, itraconazole (a US FDA approved antifungal drug), that was recently found to exhibit potent Hh inhibitory effect by targeting SMO in multiple cancers [31,32]. More importantly, both ATO and itraconazole were reported to be able to overcome SMO mutation-derived drug resistance [33], making the clinical trial results of use alone, or in conjunction with other drugs, to be expected. Combined inhibition of Hh signaling and other oncogenic pathways has been suggested as a promising strategy to improve Hh inhibition effect and overcome drug resistance. The results of a phase I clinical trial that combined saridegib with cetuximab (NCT01255800) have shown satisfactory toxicity profile and signs of antitumor activity of the combination [34]. Further clinical evaluation of this combination, as well as results of other clinical trials listed in Table 1, will be very anticipated.
3. Autophagy in Cancer
3.1. Regulation of Autophagy in Cancer Cells
Macroautophagy (autophagy hereafter), a highly conserved intracellular process that degrades harmful or superfluous cellular materials to maintain homeostasis or respond to stimuli, involves three major steps: (1) formation of the autophagosomes, double-membraned vesicles which engulf proteins or organelles need to be degraded; (2) fusion of autophagosomes with lysosomes; and (3) degradation of engulfed materials by enzymes in lysosomes. To date, there are more than 30 autophagy-related genes (ATGs) have been found to orchestrate these processes, and the underlying molecular mechanisms have been comprehensively described in most recent reviews [14,35]. Briefly, in the initial autophagosome formation, the Unc-51-like kinase (ULK) complex (consists of ULK1, ULK2, ATG13, RB1-inducible coiled-coil protein 1 (RBCC1), and ATG101) is activated by upstream signals, which further activates class III PI3K complex (includes Beclin1, ATG14, VPS34, and p150). Besides, ATG9, ATG3, ATG7, and ATG12–ATG5–ATG16L1 complex also contribute to membrane elongation and autophagosome growth. Of note, in the autophagy formation process, the conversion of LC3B-I to LC3B-II is a hallmark of autophagosome formation and is widely used as a monitoring marker to track autophagy initiation [35]. It is well established that basal level autophagy is required for almost all mammalian cells, and a variety of stimuli or stress, such as hypoxia [36], nutrient shortage [37,38], and drug treatment [39,40], can upregulate autophagy levels in various types of normal or cancer cells.
3.2. Context-Dependent Roles of Autophagy in Cancer
A large volume of literature has demonstrated the role and mechanism of autophagy in tumorigenesis, progression, treatment, and drug resistance in cancers. It is now clear that autophagy plays varied roles in different scenarios. Evidence supports various lines of stories, such as that autophagy prevents/initiates cancer and autophagy leads to cell survival/cell death. Autophagy is required for normal antitumor immunosurveillance and is therefore useful for preventing cancer initiation before neoplasms are established [41,42]. Conversely, in established tumors, upregulated autophagy helps cancer cells to cope with intracellular stress and unfavorable environmental conditions resulting from rapid cell proliferation and tumor growth, representing a mechanism of cancer progression. Besides, countless evidence indicates that autophagy helps cancer cells to escape or resist a wide range of anticancer drugs. Based on these facts, a variety of clinical trials have been proposed to enhance anticancer efficacy and circumvent drug resistance of targeted drugs (as shown in Table 2) and chemotherapeutics by combining autophagy inhibitors. Detailed clinical updates of combining autophagy in cancer therapies have been discussed in recent reviews [16,43]. Of course, autophagy does not always act as a bad guy in cancer treatment. Autophagy plays critical roles in activating anticancer immune responses in radiotherapy [44] and immunogenic chemotherapy [45]. Taken together, current evidence revealed that roles of autophagy in cancers are context-dependent, and further research on these topics will be helpful to accelerate the clinical application of modulating autophagy in cancer therapy.
4. Crosstalk between Hedgehog Signaling Pathway and Autophagy
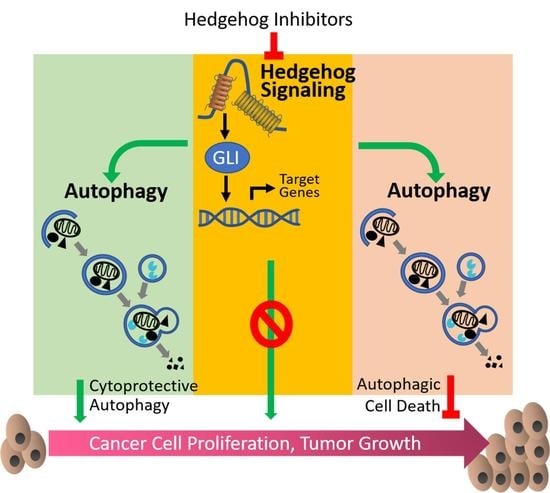
As one of the major players in the cell signaling system, Hh pathway interplays with multiple oncogenic pathways, such as PI3K/AKT/mTOR, RAF/MAPK/ERK, and KRAS pathways [46,47,48]. Although roles and mechanisms of autophagy or Hh signaling alone in human health and diseases have been intensively studies since decades ago, crosstalk between them has not been revealed until recent years. In the following sections, we will summarize current evidence of crosstalk between Hh pathway and autophagy by two categories (as depicted in Figure 1): Hh signaling inhibits autophagy and Hh signaling promotes autophagy.
4.1. Hh Signaling Inhibits Autophagy
The inhibitory effect of Hh signaling on autophagy has been found in normal and cancer cells from various tissue origins. Jimenez-Sanchez et al. systematically demonstrated that the inhibitory effect of Hh signaling on autophagy is governed by the GLI2–PERK–eIF2α axis. Inhibiting Hh signaling by overexpression of PTCH1 or PTCH2 increased autophagy level in both HeLa cells and mouse embryonic fibroblasts (MEFs). Similar results were also observed under the SMO knockdown condition. Accordingly, activating Hh signaling by overexpression of SMO or treatment with SMO agonist purmorphamine (all discussed Hh modulators have been summarized in Table 3) impaired autophagy. These data collectively confirmed that Hh signaling inhibits autophagy. Consistently, this inhibitory effect was also confirmed in Drosophila. Moreover, the authors further revealed that Hh signaling exerts its inhibitory effect by inhibiting the Protein kinase R (PRK)-like endoplasmic reticulum kinase (PERK) and reducing the phosphorylation of eukaryotic initiation factor 2α (eIF2α) [49]. Indeed, PERK–eIF2α has been implicated to be essential for autophagy induction under several conditions, such as ER stress and hypoxia [50,51]. Interestingly, purmorphamine impaired autophagy in GLI1−/− or GLI3−/− mice MEFs in the same way as in GLI wild-type counterparts, whereas it failed to inhibit autophagy in GLI2−/− MEFs, indicating that such inhibitory effect of Hh signaling on autophagy depends on GLI2, but not GLI1 or GLI3. Importantly, this evidence highlighted the central role of GLI2 in Hh–autophagy crosstalk [49].
In several cancer cell lines: H4 (glioma), ES2 (ovarian cancer), MKN45 (gastric cancer), and HT29 (colon cancer cells), by indiscriminate analysis of more than 30,000 transcripts using microarray chips, many ATGs were found to be differentially upregulated after inhibiting Hh signaling with GANT61 treatment. Further experiments validated the activation of autophagy induced by Hh signaling inhibition in these four cell lines. Moreover, activation Hh signaling by N-SHH, a recombinant SHH peptide, subdued autophagy level and partially rescued cancer cells from Hh inhibition-induced cytotoxicity, suggesting the role of autophagic cell death [64].
It has also been reported that Hh signaling was active in hepatocellular carcinoma (HCC) cells, inhibition of Hh signaling by GLI1/2 inhibitor GANT61-induced autophagy, while activation of Hh signaling by Hh ligand protein or Hh agonist prevented the autophagy induction. Interestingly, inhibiting GANT61-induced autophagy could hamper GANT61-induced apoptosis, cytotoxicity, and in vivo tumor suppression effect, indicating the cell-killing role of GANT61-induced autophagy in HCC cells [65].
Abnormal activation of Hh signaling has been observed in breast cancer and implicated in drug resistance [66,67]. Itraconazole, a clinically used antifungal drug which is repurposed for cancer therapy, has been demonstrated to be able to kill cancer cells via inhibiting Hh signaling [31] and inducing apoptosis and cell cycle arrest [32]. A recent study showed that inhibition of Hh signaling by itraconazole induced cytotoxicity, apoptosis, tumor shrinkage, and autophagy in breast cancer in vitro cell lines or in vivo mouse models. Moreover, inhibiting autophagy by 3-MA or ATG5 silencing attenuated itraconazole-induced cell death, suggesting that Hh inhibition-induced autophagy played a role in autophagic cell death [66]. In addition, another study demonstrated that itraconazole induced autophagy in glioblastoma cell lines U87 and C6, via repressing AKT1–mTOR signaling. Inhibiting autophagy by ATG5 or Beclin1 silencing rescued cells from itraconazole-induced cell death, indicating the cell-killing role of autophagy in this condition. However, the authors did not determine the change of Hh signaling activity during these processes [68].
