Onset and Progression of Human Osteoarthritis—Can Growth Factors, Inflammatory Cytokines, or Differential miRNA Expression Concomitantly Induce Proliferation, ECM Degradation, and Inflammation in Articular Cartilage?


Abstract
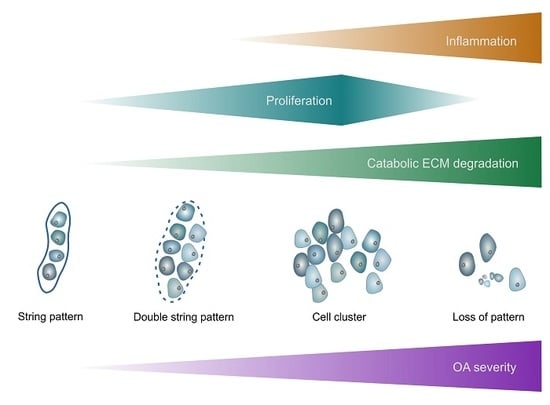
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Fibroblast Growth Factor 2 Signaling
3. Transforming Growth Factor β Signaling
4. Additional Growth Factor Signaling Pathways in Human Adult AC
4.1. WNT Signaling
4.2. Hedgehog Signaling
4.3. Bone Morphogenetic Protein Signaling
4.4. NOTCH Signaling
4.5. Insulin-Like Growth Factor Signaling
4.6. Vascular Endothelial Growth Factor Signaling
5. Inflammatory Cytokine Signaling
6. MiRNA Regulation of Fibroblast Growth Factor 2, Transforming Growth Factor β and Inflammatory Cytokine Signaling Pathways
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| ABCA1 | ATP-binding cassette transporter A1 |
| AC | articular cartilage |
| ACAN | Aggrecan |
| ACVRL1 | activin A receptor like type 1 |
| ADAM17 | A disintegrin and metalloproteinase domain-containing protein 17 |
| ADAMTS | A disintegrin and metalloproteinase with thrombospondin motifs |
| ADPN | adiponectin |
| ALK | activin receptor-like kinase |
| AML | acute myeloid leukemia |
| AP-1 | activator protein 1 |
| APOA1 | apolipoprotein A1 |
| ASPN | asporin |
| BMI | body mass index |
| BMP | bone morphogenetic protein |
| CaMK2 | calmodulin-dependent protein kinase 2 |
| CCL2 | C-C motif chemokine ligand 2 |
| CCDN1 | cyclin D1 |
| CDK6 | cyclin dependent kinase 6 |
| cFOS | cellular oncogene Fos |
| CIR | cartilage injury-related |
| COL2A1 | collagen type II α 1 chain |
| COL4A2 | collagen type IV α 2 chain |
| COL10A1 | collagen type X α 1 chain |
| CREB3L1 | CAMP Responsive Element Binding Protein 3 Like 1 |
| CSE | cystathionine-γ-lyase |
| CTGF | connective tissue growth factor |
| CYR61 | cysteine-rich angiogenic inducer 61 |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 |
| DKK1 | dickkopf WNT signaling pathway inhibitor 1 |
| DKK3 | dickkopf WNT signaling pathway inhibitor 3 |
| DLL1 | Delta-like 1 |
| DZ | deep zone |
| ECM | extracellular matrix |
| ELF3 | E74-like factor 3 |
| ELK1 | ETS-domain protein ELK1 |
| ERG | ETS-related gene |
| ERK | extracellular signal-regulated kinase |
| ETS | avian erythroblastosis virus E26 oncogene homolog |
| FGF2 | fibroblast growth factor 2 |
| FGFBP1 | fibroblast growth factor binding protein 1 |
| FGFR | fibroblast growth factor receptor |
| FLT-1 | Fms related tyrosine kinase 1 |
| FLT-4 | Fms related tyrosine kinase 4 |
| FOSB | FBJ murine osteosarcoma viral oncogene homolog B |
| FRZB | frizzled related protein |
| GDF5 | growth differentiation factor 5 |
| GLI1 | glioma-associated oncogene homolog 1 |
| HDAC4 | histone deacetylase 4 |
| HES | hairy and enhancer of split |
| HES1 | hairy and enhancer of split 1 |
| HES5 | hairy and enhancer of split 5 |
| Hh | Hedgehog |
| HHIP | hedgehog interacting protein |
| HIF-1α | hypoxia-inducible factor-1α |
| HMGA1 | high mobility group protein A1 |
| HSPG | heparan sulfate proteoglycan |
| IGF | insulin like growth factor |
| IHH | indian hedgehog |
| IL | interleukin |
| IL-1β | IL-1 beta |
| IL-1R | IL-1 receptor |
| IL-1Ra | IL-1 receptor antagonist |
| IRAK1 | IL-1 receptor-associated kinase 1 |
| LncRNA | long non-coding RNA |
| I-TAC | interferon-inducible T-cell alpha chemoattractant |
| JAG1 | jagged 1 |
| JAK | janus kinase |
| JNK | JUN N-terminal kinase |
| KDR | kinase insert domain receptor |
| KPNA3 | karyopherin subunit alpha 3 |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| MALT1 | mucosa-associated lymphoid tissue lymphoma translocation protein 1 |
| MAPK | mitogen activated protein kinase |
| MAP3K7 | mitogen-activated protein kinase kinase kinase 7 |