Also, in lung cancer, the Hh signaling activation has been detected in several cell lines and patient samples, and therefore has been evaluated as a promising therapeutic target [69,70,71]. A recent study observed the Hh–autophagy relationship in A549 and 95D lung cancer cell lines. Bisdemethoxycurcumin (BDMC), a phytochemical that exhibits potent anticancer effect, reduced cell viability and induced apoptosis in A549 and 95D cells. To further probe the underlying mechanism, this study found that BDMC induced autophagy. Importantly, inhibiting Hh signaling by GLI1 siRNA or cyclopamine treatment significantly enhanced BDMC-induced autophagy, indicating that the induction of autophagy was partially inhibited by Hh signaling under BDMC treatment [72]. Most recently, results from our own lab described the interaction between Hh signaling and autophagy in lung adenocarcinoma. Our data indicated that SMO antagonist vismodegib failed to induce obvious cell death in A549 and NCI-H1975 cells and tumor shrinkage in xenograft mouse model but triggered marked autophagy [73]. Our further experiments showed that inhibiting vismodegib-induced autophagy by pharmacological inhibitors or gene knockdown could reboot the sensitivity of lung adenocarcinoma to vismodegib both in cell lines and in vivo models, suggesting autophagy might be a compensatory mechanism of Hh inhibition in lung cancer [74].
Consuelo Amantini et al. has reported that capsaicin, a plant-derived natural product which exerts anticancer potential, induced significant autophagy and upregulated expression of PTCH2 (a negative regulator of Hh signaling) in bladder cancer cell lines. This activation of autophagy resulted in epithelial–mesenchymal transition (EMT) and chemoresistance in bladder cancer cells. Importantly, silencing the PTCH2 gene could weaken the autophagy induction, suggesting the involvement of Hh signaling in capsaicin-induced autophagy in bladder cancer cells [75].
In pancreatic cancer cells, inhibiting Hh signaling by GANT61-induced autophagy both in in vitro cells and in vivo mouse model, while activating Hh signaling by N-SHH transfection reduced autophagy. Moreover, the combination with 3-MA in vitro and in vivo showed an impaired antitumor activity of GANT61, suggesting that autophagy acted as a cell-killing role [76]. Hh signaling has also been demonstrated to suppress tumor growth by promoting the formation of fibroblast-rich stroma in pancreatic carcinoma [77]. Besides, a recent study delineated that the stromal stellate cells provoked pancreatic tumor growth by supplying alanine to overcome nutrient-poor tumor microenvironment, and the increased secretion of alanine was achieved by induction of autophagy [78]. Based on these two studies, it can be inferred that Hh signaling represses autophagy in stromal stellate cells to restrain tumor growth by subduing autophagy-induced alanine supplement. These studies also indicated that autophagy plays distinct roles in pancreatic stroma and pancreatic cancer cells.
In chondrosarcoma, the activation of the Hh signaling pathway has also been observed and has been suggested as a potential therapeutic target [79,80]. Expression of PTCH1, SMO, and GLI1 proteins was detected in chondrosarcoma patient tissues and three chondrosarcoma cell lines (HC-a, SW1353, and JJ012), but not in normal articular cartilage tissues. Interestingly, silencing of GLI1 by siRNA significantly decreased cell viabilities and induced autophagy at the same time. Of note, data from this study indicated that activation of Hh signaling in chondrosarcoma cancer cells is type I noncanonical Hh signaling (GLI1-dependent and SMO-independent) since the Hh pathway activity can be diminished by GLI1 knockdown, but not by cyclopamine treatment. The authors proposed the GLI1–mTOR–autophagy cascade by proving the requirement of mTOR dephosphorylation in this process. Further results depicted that co-treatment with 3-MA and CQ or silencing of ATG5/Beclin1 could reverse GLI1 inhibition-induced cell death, indicating the cell-killing effect of autophagy in this case [81]. In agreement with these results, another study reported that arsenic trioxide (ATO), a GLI targeted Hh signaling inhibitor, induced autophagy in SW1353 cell line [82].
Rhabdomyosarcoma, the most common soft tissue sarcoma in young children, has been characterized by abnormal activation of Hh signaling [83,84]. A recent study has depicted that inhibition of Hh signaling using LDE225 or cyclopamine could activate autophagy in RUCH-2 and Rh41 rhabdomyosarcoma cell lines [85].
Neuroblastoma (NB) is the most common extracranial childhood solid tumor, accounting for ~15% of cancer-related childhood mortality [86]. Amplification of MYCN oncogene in NB is consistently correlated with high-risk NB stage, poor clinical outcome, and therapy-resistance [87,88]. Blocking Hh signaling showed antitumor efficacy both in in vitro and in vivo models, and therefore, has been suggested as a potential NB treatment strategy [89]. Whereas several studies have suggested that compared with MYCN-non-amplified NB cells, MYCN-amplified NB cells are relatively insensitive to Hh inhibition. To explore the underlying mechanism, Jing Wang et al. proposed autophagy activation induced by Hh inhibition as a mechanism of this compromised treatment effect. GANT61, a GLI1/2 inhibitor, induced significant cytotoxicity in MYCN-non-amplified NB cells without induction of autophagy. Conversely, GANT61 failed to induce cytotoxicity in MYCN-amplified NB cells, but triggered obvious autophagy. Inhibition of autophagy by 3-MA or ATG5/ATG7 silencing could enhance GANT61-induced cell death, suggesting that the cytoprotective effect of autophagy played a critical role in this case. Collectively, this suggested that autophagy may be responsible for the insensitivity of MYCN-amplified NB cells to Hh inhibition [90]. Most recently, data from the same research lab indicated that PERK is a key mediator of GANT61-induced autophagy in MYCN-amplified NB cells [91], which is in line with results from MEF models [49].
The development of medulloblastoma (MB), another prevalent brain tumor in young children, can be generally attributed to four types of dysregulations: WNT/β-catenin, SHH, MYCN, and heterogeneous genes [92]. Amplification of Hh signaling has been well characterized, and several clinical trials have been initiated to evaluate the efficacy of the SMO antagonist vismodegib on MB patients (Table 3). Moreover, the PI3K/mTOR pathway has been demonstrated to play crucial roles in SHH- and MYCN-driven MB [13,93]. Importantly, Hh signaling can be activated by PI3K/mTOR in several cancers [18,19]. A most recent study reported that the conjunction of Hh inhibitor and PI3K/mTOR inhibitor exhibited significantly enhanced antitumor effects both in cell lines and xenograft mouse model of Hh-driven and MYCN-driven MB, underpinning the importance of Hh and PI3K/mTOR in MB [94]. In addition, it is well documented that the induction of PI3K/mTOR pathway halts autophagy, while inhibition of the PI3K/mTOR pathway stimulates autophagy [35]. Taken together, autophagy might be involved in the crosstalk between Hh and PI3K/mTOR in MB.
Resistance to ABL tyrosine kinase inhibitors is a major obstacle for improving clinical outcomes of chronic myeloid leukemia (CML) treatment. Both deregulations of Hh signaling and autophagy have been demonstrated to be involved in CML drug resistance [95,96,97,98]. Our study indicated that SMO antagonist vismodegib induced autophagy in several BCR-ABL+ CML cell lines, including K562 and BaF3 cell lines harboring the T315I mutation of BCR-ABL fusion gene (BCR-ABL) (an infamous drug resistance mutation in CML). Knockdown of SMO also led to an obvious elevation of autophagy level. Data from this study also suggested that inhibition of AKT/mTOR signaling pathway was involved in the autophagy induction. Most importantly, co-inhibiting Hh signaling and autophagy by the combination of vismodegib and CQ could induce robust apoptosis in BaF3-BCR-ABLT315I cells which are resistant to CML drugs, such as imatinib and dasatinib, highlighting a possible way to overcome drug resistance in BCR-ABL+ CML patients [99]. Apart from CML cells, our recent study also suggested that in B-cell non-Hodgkin’s lymphoma (B-NHL) cell line Raji, inhibition of Hh signaling by vismodegib also triggered significant autophagy and blocking autophagy could sensitize B-NHL cells to vismodegib [100]. Moreover, a study from another group observed that inhibition of Hh signaling by LDE225 evoked autophagy in mantle cell lymphoma. Data from this study supported that upregulation of CXCR4 expression was involved in the autophagy induction, as CXCR4 knockout cells failed to initiate autophagy upon LDE225 treatment. At the same time, the combination with 3-MA significantly enhanced LDE225-induced cell cytotoxicity, revealing the cytoprotective role of autophagy in this case [101]. These data collectively suggested that in addition to solid tumors, modulating Hh–autophagy interplay may be also useful for improving the anticancer effect and overcoming drug resistance in hematologic malignancies.
In addition to cancer cells, Hh-regulated autophagy has also been reported in other human cell types. Junpeng Huang et al. reported that exposure to hexavalent chromium (Cr(VI)), a class of compounds which are recognized as human carcinogens and related to lung cancer risk, upregulated GLI2 gene expression, and such upregulation prevented the induction of autophagy in human bronchial epithelial cells. Inhibition of GLI2 by GANT61 or GLI2 siRNA transfection reversed the autophagy inhibition. These results underscored the impact of GLI2 on autophagy inhibition [102]. Besides, activation of GLI2 has been reported to be involved in fibrogenesis, a process involves the transdifferentiation of human primary fibroblasts into myofibroblasts. In this process, the reduced autophagy level co-occurred with GLI2 activation, accordingly, inhibition of GLI2 with GANT61 counteracted the autophagy inhibition in human primary fibroblasts. These data supported the idea that GLI2 inhibits autophagy. This study also proposed that eIF2α may link Hh signaling and autophagy, since the inhibition of eIF2α phosphorylation was correlated with GLI2 activation [103]. Hepatic stellate cells (HSCs) are critical for fibrogenic progression in non-alcoholic steatohepatitis. Recent studies have reported a distinct relationship between Hh signaling and autophagy in HSCs. Xuyou Liu et al. reported that inhibition of Hh signaling induced autophagy in HSCs. Inhibition of Hh signaling by cyclopamine or PTCH1/GLI1 siRNA led to increased autophagy level, and stimulation of Hh signaling by purmorphamine reduced autophagy, confirming the negative regulation of Hh signaling on autophagy in HSCs [104]. In supporting this observation, another study has reported that, in hepatic stellate cell line LX-2, inhibition of Hh signaling by GANT61 induced autophagy, which plays a cytoprotective role and relies on the induction of endoplasmic reticulum stress [105]. In contrast to these two studies, Nana Duan et al. found that the palmitic acid exposure led to activation of both Hh signaling and autophagy in human immortalized HSC, rat BSC-C10, and primary rat HSC cells. Accordingly, inhibition of Hh signaling by LDE225 suppressed autophagy induction [106]. These controversial results may be attributed to varied study conditions, such as different cell origins and perturbation agents.
Furthermore, it has been suggested that the crosstalk between Hh signaling and autophagy also plays roles in pathogen–host interactions in infective diseases. Autophagy plays a crucial role in both innate and adaptive immune systems [41,107]. Host cells can elevate autophagy to restrict intracellular pathogens and promote the major histocompatibility complex (MHC) class II presentation of the pathogen-derived antigens, servicing as a major mechanism of infection prevention [108,109,110]. However, pathogens can evade this protection mechanism by inhibiting autophagy level in host cells. Sahana Holla et al. reported that some bacteria species, such as Mycobacteria, Shigella, and Listeria, could selectively inhibit autophagy in macrophages by provoking robust Hh signaling activation. Their data demonstrated that Hh signaling pathway repressed autophagy via the upregulation of arachidonate 5-lipoxygenase (ALOX5) or arachidonate 15-lipoxygenase (ALOX15) gene expression, which negatively regulates autophagy. The authors proposed the mTOR–GSK3β–GLI1–ALOX5/ALOX15–autophagy axis in this regulation [111].
4.2. Hh Signaling Upregulates Autophagy
Many studies have observed the activating effect of Hh signaling on autophagy. For example, Luis Milla et al. reported that Hh antagonist cyclopamine prevented autophagy activation in neuroblastoma cell line SHSY5Y. Moreover, in the SHH-sensitive cell line C3H10T1/2, deprivation of SHH ligand protein in culture medium abolished Hh signaling activation and, at the same time, led to decreased protein expression of ATG5, a key protein for autophagy activation. These findings suggested that Hh signaling mediated the activation of autophagy in neuroblastoma cell lines [112]. Oncogene KRAS could induce autophagy to promote tumorigenesis and cancer progression and the KRAS–PI3K–AKT1–GLI3–VMP1 axis has been postulated as an underlying mechanism [113]. In this case, GLI3 was able to activate autophagy via upregulating expression of the VMP1, a protein essential for autophagy activation, whereas, GLI3-regulated autophagy was SMO-independent.
Tumor-initiating cells (TICs), also known as cancer stem cells (CSCs), are a small subpopulation of cells in the tumor which can be recognized by several cell surface markers, such as CD133, CD24, CD44, and Bmi-1 [114]. TICs exert the capacity of self-renewal, differentiation, and tumor initiation, and play critical roles in tumorigenesis, tumor growth and maintenance, radio- and chemotherapy resistance, and tumor relapse [115,116,117]. Importantly, Hh signaling regulates TICs in many cancers, including breast cancer, pancreatic cancer, gastric cancer, and brain cancers [118,119,120,121,122], representing a promising therapeutic target by controlling TICs. For example, it has been reported that activated Hh signaling suppressed Fas and DR4/5 expression, stimulated Bcl-2 and PDGFR expression in pancreatic TICs, and thus promoted cell growth, while inhibition of Hh led to apoptosis [118]. Activation of Hh signaling promoted TIC growth via modulating Bmi-1 gene expression in human breast cancer [119]. Moreover, it has been revealed that autophagy plays critical roles in TIC maintenance and growth. For instance, in breast cancer, activation of autophagy suppressed tumor development in a TIC-poor enriched tumor model but played a promotional role in TIC maintenance [123], revealing the unique role of autophagy in breast cancer TICs for growth and survival. In pancreatic cancer, Haitao et al. reported that under hypoxia, the upregulated HIF-1α induced autophagy which mediated the conversion of non-stem pancreatic cancer cells to stem-like cells to maintain the equilibrium of TICs [124]. In addition, another study revealed that hypoxia activated HIF-1α expression, which subsequently led to upregulated of Hh signaling, and finally made pancreatic cancer more aggressive and resistant to treatment [125]. Taken together, it might be speculated that Hh signaling promotes autophagy in pancreatic TICs, and both of them collectively contribute to growth and survival of pancreatic cancer TICs.
Strictly controlled Hh signaling activity is essential for development, repair, and homeostasis of many tissues, including cardiovascular tissue, intestinal epithelium, and neurons [2]. In mouse vascular smooth muscle cells, a research group reported that adding SHH ligands or overexpressing SHH gene instigated autophagy. Activated Hh signaling promoted cell proliferation, and this effect partially depended on the activation of autophagy [126]. By establishing an SHH intestinal epithelial conditional knockout mice model, Jessica G.S. et al. indicated that Hh signaling plays a critical role in homeostasis of intestinal ileum, and loss of SHH signaling lead to the decrease of autophagy in intestinal ileum [127]. Qing Xiao et al. reported that Hh signaling activation by SHH agonist SAG protected cardiomyocytes H9C2 cells from injury under oxygen-glucose deprivation (OGD) condition. This protective role of SHH signaling was achieved by upregulation of autophagy through activating AMPK/UlK1 signaling [128]. Ronald S.P. and colleagues revealed that in hippocampal neurons, exposure to SHH ligand proteins led to significant upregulation of autophagy. Their further results demonstrated that the Class III PI3K was required for this autophagy activation since 3-MA could effectively prevent SHH ligand-induced autophagy [129]. However, the further molecular mechanism under this crosstalk in these cell types remains unclear.
It is worth noting that, in a patient-based analysis, key components of Hh signaling and autophagy have been suggested as significant prognostic biomarkers. In a study of 108 gastric patients, immunohistochemical staining indicated that both Beclin1 and GLI2 are highly expressed in adjacent normal gastric mucosa samples (86.5% and 100%, respectively) and adenocarcinoma samples (60.2% and 72.2%, respectively) [130]. Higher expression of GLI2 was correlated with lower primary tumor stage (T1-2 vs. T3-4, p = 0.036), no lymphatic invasion (absent vs. present, p = 0.034), and no tumor recurrence (absent vs. present, p = 0.011). Similarly, increased expression of Beclin1 was correlated with favorable prognostic variables. Moreover, the expression of GLI2 was highly correlated with Beclin1 expression level in patient samples. Hence, this study postulated that autophagy may be related to Hh signaling in gastric cancer, and together, Beclin1 and GLI2 expression level is a possible prognostic biomarker. However, the interaction between HH signaling and autophagy has not been investigated in this study. Importantly, this study provided unexcepted evidence that higher GLI2 expression could be a favorable prognostic marker, and Beclin1 acted as a tumor suppressor in gastric cancer. This is opposed to our conventional impression that activation of Hh signaling is related to cancer initiation, progression, and metastasis, indicating the complex roles of Hh signaling and autophagy in cancers again.