| MCP-1 | monocyte chemotactic protein 1 |
| MCPIP-1 | monocyte chemoattractant protein-induced protein 1 |
| MIG | monokine induced by interferon-gamma |
| miRNA | Micro RNA |
| MMP | matrix metalloproteinase |
| MSPC | mesenchymal stem and progenitor cells |
| Matn3 | matrilin 3 |
| Mtf-1 | metal regulatory transcription factor-1 |
| MZ | middle zone |
| NFAT5 | CaMK2 nuclear factor of activated T-cells 5, tonicity-responsive |
| NFATC2 | nuclear factor of activated T-cells 2 |
| NF-κB | nuclear factor kappa B |
| NOTCH | notch homolog |
| NRAS | neuroblastoma RAS viral oncogene homolog |
| OA | osteoarthritis |
| OSM | oncostatin M |
| PBMC | peripheral blood mononuclear cells |
| PCM | pericellular matrix |
| PI3K | phosphatidylinositol 3-kinase |
| PKC δ | protein kinase C δ |
| PTCH1 | patched 1 |
| R | recombinant |
| RAS | rat sarcoma viral oncogene homolog |
| RUNX2 | runt related transcription factor 2 |
| SCSO | superficial cell spatial organization |
| SDF1 | Stromal cell derived factor 1 |
| SF | synovial fluid |
| SHH | sonic hedgehog |
| SIRT1 | sirtuin 1 |
| SMAD | SMA- and MAD-related protein |
| SNP | single nucleotide polymorphism |
| SOX9 | SRY-related HMG box-containing |
| SREBP-2 | sterol regulatory element-binding protein 2 |
| STAT3 | signal transducer and activator of transcription 3 |
| SZ | superficial zone |
| TAB3 | TGF-β activated kinase 1 and MAP3K7 binding protein 3 |
| TAK1 | TGF-β-activated kinase-1 |
| TGF-β | transforming growth factor β |
| TGFBR | transforming growth factor β receptor |
| TIMP | tissue inhibitor of metalloproteinases |
| TMJ | temporomandibular joint |
| TNF-α | tumor necrosis factor α |
| TNFRSF11B | tumor necrosis factor receptor superfamily member 11b |
| TRAF6 | TNF receptor-associated factor 6 |
| VEGF | vascular endothelial growth factor |
| WNT | wingless-type MMTV integration site family |
| ZIP8 | Zrt- and Irt-like protein 8 |
References
- McAlindon, T.E.; Bannuru, R.R.; Sullivan, M.C.; Arden, N.K.; Berenbaum, F.; Bierma-Zeinstra, S.M.; Hawker, G.A.; Henrotin, Y.; Hunter, D.J.; Kawaguchi, H.; et al. Oarsi guidelines for the non-surgical management of knee osteoarthritis. Osteoarthr. Cartil. 2014, 22, 363–388. [Google Scholar] [CrossRef] [PubMed]
- Ondresik, M.; Azevedo Maia, F.R.; da Silva Morais, A.; Gertrudes, A.C.; Dias Bacelar, A.H.; Correia, C.; Goncalves, C.; Radhouani, H.; Amandi Sousa, R.; Oliveira, J.M.; et al. Management of knee osteoarthritis. Current status and future trends. Biotechnol. Bioeng. 2017, 114, 717–739. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.J.; Eyles, J.P.; Hunter, D.J. Hip osteoarthritis: Etiopathogenesis and implications for management. Adv. Ther. 2016, 33, 1921–1946. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, X.; Eymard, F.; Richette, P. Biologic agents in osteoarthritis: Hopes and disappointments. Nat. Rev. Rheumatol. 2013, 9, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.R.; Williams, A.A.; Coyle, C.H.; Bowers, M.E. Early diagnosis to enable early treatment of pre-osteoarthritis. Arthritis Res. Ther. 2012, 14, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobasheri, A. The future of osteoarthritis therapeutics: Targeted pharmacological therapy. Curr. Rheumatol. Rep. 2013, 15, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marles, P.J.; Hoyland, J.A.; Parkinson, R.; Freemont, A.J. Demonstration of variation in chondrocyte activity in different zones of articular cartilage: An assessment of the value of in-situ hybridization. Int. J. Exp. Pathol. 1991, 72, 171–182. [Google Scholar] [PubMed]