5. Combined Targeting Hh Pathway and Autophagy: A Therapeutic Opportunity for Cancer Therapy
As discussed in preceding sections, current studies indicated that how Hh signaling affects autophagy depends on specific research conditions, and Hh signaling exerts negative regulation on autophagy in the majority of these studies. As shown in Table 4, in some cases, such as CML [99] and lung cancer [73,74], Hh inhibition could induce cytoprotective autophagy, indicating the benefits of the combination of Hh pathway inhibitors with autophagy inhibitors. In such cases, inducing autophagy seems to be a bypass or compensatory mechanism when Hh signaling is inhibited, whereas other studies, such as HCC [65] and cases of chondrosarcoma [81], implicated the cell-killing role of Hh-related autophagy, suggesting that autophagy activators may be helpful. Collectively, these results presented the complex roles of Hh-related autophagy in cancers and whether inhibiting or activating, Hh-related autophagy is not a “one-size-fits-all” paradigm. In fact, the context-dependent roles of autophagy activation in cancer therapy have attracted intense attention and raised hot debates. Both inhibiting and activating autophagy, therefore, have been proposed to improve the anticancer activity of many drugs. Fortunately, in the view of the forthcoming precision/personalized medicine, the context-dependent roles of autophagy will not be an obstacle for manipulating autophagy in cancer therapy, because patients will be well stratified into subpopulations based on some indicative biomarkers which correspond to different optimal treatment strategies [131]. In such a desirable scenario in the future, autophagy may become one of these indicative biomarkers.
To this end, much attention has been paid to evaluate the potential applications of autophagy proteins, including Beclin1, LC3B, ATG7, MAPK8IP1, and SH3GLB1, as prognostic biomarkers in several cancer types, such as breast cancer, colon cancer, melanoma, and glioma [132,133,134,135,136,137,138]. For example, Park et al. analyzed the correlation of Beclin1 expression and overall survival of 178 colon cancer patients who were receiving 5-fluorouracil therapy, and their results demonstrated that Beclin1 expression (hazard ratio was 1.82) could be a prognostic biomarker to guide optimal patient stratification [134]. LC3B has also been suggested as a prognostic biomarker for both relapse-free survival and OS in breast cancer based on an immunochemistry analysis of 229 breast cancer patient specimens [135]. These studies collectively indicated the exciting prospect of integrating autophagy as a biomarker in clinical applications. Nevertheless, the detection of almost all these biomarkers will heavily rely on tumor specimen or biopsy, which limits their practicability in clinical usage given that the autophagy in cancer progression and treatment is a dynamic process and thus needs dynamic monitoring. Therefore, the development of new biomarkers which can be monitored in blood or other bodily fluids using noninvasive detection methods is extremely significant, and will largely improve the applicability of monitoring autophagy in clinical scenarios.
Although the role of Hh-related autophagy seems to be confused currently, encouraging results from MYCN-amplified neuroblastoma and BCR-ABL mutation CML studies underscored the possibility of co-modulating Hh signaling and autophagy to overcome drug resistance in different cancer domains. Acquired drug resistance is emerging as the main challenge for achieving optimal clinical outcomes of SMO-targeted drugs. Currently proposed concepts mainly focus on developing drugs with new chemical properties, designing combination strategies that co-target multiple components of Hh signaling, and combined targeting of Hh signaling and other oncogenic pathways. Given the wide occurrence of Hh–autophagy crosstalk and its critical roles in cancer cells, we suggest that taking Hh–autophagy crosstalk into consideration may represent an alternative way.
However, only a few studies have attempted to delineate the underlying molecular mechanisms of Hh–autophagy association. Lack of mechanistic understanding of crosstalk between Hh signaling and autophagy limits the therapeutic applications. Hopefully, with the advances in the understanding of how Hh–autophagy crosstalk can be utilized towards an improve anticancer effect, the concept of combined targeting Hh signaling and autophagy will be validated in more preclinical or clinical studies, which will ultimately benefit cancer therapy.
6. Conclusions
Much evidence indicates that cancer progression and relapse may not be abolished by inhibiting the Hh signaling pathway alone, and redundant bypass and crosstalk with other pathways should be taken into consideration in future therapeutics development. Hh signaling regulates autophagy in various cell or animal models, and such crosstalk exhibits distinct pathological or pharmacological roles. In some cases, autophagy likely acts as a bypass or compensatory mechanism, which will be activated to support cancer cell growth when Hh is blocked. In these scenarios, co-targeting Hh signaling and autophagy represents a promising option to improve clinical outcomes of Hh-targeted agents and circumvent drug resistance. Some of these co-targeting attempts have shown encouraging results in in vitro or in vivo models. However, induction of autophagy could also be useful for Hh-targeted therapy in other cases, since activation of autophagy could lead to autophagic cell death, indicating that stimulating autophagy may result in favorable outcomes. Before we can harness the Hh–autophagy crosstalk to design improved anticancer strategies, considerable research efforts are needed to gain a deeper understanding of the underlying molecular mechanisms.
Author Contributions
X.Z. and D.J. wrote the paper.
Funding
This research was funded by the National Key Basic Research Program of China under grant 2015CB931800 and the National Natural Science Foundation of China under grant 81573332 and 81773620.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 3-MA | 3-Methyladenine |
| AKT | Protein kinase B |
| AR | Androgen receptor |
| ATG | Autophagy-related protein |
| BCC | Basal cell carcinoma |
| BCL | B-cell lymphoma |
| BCR-ABL | BCR-ABL fusion gene |
| BDMC | Bisdemethoxycurcumin |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
| CDK | Cyclin-dependent kinase |
| CML | Chronic myeloid leukemia |
| CQ | Chloroquine |
| DHH | Desert hedgehog |
| EGFR | Epidermal growth factor receptor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FOLFOX | A chemotherapy regimen that consists of folinic acid, fluorouracil, and oxaliplatin |
| GLI | Glioma-associated oncogene |
| GSK | Glycogen synthase kinase |
| HCC | Hepatocellular carcinoma |
| HCQ | Hydroxychloroquine |
| HDAC | Histone deacetylases |
| Hh | Hedgehog |
| IHH | Indian hedgehog |
| JAK | Janus kinase |
| KRAS | Kirsten rat sarcoma 2 viral oncogene homolog |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| MAPK | Mitogen-activated protein kinase |
| MB | Medulloblastoma |
| MEF | Mouse embryonic fibroblasts |
| MEK | Meiosis-specific serine/threonine-protein kinase |
| mTOR | Mammalian target of rapamycin |
| OGD | Oxygen-glucose deprivation |
| PD-1 | Programmed death-ligand 1 |
| PDGFR | Platelet-derived growth factor receptor |
| PERK | PKR-like endoplasmic reticulum kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| PTCH | Patched |
| RAF | Raf serine/threonine kinase |
| RBCC1 | RB1-inducible coiled-coil protein 1 |
| SHH | Sonic hedgehog |
| SMO | Smoothened |
| SUFU | Suppressor of fused |
| SMAD3 | Sma- and Mad-related Protein 3 |
| SRC | Src kinase |
| SUFU | Suppressor of fused |
| TGFβ | Transforming growth factor-β |
| ULK | Unc-51-like kinase |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| WNT | Wingless-related integration site |
| XELOX | A chemotherapy combination that combines capecitabine and oxaliplatin |
References
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004, 432, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Ji, X.; De La Cruz, L.K.; Thareja, S.; Wang, B. Strategies to target the Hedgehog signaling pathway for cancer therapy. Med. Res. Rev. 2018, 38, 870–913. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 1 July 2018).