- Asari, A.; Miyauchi, S.; Kuriyama, S.; Machida, A.; Kohno, K.; Uchiyama, Y. Localization of hyaluronic acid in human articular cartilage. J. Histochem. Cytochem. 1994, 42, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Kuettner, K.E.; Aydelotte, M.B.; Thonar, E.J. Articular cartilage matrix and structure: A minireview. J. Rheumatol. Suppl. 1991, 27, 46–48. [Google Scholar] [PubMed]
- Aicher, W.K.; Rolauffs, B. The spatial organisation of joint surface chondrocytes: Review of its potential roles in tissue functioning, disease and early, preclinical diagnosis of osteoarthritis. Ann. Rheum. Dis. 2014, 73, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Rolauffs, B.; Williams, J.M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Distinct horizontal patterns in the spatial organization of superficial zone chondrocytes of human joints. J. Struct. Biol. 2008, 162, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolauffs, B.; Williams, J.M.; Aurich, M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Proliferative remodeling of the spatial organization of human superficial chondrocytes distant from focal early osteoarthritis. Arthritis Rheumtol. 2010, 62, 489–498. [Google Scholar]
- Felka, T.; Rothdiener, M.; Bast, S.; Uynuk-Ool, T.; Zouhair, S.; Ochs, B.G.; De Zwart, P.; Stoeckle, U.; Aicher, W.K.; Hart, M.L.; et al. Loss of spatial organization and destruction of the pericellular matrix in early osteoarthritis in vivo and in a novel in vitro methodology. Osteoarthr. Cartil. 2016, 24, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.K.; Otsuki, S.; Grogan, S.P.; Sah, R.; Terkeltaub, R.; D’Lima, D. Cartilage cell clusters. Arthritis Rheumtol. 2010, 62, 2206–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldring, M.B. The role of the chondrocyte in osteoarthritis. Arthritis Rheumtol. 2000, 43, 1916–1926. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, B.L.; Su, J.L.; Lindley, K.M.; Kuettner, K.E.; Cole, A.A. Horizontally oriented clusters of multiple chondrons in the superficial zone of ankle, but not knee articular cartilage. Anat. Rec. 2002, 266, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetlow, L.C.; Adlam, D.J.; Woolley, D.E. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: Associations with degenerative changes. Arthritis Rheumtol. 2001, 44, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Bush, J.R.; Beier, F. TGF-beta and osteoarthritis—The good and the bad. Nat. Med. 2013, 19, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Cromme, C.; Umlauf, D.; Frank, S.; Pap, T. Molecular mechanisms of cartilage remodelling in osteoarthritis. Int. J. Biochem. Cell Biol. 2010, 42, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.I.; Argyle, D.J.; Clements, D.N. In vitro models for the study of osteoarthritis. Vet. J. 2016, 209, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Yun, Y.R.; Won, J.E.; Jeon, E.; Lee, S.; Kang, W.; Jo, H.; Jang, J.H.; Shin, U.S.; Kim, H.W. Fibroblast growth factors: Biology, function, and application for tissue regeneration. J. Tissue Eng. 2010, 2010, 218142. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.A.; Schleicher, S.B.; Traub, F.; Rolauffs, B. Chondrosarcoma: A rare misfortune in aging human cartilage? The role of stem and progenitor cells in proliferation, malignant degeneration and therapeutic resistance. Int. J. Mol. Sci. 2018, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Zhai, G.; Dore, J.; Rahman, P. TGF-beta signal transduction pathways and osteoarthritis. Rheumatol. Int. 2015, 35, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.L.; McLean, C.J.; Full, L.E.; Peston, D.; Saklatvala, J. FGF-2 is bound to perlecan in the pericellular matrix of articular cartilage, where it acts as a chondrocyte mechanotransducer. Osteoarthr. Cartil. 2007, 15, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Hermansson, M.; Bolton, M.; Wait, R.; Saklatvala, J. Basic fgf mediates an immediate response of articular cartilage to mechanical injury. Proc. Natl. Acad. Sci. USA 2002, 99, 8259–8264. [Google Scholar] [CrossRef] [PubMed]