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Caiping, S.; Qing, Z.; Xiaojing, W. Sonic hedgehog-Gli1 signals promote epithelial-mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med. Oncol. 2015, 32, 368. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Pedone, C.A.; del Valle, L.; Reiss, K.; Holland, E.C.; Fults, D.W. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene 2004, 23, 6156–6162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Rajurkar, M.; de Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Ingram, L.; Tolosa, E.J.; Vera, R.E.; Li, Q.; Kim, S.; Ma, Y.; Spyropoulos, D.D.; Beharry, Z.; Huang, J.; et al. Gli Transcription Factors Mediate the Oncogenic Transformation of Prostate Basal Cells Induced by a Kras-Androgen Receptor Axis. J. Biol. Chem. 2016, 291, 25749–25760. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.L.; Schumacher, D.; Staudte, S.; Steffen, A.; Haybaeck, J.; Keilholz, U.; Schweiger, C.; Golob-Schwarzl, N.; Mumberg, D.; Henderson, D.; et al. Non-Canonical Hedgehog Signaling Is a Positive Regulator of the WNT Pathway and Is Required for the Survival of Colon Cancer Stem Cells. Cell Rep. 2017, 21, 2813–2828. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Shah, A.S.; Moninger, T.O.; Ostedgaard, L.S.; Lu, L.; Tang, X.X.; Thornell, I.M.; Reznikov, L.R.; Ernst, S.E.; Karp, P.H.; et al. Motile cilia of human airway epithelia contain hedgehog signaling components that mediate noncanonical hedgehog signaling. Proc. Natl. Acad. Sci. USA 2018, 115, 1370–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blotta, S.; Jakubikova, J.; Calimeri, T.; Roccaro, A.M.; Amodio, N.; Azab, A.K.; Foresta, U.; Mitsiades, C.S.; Rossi, M.; Todoerti, K.; et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood 2012, 120, 5002–5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Hou, Y.C.; Huang, J.; Fang, J.Y.; Xiong, H. Itraconazole induces apoptosis and cell cycle arrest via inhibiting Hedgehog signaling in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 50. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.W.; Keysar, S.B.; Eagles, J.R.; Wang, G.; Glogowska, M.J.; McDermott, J.D.; Le, P.N.; Gao, D.; Ray, C.E.; Rochon, P.J.; et al. A pilot study of cetuximab and the hedgehog inhibitor IPI-926 in recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2016, 53, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybstein, M.D.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 2018, 20, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, Y.; Fan, J.; Zhao, H.; Xian, Z.; Sun, Y.; Wang, Z.; Wang, S.; Zhang, G.; Ju, D. Recombinant human arginase induced caspase-dependent apoptosis and autophagy in non-Hodgkin’s lymphoma cells. Cell Death Dis. 2013, 4, e840. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Ye, L.; Fan, J.; Li, Y.; Zeng, X.; Wang, Z.; Wang, S.; Zhang, G.; Yang, P.; Cao, Z.; et al. Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget 2015, 6, 3861–3873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, W.; Fan, J.; Wang, S.; Xian, Z.; Luan, J.; Li, Y.; Wang, Y.; Nan, Y.; Luo, M.; et al. Disrupting CD47-SIRPalpha axis alone or combined with autophagy depletion for the therapy of glioblastoma. Carcinogenesis 2018, 39, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, S.; Wang, Z.; Qian, X.; Fan, J.; Zeng, X.; Sun, Y.; Song, P.; Feng, M.; Ju, D. Cationic poly(amidoamine) dendrimers induced cyto-protective autophagy in hepatocellular carcinoma cells. Nanotechnology 2014, 25, 365101. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Munz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.Y.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Marinković, M.; Šprung, M.; Buljubašić, M.; Novak, I. Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.; Kanehisa, A.; Martins, I.; Senovilla, L.; Chargari, C.; Dugue, D.; Marino, G.; Kepp, O.; Michaud, M.; Perfettini, J.L.; et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 2014, 21, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.T.; Pellegatti, P.; Shen, S.S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Brechbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: Implications for cancer therapy. Expert Rev. Mol. Med. 2015, 17, e5. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Stecca, B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin. Cancer Biol. 2015, 35, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Menzies, F.M.; Chang, Y.Y.; Simecek, N.; Neufeld, T.P.; Rubinsztein, D.C. The Hedgehog signalling pathway regulates autophagy. Nat. Commun. 2012, 3, 1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.A.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del, C.C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the HedgehogGLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Deng, L.; Chen, Q.; Wang, Y.; Xu, R.; Shi, C.; Shao, J.; Hu, G.; Gao, M.; Rao, H.; et al. Inhibition of Hedgehog signaling pathway impedes cancer cell proliferation by promotion of autophagy. Eur. J. Cell Biol. 2015, 94, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013, 58, 995–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wei, S.; Zhao, Y.; Shi, C.; Liu, P.; Zhang, C.; Lei, Y.; Zhang, B.; Bai, B.; Huang, Y.; et al. Anti-proliferation of breast cancer cells with itraconazole: Hedgehog pathway inhibition induces apoptosis and autophagic cell death. Cancer Lett. 2017, 385, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Villegas, V.E.; Rondon-Lagos, M.; Annaratone, L.; Castellano, I.; Grismaldo, A.; Sapino, A.; Zaphiropoulos, P.G. Tamoxifen Treatment of Breast Cancer Cells: Impact on Hedgehog/GLI1 Signaling. Int. J. Mol. Sci. 2016, 17, 308. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer 2016, 99, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. BioMed Res. Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Yang, H.P.; Zhou, X.D.; Wang, H.J.; Gong, L.; Tang, C.L. Autophagy Accompanied with Bisdemethoxycurcumin-induced Apoptosis in Non-small Cell Lung Cancer Cells. Biomed. Environ. Sci. 2015, 28, 105–115. [Google Scholar] [PubMed]
- Fan, J.; Ju, D.; Li, Y.; Wang, S.; Wang, Z. A novel approach to overcome non-small-cell lung cancer: Co-inhibition of autophagy and Hedgehog pathway. Ann. Oncol. 2015, 26 (Suppl. 7), vii106–vii151. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, X.; Wang, S.; Chen, W.; Li, Y.; Zeng, X.; Wang, Y.; Luan, J.; Li, L.; Sun, X.; et al. Regulating autophagy facilitated therapeutic efficacy of Sonic hedgehog pathway inhibition on lung adenocarcinoma through Gli 2 suppression and ROS production. Cell Death Dis. 2018. under review. [Google Scholar]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Cardinali, C.; Santoni, M.; Gismondi, A.; Santoni, G. Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an Hedgehog-dependent manner. Oncotarget 2016, 7, 50180–50194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; An, Y.; Wang, X.; Zha, W.; Li, X. Inhibition of the Hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol. Rep. 2014, 31, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Campbell, V.T.; Nadesan, P.; Ali, S.A.; Wang, C.Y.; Whetstone, H.; Poon, R.; Wei, Q.; Keilty, J.; Proctor, J.; Wang, L.W.; et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Mol. Cancer Ther. 2014, 13, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Guo, W.; Ren, T.; Liang, W.; Zhou, W.; Lu, Q.; Jiao, G.; Yan, T. Gli1 inhibition suppressed cell growth and cell cycle progression and induced apoptosis as well as autophagy depending on ERK1/2 activity in human chondrosarcoma cells. Cell Death Dis. 2014, 5, e979. [Google Scholar] [CrossRef] [PubMed]
- Jiao, G.; Ren, T.; Guo, W.; Ren, C.; Yang, K. Arsenic trioxide inhibits growth of human chondrosarcoma cells through G2/M arrest and apoptosis as well as autophagy. Tumour Biol. 2015, 36, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Zibat, A.; Missiaglia, E.; Rosenberger, A.; Pritchard-Jones, K.; Shipley, J.; Hahn, H.; Fulda, S. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene 2010, 29, 6323–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitzki, F.; Cuvelier, N.; Drager, J.; Schneider, A.; Braun, T.; Hahn, H. Hedgehog/Patched-associated rhabdomyosarcoma formation from delta1-expressing mesodermal cells. Oncogene 2016, 35, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Ridzewski, R.; Rettberg, D.; Dittmann, K.; Cuvelier, N.; Fulda, S.; Hahn, H. Hedgehog Inhibitors in Rhabdomyosarcoma: A Comparison of Four Compounds and Responsiveness of Four Cell Lines. Front. Oncol. 2015, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Shohet, J.M. Neuroblastoma: Molecular pathogenesis and therapy. Annu. Rev. Med. 2015, 66, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.X.; Huang, C.; Gao, C.; Xing, T.Y.; Liu, S.G.; Li, X.J.; Zhao, Q.; Wang, X.S.; Zhao, W.; Jin, M.; et al. MYCN amplification predicts poor prognosis based on interphase fluorescence in situ hybridization analysis of bone marrow cells in bone marrow metastases of neuroblastoma. Cancer Cell. Int. 2017, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.S.; Wang, X.W.; Wan, J.H.; Li, T.; Gong, X.Y.; Zhang, K.; Yi, L.; Xiang, Z.H.; Xu, M.H.; Cui, H.J. Sonic Hedgehog pathway is essential for neuroblastoma cell proliferation and tumor growth. Mol. Cell Biochem. 2012, 364, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, S.; Huang, J.; Chen, S.; Zhang, Z.; Xu, M. Inhibition of autophagy potentiates the efficacy of Gli inhibitor GANT-61 in MYCN-amplified neuroblastoma cells. BMC Cancer 2014, 14, 768. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, S.; Tian, R.; Chen, J.; Gao, H.; Xie, C.; Shan, Y.; Zhang, Z.; Gu, S.; Xu, M. The protective autophagy activated by GANT-61 in MYCN amplified neuroblastoma cells is mediated by PERK. Oncotarget 2018, 9, 14413–14427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, K.W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.K.; Kling, M.J.; Coulter, D.W.; McGuire, T.R.; Ray, S.; Kesherwani, V.; Joshi, S.S.; Sharp, J.G. Improved therapy for medulloblastoma: Targeting hedgehog and PI3K-mTOR signaling pathways in combination with chemotherapy. Oncotarget 2018, 9, 16619–16633. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.; Rothe, K.; Woolfson, A.; Jiang, X. SMO and GLI2 are key regulators mediating resistance of CML stem/progenitor cells to tyrosine kinase inhibitors. Exp. Hematol. 2017, 53, S62. [Google Scholar] [CrossRef]