- Cucchiarini, M.; Terwilliger, E.F.; Kohn, D.; Madry, H. Remodelling of human osteoarthritic cartilage by FGF-2, alone or combined with SOX9 via RAAV gene transfer. J. Cell. Mol. Med. 2009, 13, 2476–2488. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Andres, G.; Leali, D.; Dell’Era, P.; Ronca, R. Inflammatory cells and chemokines sustain FGF2-induced angiogenesis. Eur. Cytokine Netw. 2009, 20, 39–50. [Google Scholar] [PubMed]
- Honsawek, S.; Yuktanandana, P.; Tanavalee, A.; Saetan, N.; Anomasiri, W.; Parkpian, V. Correlation between plasma and synovial fluid basic fibroblast growth factor with radiographic severity in primary knee osteoarthritis. Int. Orthop. 2012, 36, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Chen, D.; Cool, S.M.; van Wijnen, A.J.; Mikecz, K.; Murphy, G.; Im, H.J. Fibroblast growth factor receptor 1 is principally responsible for fibroblast growth factor 2-induced catabolic activities in human articular chondrocytes. Arthritis Res. Ther. 2011, 13, R130. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ellman, M.B.; Kroin, J.S.; Chen, D.; Yan, D.; Mikecz, K.; Ranjan, K.C.; Xiao, G.; Stein, G.S.; Kim, S.G.; et al. Species-specific biological effects of FGF-2 in articular cartilage: Implication for distinct roles within the fgf receptor family. J. Cell. Biochem. 2012, 113, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Im, H.J.; Muddasani, P.; Natarajan, V.; Schmid, T.M.; Block, J.A.; Davis, F.; van Wijnen, A.J.; Loeser, R.F. Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase cdelta pathways in human adult articular chondrocytes. J. Biol. Chem. 2007, 282, 11110–11121. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Chen, D.; Im, H.J. Fibroblast growth factor-2 promotes catabolism via FGFR1-Ras-Raf-MEK1/2-ERK1/2 axis that coordinates with the PKCdelta pathway in human articular chondrocytes. J. Cell. Biochem. 2012, 113, 2856–2865. [Google Scholar] [CrossRef] [PubMed]
- Ellman, M.B.; Yan, D.; Ahmadinia, K.; Chen, D.; An, H.S.; Im, H.J. Fibroblast growth factor control of cartilage homeostasis. J. Cell. Biochem. 2013, 114, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muddasani, P.; Norman, J.C.; Ellman, M.; van Wijnen, A.J.; Im, H.J. Basic fibroblast growth factor activates the MAPK and NFkappaB pathways that converge on Elk-1 to control production of matrix metalloproteinase-13 by human adult articular chondrocytes. J. Biol. Chem. 2007, 282, 31409–31421. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Y.; Hu, K.Z.; Yin, Z.S. Inhibition of the p38-mapk signaling pathway suppresses the apoptosis and expression of proinflammatory cytokines in human osteoarthritis chondrocytes. Cytokine 2016, 90, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Takebe, K.; Nishiyama, T.; Hayashi, S.; Hashimoto, S.; Fujishiro, T.; Kanzaki, N.; Kawakita, K.; Iwasa, K.; Kuroda, R.; Kurosaka, M. Regulation of p38 MAPK phosphorylation inhibits chondrocyte apoptosis in response to heat stress or mechanical stress. Int. J. Mol. Med. 2011, 27, 329–335. [Google Scholar] [PubMed]
- Fan, Z.; Soder, S.; Oehler, S.; Fundel, K.; Aigner, T. Activation of interleukin-1 signaling cascades in normal and osteoarthritic articular cartilage. Am. J. Pathol. 2007, 171, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Nummenmaa, E.; Hamalainen, M.; Moilanen, T.; Vuolteenaho, K.; Moilanen, E. Effects of FGF-2 and FGF receptor antagonists on mmp enzymes, aggrecan, and type II collagen in primary human OA chondrocytes. Scand. J. Rheumatol. 2015, 44, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Crepin, M.; Liu, J.M.; Barritault, D.; Ledoux, D. FGF-2 and TPA induce matrix metalloproteinase-9 secretion in MCF-7 cells through PKC activation of the RAS/ERK pathway. Biochem. Biophys. Res. Commun. 2002, 293, 1174–1182. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Aoyama, E.; Janune, D.; Maeda, A.; Takigawa, M. Effect of CCN2 on FGF2-induced proliferation and MMP9 and MMP13 productions by chondrocytes. Endocrinology 2011, 152, 4232–4241. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Manner, P.A.; Horner, A.; Shum, L.; Tuan, R.S.; Nuckolls, G.H. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 963–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamekura, S.; Kawasaki, Y.; Hoshi, K.; Shimoaka, T.; Chikuda, H.; Maruyama, Z.; Komori, T.; Sato, S.; Takeda, S.; Karsenty, G.; et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheumtol. 2006, 54, 2462–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.G.; Kim, H.J.; Park, H.J.; Kim, Y.J.; Yoon, W.J.; Lee, S.J.; Ryoo, H.M.; Cho, J.Y. RUNX2 phosphorylation induced by fibroblast growth factor-2/protein kinase c pathways. Proteomics 2006, 6, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Tetsunaga, T.; Nishida, K.; Furumatsu, T.; Naruse, K.; Hirohata, S.; Yoshida, A.; Saito, T.; Ozaki, T. Regulation of mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2 transcriptional factor in SW1353 chondrocyte-like cells. Osteoarthr. Cartil. 2011, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, Y.; Jacobi, J.L.; Tuan, R.S. Inhibition of histone deacetylases antagonized FGF2 and IL-1beta effects on MMP expression in human articular chondrocytes. Growth Factors 2009, 27, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.L.; Sawaji, Y.; Burleigh, A.; McLean, C.; Inglis, J.; Saklatvala, J.; Vincent, T. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheumtol. 2009, 60, 2019–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Wakitani, S.; Imoto, K.; Hattori, T.; Nakaya, H.; Saito, M.; Yonenobu, K. Fibroblast growth factor-2 promotes the repair of partial thickness defects of articular cartilage in immature rabbits but not in mature rabbits. Osteoarthr. Cartil. 2004, 12, 636–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, R.L.; Chen, A.C.; Grodzinsky, A.J.; Trippel, S.B. Differential effects of bFGF and IGF-I on matrix metabolism in calf and adult bovine cartilage explants. Arch. Biochem. Biophys. 1994, 308, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Nakamura, Y.; Maruyama, M.; Abe, K.; Watanapokasin, R.; Kato, H. Expression profiles of human CCN genes in patients with osteoarthritis or rheumatoid arthritis. J. Orthop. Sci. 2015, 20, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Nishida, K.; Yamaai, Y.; Shibahara, M.; Nishida, T.; Doi, T.; Asahara, H.; Nakanishi, T.; Inoue, H.; Takigawa, M. Expression and localization of connective tissue growth factor (CTGF/Hcs24/CCN2) in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Verdier, M.P.; Seite, S.; Guntzer, K.; Pujol, J.P.; Boumediene, K. Immunohistochemical analysis of transforming growth factor beta isoforms and their receptors in human cartilage from normal and osteoarthritic femoral heads. Rheumatol. Int. 2005, 25, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Pombo-Suarez, M.; Castano-Oreja, M.T.; Calaza, M.; Gomez-Reino, J.; Gonzalez, A. Differential upregulation of the three transforming growth factor beta isoforms in human osteoarthritic cartilage. Ann. Rheum. Dis. 2009, 68, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, W.; Bemis, A.; Wang, E.; Qiu, Y.; Morris, E.A.; Flannery, C.R.; Yang, Z. Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheumtol. 2007, 56, 3675–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kizawa, H.; Kou, I.; Iida, A.; Sudo, A.; Miyamoto, Y.; Fukuda, A.; Mabuchi, A.; Kotani, A.; Kawakami, A.; Yamamoto, S.; et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat. Genet. 