- Liu, X.; Rothe, K.; Yen, R.; Fruhstorfer, C.; Maetzig, T.; Chen, M.; Forrest, D.L.; Humphries, R.K.; Jiang, X. A novel AHI-1-BCR-ABL-DNM2 complex regulates leukemic properties of primitive CML cells through enhanced cellular endocytosis and ROS-mediated autophagy. Leukemia 2017, 31, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.A.; Copland, M. Targeting hedgehog in hematologic malignancy. Blood 2012, 119, 2196–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Zhao, H.; Li, Y.; Fan, J.; Sun, Y.; Wang, S.; Wang, Z.; Song, P.; Ju, D. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABL-positive chronic myeloid leukemia. Autophagy 2015, 11, 355–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Zeng, X.; Li, Y.; Wang, S.; Yang, P.; Cao, Z.; Wang, Z.; Song, P.; Mei, X.; Ju, D. A novel therapeutic approach against B-cell non-Hodgkin’s lymphoma through co-inhibition of Hedgehog signaling pathway and autophagy. Tumour Biol. 2016, 37, 7305–7314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, Z.; Sattva, S.N.; Jorge, R.; Nami, M. Hedgehog inhibitors selectively target cell migration and adhesion of mantle cell lymphoma in bone marrow microenvironment. Oncotarget 2016, 7, 14350–14365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Wu, G.; Zeng, R.; Wang, J.; Cai, R.; Ho, J.C.-M.; Zhang, J.; Zheng, Y. Chromium contributes to human bronchial epithelial cell carcinogenesis by activating Gli2 and inhibiting autophagy. Toxicol. Res. 2017, 6, 324–332. [Google Scholar] [CrossRef]
- Granato, M.; Zompetta, C.; Vescarelli, E.; Rizzello, C.; Cardi, A.; Valia, S.; Antonelli, G.; Marchese, C.; Torrisi, M.R.; Faggioni, A.; et al. HCV derived from sera of HCV-infected patients induces pro-fibrotic effects in human primary fibroblasts by activating GLI2. Sci. Rep. 2016, 6, 30649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Y.; He, Y.J.; Yang, Q.H.; Huang, W.; Liu, Z.H.; Ye, G.R.; Tang, S.H.; Shu, J.C. Induction of autophagy and apoptosis by miR-148a through the sonic hedgehog signaling pathway in hepatic stellate cells. Am. J. Cancer Res. 2015, 5, 2569–2589. [Google Scholar] [PubMed]
- Li, J.; Zhang, L.; Xia, Q.; Fu, J.; Zhou, Z.; Lin, F. Hedgehog signaling inhibitor GANT61 induces endoplasmic reticulum stress-mediated protective autophagy in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2017, 493, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.N.; Liu, X.J.; Wu, J. Palmitic acid elicits hepatic stellate cell activation through inflammasomes and hedgehog signaling. Life Sci. 2017, 176, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.K.; Baena, A.; Ng, T.W.; Venkataswamy, M.M.; Kennedy, S.C.; Kunnath-Velayudhan, S.; Carreno, L.J.; Xu, J.; Chan, J.; Larsen, M.H.; et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 2016, 1, 16133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, P.A.; Furuta, K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat. Rev. Immunol. 2015, 15, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483. [Google Scholar] [CrossRef] [PubMed]
- Holla, S.; Kurowska-Stolarska, M.; Bayry, J.; Balaji, K.N. Selective inhibition of IFNG-induced autophagy by Mir155- and Mir31-responsive WNT5A and SHH signaling. Autophagy 2014, 10, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Milla, L.A.; Gonzalez-Ramirez, C.N.; Palma, V. Sonic Hedgehog in cancer stem cells: A novel link with autophagy. Biol. Res. 2012, 45, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, A.E.; Fernandez-Barrena, M.G.; Almada, L.L.; Mills, L.D.; Elsawa, S.F.; Lund, G.; Ropolo, A.; Molejon, M.I.; Vaccaro, M.I.; Fernandez-Zapico, M.E. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J. Biol. Chem. 2012, 287, 25325–25334. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venugopal, C.; Manoranjan, B.; McFarlane, N.; O’Farrell, E.; Nolte, S.; Gunnarsson, T.; Hollenberg, R.; Kwiecien, J.; Northcott, P.; et al. Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene 2012, 31, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Po, A.; Ferretti, E.; Miele, E.; de Smaele, E.; Paganelli, A.; Canettieri, G.; Coni, S.; di Marcotullio, L.; Biffoni, M.; Massimi, L.; et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010, 29, 2646–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Bauvy, C.; Tonelli, G.; Yue, W.; Delomenie, C.; Nicolas, V.; Zhu, Y.; Domergue, V.; Marin-Esteban, V.; Tharinger, H.; et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013, 32, 2261–2272, 2272e1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell. Int. 2013, 13, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak-Kroizman, T.R.; Hostetter, G.; Posner, R.; Aziz, M.; Hu, C.; Demeure, M.J.; Von Hoff, D.; Hingorani, S.R.; Palculict, T.B.; Izzo, J.; et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 2013, 73, 3235–3247. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Li, J.J.; Li, Y.N.; Singh, P.; Cao, L.; Xu, L.J.; Li, D.; Wang, Y.B.; Xie, Z.P.; Gui, Y.; et al. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. Am. J. Physiol.-Heart C 2012, 303, H1319–H1331. [Google Scholar] [CrossRef] [PubMed]
- Gagne-Sansfacon, J.; Allaire, J.M.; Jones, C.; Boudreau, F.; Perreault, N. Loss of Sonic hedgehog leads to alterations in intestinal secretory cell maturation and autophagy. PLoS ONE 2014, 9, e98751. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yang, Y.; Qin, Y.; He, Y.H.; Chen, K.X.; Zhu, J.W.; Zhang, G.P.; Luo, J.D. AMP-activated protein kinase-dependent autophagy mediated the protective effect of sonic hedgehog pathway on oxygen glucose deprivation-induced injury of cardiomyocytes. Biochem. Biophys. Res. Commun. 2015, 457, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Petralia, R.S.; Schwartz, C.M.; Wang, Y.X.; Kawamoto, E.M.; Mattson, M.P.; Yao, P.J. Sonic hedgehog promotes autophagy in hippocampal neurons. Biol. Open 2013, 2, 499–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, K.Y.; Kim, G.Y.; Lim, S.J.; Sung, J.Y.; Kim, Y.W.; Park, Y.K.; Lee, J.; Choi, H.S. Autophagy is related to the hedgehog signaling pathway in human gastric adenocarcinoma: Prognostic significance of Beclin-1 and Gli2 expression in human gastric adenocarcinoma. Pathol. Res. Pract. 2015, 211, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, C.B.; DeVorkin, L.; Bosc, D.; Rothe, K.; Singh, J.; Bally, M.; Jiang, X.; Young, R.N.; Lum, J.J.; Gorski, S.M. Precision autophagy: Will the next wave of selective autophagy markers and specific autophagy inhibitors feed clinical pipelines? Autophagy 2015, 11, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Bortnik, S.; Gorski, S.M. Clinical Applications of Autophagy Proteins in Cancer: From Potential Targets to Biomarkers. Int. J. Mol. Sci. 2017, 18, 1496. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Huang, S.; Wu, T.T.; Foster, N.R.; Sinicrope, F.A. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol. Ther. 2013, 14, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Jiang, Y.Z.; Huang, L.; Zhou, R.J.; Yu, K.D.; Liu, Y.; Shao, Z.M. The residual tumor autophagy marker LC3B serves as a prognostic marker in local advanced breast cancer after neoadjuvant chemotherapy. Clin. Cancer Res. 2013, 19, 6853–6862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Hang, D.; Jiang, Y.; Chen, J.; Han, J.; Zhou, W.; Jin, G.; Ma, H.; Dai, J. Evaluation of genetic variants in autophagy pathway genes as prognostic biomarkers for breast cancer. Gene 2017, 627, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.Y.; Ellis, R.A.; Lovat, P.E. Prognostic Impact of Autophagy Biomarkers for Cutaneous Melanoma. Front. Oncol. 2016, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, X.; Wang, N.; Wang, J.; Cao, Y.; Wang, T.; Zhou, X.; Jiao, Y.; Yang, L.; Wang, X.; et al. Autophagy-related gene expression is an independent prognostic indicator of glioma. Oncotarget 2017, 8, 60987–61000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Crosstalk between Hh signaling and autophagy based on current evidence. Green arrow: upregulate; Red line: inhibit; Box with dash line: both GLI1 and GLI2 were involved in Bnip3 inhibition. Gray arrows: dissociation of Bcl-2/Beclin1 complex.
Figure 1.
Crosstalk between Hh signaling and autophagy based on current evidence. Green arrow: upregulate; Red line: inhibit; Box with dash line: both GLI1 and GLI2 were involved in Bnip3 inhibition. Gray arrows: dissociation of Bcl-2/Beclin1 complex.


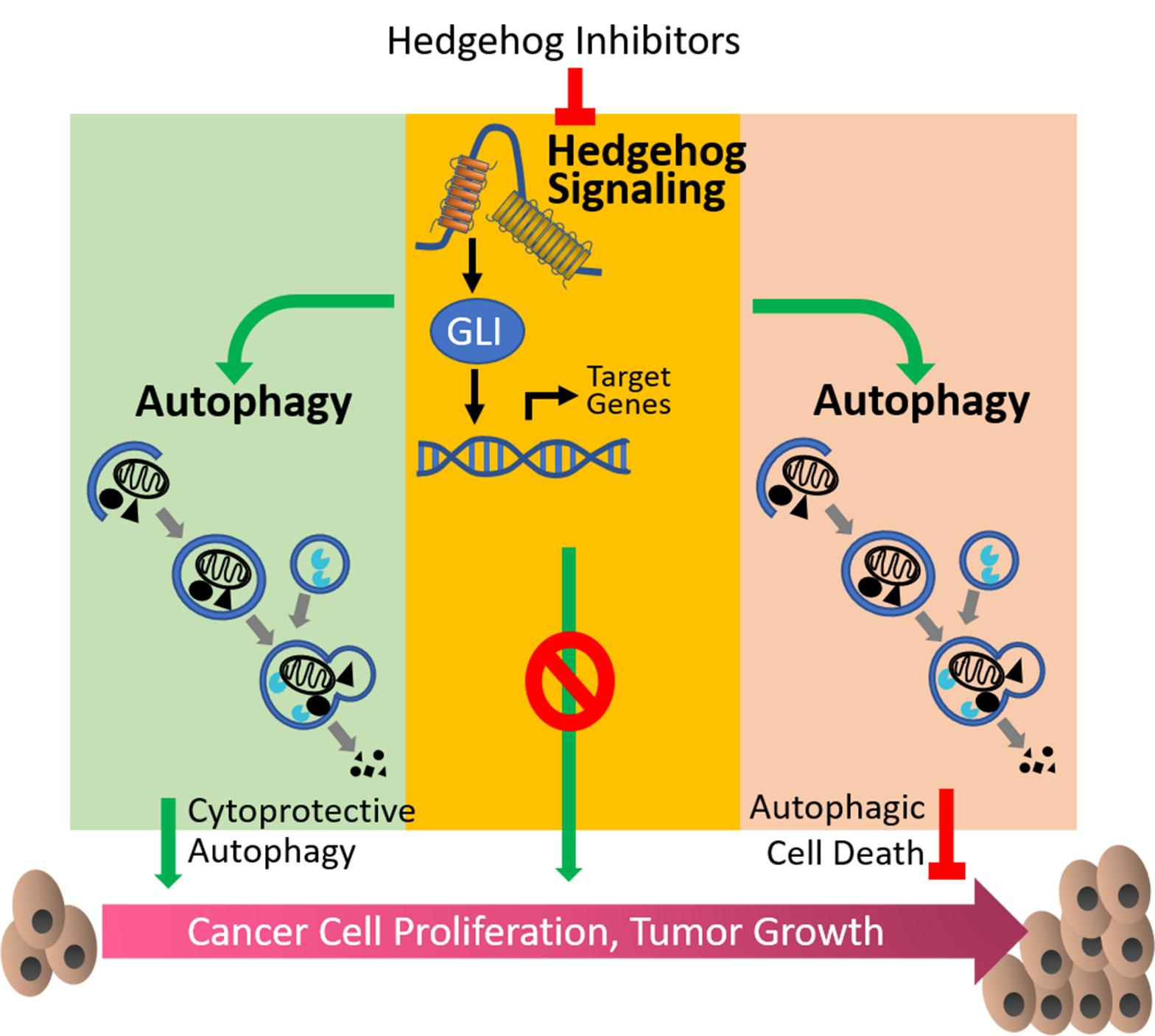{kind=link}
{kind=link}
Table 1.
Clinical trials of combination of Hedgehog (Hh) inhibitors and other targeted therapeutics. (Data from ClinicalTrials.gov [17]).
Table 1.
Clinical trials of combination of Hedgehog (Hh) inhibitors and other targeted therapeutics. (Data from ClinicalTrials.gov [17]).
| Combined Drugs | Combined Targets | Disease Indications | Clinical Trial Stage | Clinical Trial Status | ClinicalTrials.gov Accession |
|---|---|---|---|---|---|
| Sonidegib + Ruxolitinib | SMO + JAK1/2 | Myelofibrosis | Phase 1/2 | Active, not recruiting | NCT01787552 |
| Sonidegib + Buparlisib | SMO + PI3K | Advanced Solid Tumors | Phase 1 | Completed | NCT01576666 |
| Sonidegib + Ribociclib | SMO + CDK4/6 | Refractory or Recurrent Medulloblastoma | Phase 1 | Recruiting | NCT03434262 |
| Vismodegib + RO4929097 | SMO + Gamma-Secretase | Advanced or Metastatic Sarcoma | Phase 1/2 | Completed | NCT01154452 |
| Vismodegib + Bevacizumab | SMO + VEGF | Metastatic Colorectal Cancer | Phase 2 | Completed | NCT00636610 |
| Vismodegib + Sirolimus | SMO + mTOR | Metastatic Pancreatic Cancer | Phase 1 | Suspended | NCT01537107 |
| Vismodegib + Pembrolizumab | SMO + PD-1 | Metastatic or Unresectable Basal Cell Skin Cancer | Phase 1/2 | Active, not recruiting | NCT02690948 |
| Vismodegib + Erlotinib + Gemcitabine | SMO + EGFR | Metastatic Pancreatic Cancer | Phase 1 | Active, not recruiting | NCT00878163 |
| Saridegib + Cetuximab | SMO + EGFR | Recurrent Head and Neck Cancer | Phase 1 | Completed | NCT01255800 |
| Sonidegib + Nilotinib | SMO + BCR-ABL | Chronic or Accelerated Phase Myeloid Leukemia | Phase 1 | Completed | NCT01456676 |
| BMS-833923 + Dasatinib | SMO + BCR-ABL/SRC | Chronic Myeloid Leukemia | Phase 1/2 | Completed | NCT01218477 |
| BMS-833923 + Dasatinib | SMO + BCR-ABL/SRC | Chronic Myeloid Leukemia | Phase 2 | Terminated | NCT01357655 |
| ATO + Icotinib | GLI + EGFR | EGFR-TKI Resistant Non-Small Cell Lung Cancer | Phase 1 | Unknown | NCT02066870 |
| ATO + Gleevec | GLI + BCR-ABL | CML Who Fail Gleevec | Phase 2 | Completed | NCT00250042 |
| ATO + GO | GLI + CD33 | Advanced Myelodysplastic Syndromes | Phase 2 | Completed | NCT00274781 |
| ATO + GO + ATRA | GLI + CD33 | Acute Promyelocytic Leukemia | Phase 2 | Recruiting | NCT01409161 |
Abbreviations: SMO: smoothened; JAK: Janus kinase; PI3K: phosphatidylinositol 3-kinase; CDK: cyclin-dependent kinase; VEGF: vascular endothelial growth factor; mTOR: mammalian target of rapamycin; PD-1: programmed death-ligand 1; EGFR: epidermal growth factor receptor; BCR-ABL: BCR-ABL fusion gene; SRC: Src kinase; GLI: glioma-associated oncogene; SRC: Src kinase family; GO: gemtuzumab ozogamicin; ATRA: all-trans retinoic acid; ATO: arsenic trioxide.
Table 2.
Clinical trials of combination of autophagy inhibitors and other targeted therapeutics. (Data from ClinicalTrials.gov [17]).
Table 2.
Clinical trials of combination of autophagy inhibitors and other targeted therapeutics. (Data from ClinicalTrials.gov [17]).
| Combined Drugs | Combined Targets | Disease Indications | Clinical Trial Stage | Clinical Trial Status | ClinicalTrials.gov Accession |
|---|---|---|---|---|---|
| Vorinostat + HCQ | HDAC + Autophagy | Advanced Solid Tumors | Phase 1 | Recruiting | NCT01023737 |
| Vorinostat + HCQ | HDAC + Autophagy | Advanced Cancer | Phase 1 | Active, not recruiting | NCT01266057 |
| Vorinostat + HCQ | HDAC + Autophagy | Colorectal Cancer | Phase 2 | Recruiting | NCT02316340 |
| Sorafenib + HCQ | VEGFR/PDGFR/RAF + Autophagy | Refractory or Relapsed Solid Tumors | Phase 1 | Completed | NCT01634893 |
| Sorafenib + HCQ | VEGFR/PDGFR/RAF + Autophagy | Hepatocellular Cancer | Phase 2 | Recruiting | NCT03037437 |
| RAD001 + HCQ | MTOR + Autophagy | Renal Cell Carcinoma | Phase 1/2 | Active, not recruiting | NCT01510119 |
| MK2206 + HCQ | AKT + Autophagy | Advanced Solid Tumors, Melanoma, Prostate or Kidney Cancer | Phase 1 | Active, not recruiting | NCT01480154 |
| Trametinib + HCQ | MEK1/2 + Autophagy | Advanced BRAF Mutant Melanoma | Phase 1/2 | Unknown | NCT02257424 |
| FOLFOX6/XELOX + Bevacizumab + HCQ | VEGF + Autophagy | Metastatic Colorectal Cancer | Phase 2 | Completed | NCT01006369 |
| Abiraterone + Navitoclax + HCQ | BCL-2/BCL-xL/BCL-w + Autophagy | Progressive Metastatic Castrate Refractory Prostate Cancer | Phase 2 | Terminated | NCT01828476 |
Abbreviations: HCQ: hydroxychloroquine; HDAC: histone deacetylases; VEGFR: vascular endothelial growth factor receptor; PDGFR: platelet-derived growth factor receptor; FOLFOX: a chemotherapy regimen that consists of folinic acid, fluorouracil, and oxaliplatin; XELOX: a chemotherapy combination that combines capecitabine and oxaliplatin; BCL: B-cell lymphoma.
Table 3.
Hh pathway modulators discussed in the current context.
| Chemical Modulator Name | Target (Mode of Action) | Clinical Indication Examples | Maximum Developmental Stage |
|---|---|---|---|
| Vismodegib (GDC-0449) | SMO (Antagonist) | Approved: BCC Clinical Trials: Pancreatic ductal adenocarcinoma (NCT01096732); MB (NCT00939484, NCT01239316, NCT00822458); Advanced/metastatic sarcoma (NCT01154452); Ovarian cancer (NCT00739661, NCT00959647); AML (NCT01880437); | Approved |
| Sonidegib (Erismodegib, NVP-LDE225, LDE-225) | SMO (Antagonist) | Approved: BCC Clinical Trials: Prostate Cancer (NCT02111187); Pancreatic Adenocarcinoma (NCT01431794); Multiple Myeloma (NCT02254551, NCT02086552); Ovarian Cancer (NCT02195973); Breast Cancer (NCT01757327); Small Cell Lung Cancer (NCT01579929); | Approved |
| Saridegib (IPI-926, Patidegib) | SMO (Antagonist) | Clinical Trials: Solid Tumors (NCT00761696); Myelofibrosis (NCT01371617); Chondrosarcoma (NCT01310816); Metastatic Pancreatic Cancer (NCT01130142); Gorlin Syndrome (NCT02762084); | Phase 2 |
| Arsenic Trioxide (ATO) | GLI (Antagonist) | Approved: Acute promyelocytic leukemia (APL) Clinical Trials: Non-Small Cell Lung Cancer (NCT00075426); CML (NCT00250042); AML (NCT00005795); Myelodysplastic Syndrome (NCT00225992); | Approved 1 |
| Itraconazole | SMO (Antagonist) | Clinical Trials: BCC (NCT01108094); | Approved 2 |
| Cyclopamine | SMO (Antagonist) | Indication Evidence From In Vivo Studies: Small Cell Lung Cancer [52]; Glioblastoma [53]; CML [54]; Medulloblastoma [55]; Prostate [56]; Digestive tract tumors [57]; Pancreatic cancer [58]; | Experimental Stage |
| GANT61 | GLI (Antagonist) | Indication Evidence From In Vivo Studies: Pancreatic cancer [59]; Breast cancer [60]; Prostate Cancer [61] | Experimental Stage |
| Glabrescione B (GlaB) | GLI1 3 (Antagonist) | Indication Evidence From In Vivo Studies: MB [62] | Experimental Stage |
| SAG | SMO (Agonist) | Indication Evidence From In Vivo Studies: Pancreatic cancer [63] 4 | Experimental Stage |
| Purmorphamine | SMO (Agonist) | -- | -- |
1 ATO has been approved by US FDA for APL but was not based on a mechanism of Hh inhibition; 2 Itraconazole has been approved by US FDA for antifungal purpose, while recent studies revealed its Hh inhibitory effect; 3 GlaB achieves inhibitory effect on GLI1 activity through binding to GLI1 zinc finger so that impairs its interaction with DNA; 4 Activation of Hh signaling by SAG in pancreatic stroma to restrain tumorigenesis and progression.
Table 4.
A brief summary of studies that combined targeting Hh pathway and autophagy.
| Cell Lines Used (Related Diseases) | Role of Hh-Related Autophagy and Supporting Evidence | Therapeutic Implications | References |
|---|---|---|---|
| H4 (Glioma), ES2 (Ovarian cancer), MKN45 (Gastric cancer), HT29 (Colon cancer) | Role: Autophagic cell death; Evidence: Inhibiting autophagy by 3-MA or knockout ATG5 partially rescued HH inhibition-induced cell proliferation. | Hh inhibitor + autophagy inducer | [64] |
| MCF-7, SKBR-3 (Breast cancer) | Role: Autophagic cell death Evidence: Inhibition of autophagy by 3-MA or ATG5 silencing impedes itraconazole-induced cell death. | Hh inhibitors + autophagy inducer | [66] |
| A549, NCI-H1975 (Lung cancer) | Role: Cytoprotective; Evidence: Inhibiting autophagy by inhibitors CQ or ATG5 or ATG7 siRNA strengthened vismodegib-induced cytotoxicity in cell lines. Co-administration of vismodegib-induced tumor-shrinkage in xenograft mouse model. | Hh inhibitor + autophagy inhibitor | [73,74] |
| HC-a, SW1353, JJ012 (Chondrosarcoma) | Role: Autophagic cell death; Evidence: Inhibiting autophagy by inhibitors (3-MA or CQ) or gene knockdown (ATG7 or Beclin1 siRNA) prevented GLI1 inhibition-induced cell death. | Hh inhibitor + autophagy inducer | [81] |
| Huh7, Hep3B, HepG2 (Liver cancer) | Role: Autophagic cell death; Evidence: inhibition autophagy by 3-MA or Beclin1 siRNA hampered GANT61-induced apoptosis and cytotoxicity; 3-MA co-administration weakened the tumor-shrinkage induced by GANT61 in Huh7 xenograft mouse model. | Hh inhibitor + autophagy inducer | [65] |
| MYCN-amplified NBL-W-S and SK-N-BE cell lines (neuroblastoma) | Role: Cytoprotective; Evidence: Inhibiting autophagy by 3-MA or gene knockdown (ATG5 or ATG7 siRNA) enhanced GANT61-induced apoptosis. | Hh inhibitor + autophagy inhibitor | [90] |
| K562, BaF3-BCR-ABLWT, BaF3-BCR-ABLY253F, BaF3-BCR-ABLT315I (drug resistant CML) | Role: Cytoprotective; Evidence: Inhibiting autophagy by CQ or gene knockdown (ATG5 or ATG7 siRNA) enhanced vismodegib-induced apoptosis. | Hh inhibitor + autophagy inhibitor | [99] |
| Raji (non-Hodgkin’s lymphoma) | Role: Cytoprotective; Evidence: Inhibiting autophagy by inhibitors (CQ or bafilomycin A) or ATG5 siRNA enhanced vismodegib-induced apoptosis. | Hh inhibitor + autophagy inhibitor | [100] |
| SP53 (Mantle cell lymphoma), Jeko, REC1, Pt1, Pt2 | Role: Cytoprotective; Evidence: Combination with 3-MA significantly increased LDE255-induced cytotoxicity. | Hh inhibitor + autophagy inhibitor | [101] |
| Hepatic stellate cell line LX-2 (Liver fibrosis) | Role: Cytoprotective Evidence: inhibiting autophagy with 3-MA or CQ can enhance GANT61-induced cytotoxicity. | Hh inhibitors + autophagy inhibitor | [106] |
| CFPAC-1 (pancreatic cancer) | Role: Autophagic cell death; Evidence: Inhibiting autophagy by 3-MA reversed GANT61-induced cytotoxicity in cell lines and anticancer effect in in vivo mouse model. | Hh inhibitor + autophagy inducer | [76] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zeng, X.; Ju, D. Hedgehog Signaling Pathway and Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 2279. https://doi.org/10.3390/ijms19082279
AMA Style
Zeng X, Ju D. Hedgehog Signaling Pathway and Autophagy in Cancer. International Journal of Molecular Sciences. 2018; 19(8):2279. https://doi.org/10.3390/ijms19082279
Chicago/Turabian StyleZeng, Xian, and Dianwen Ju. 2018. "Hedgehog Signaling Pathway and Autophagy in Cancer" International Journal of Molecular Sciences 19, no. 8: 2279. https://doi.org/10.3390/ijms19082279
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.