2005, 37, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Kizawa, H.; Saitoh, M.; Kou, I.; Miyazono, K.; Ikegawa, S. Mechanisms for asporin function and regulation in articular cartilage. J. Biol. Chem. 2007, 282, 32185–32192. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Spector, T.D.; Tamm, A.; Kisand, K.; Doherty, S.A.; Dennison, E.M.; Mangino, M.; Tamm, A.; KeRNA, I.; Hart, D.J.; et al. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheumtol. 2010, 62, 2347–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerne, P.A.; Blanco, F.; Kaelin, A.; Desgeorges, A.; Lotz, M. Growth factor responsiveness of human articular chondrocytes in aging and development. Arthritis Rheumtol. 1995, 38, 960–968. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, P.C.; Masi, T.L.; de Ortiz, J.L.; Binette, F.; Tubo, R.; McPherson, J.M. Synergistic action of transforming growth factor-beta and insulin-like growth factor-I induces expression of type II collagen and aggrecan genes in adult human articular chondrocytes. Exp. Cell Res. 1997, 237, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef] [PubMed]
- Finnson, K.W.; Parker, W.L.; ten Dijke, P.; Thorikay, M.; Philip, A. ALK1 opposes ALK5/SMAD3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res. 2008, 23, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Goumans, M.J.; Blaney Davidson, E.; ten Dijke, P. Age-dependent alteration of tgf-beta signalling in osteoarthritis. Cell Tissue Res. 2012, 347, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.K.; Rey-Rico, A.; Schmitt, G.; Wezel, A.; Madry, H.; Cucchiarini, M. rAAV-mediated overexpression of TGF-beta stably restructures human osteoarthritic articular cartilage in situ. J. Transl. Med. 2013, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; van der Kraan, P.M.; van den Berg, W.B. TGF-beta and osteoarthritis. Osteoarthr. Cartil. 2007, 15, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Finnson, K.W.; Chi, Y.; Bou-Gharios, G.; Leask, A.; Philip, A. TGF-b signaling in cartilage homeostasis and osteoarthritis. Front. Biosci. (Schol. Ed.) 2012, 4, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Nakerakanti, S.S.; Bujor, A.M.; Trojanowska, M. CCN2 is required for the TGF-beta induced activation of SMAD1-ERK1/2 signaling network. PLoS ONE 2011, 6, e21911. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.M.; Markova, D.; Smith, H.E.; Susarla, B.; Ponnappan, R.K.; Anderson, D.G.; Symes, A.; Shapiro, I.M.; Risbud, M.V. Regulation of CCN2/connective tissue growth factor expression in the nucleus pulposus of the intervertebral disc: Role of Smad and activator protein 1 signaling. Arthritis Rheumtol. 2010, 62, 1983–1992. [Google Scholar]
- Cheng, J.; Hu, X.; Dai, L.; Zhang, X.; Ren, B.; Shi, W.; Liu, Z.; Duan, X.; Zhang, J.; Fu, X.; et al. Inhibition of transforming growth factor beta-activated kinase 1 prevents inflammation-related cartilage degradation in osteoarthritis. Sci. Rep. 2016, 6, 34497. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Chubinskaya, S.; Liao, W.; Loeser, R.F. Wnt5a induces catabolic signaling and matrix metalloproteinase production in human articular chondrocytes. Osteoarthr. Cartil. 2017, 25, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Thorfve, A.; Dehne, T.; Lindahl, A.; Brittberg, M.; Pruss, A.; Ringe, J.; Sittinger, M.; Karlsson, C. Characteristic markers of the wnt signaling pathways are differentially expressed in osteoarthritic cartilage. Cartilage 2012, 3, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, W.; Sun, M.; Deng, Z.; Zeng, C.; Li, H.; Yang, T.; Li, L.; Luo, W.; Lei, G. The expression of osteopontin and wnt5a in articular cartilage of patients with knee osteoarthritis and its correlation with disease severity. Biomed. Res. Int. 2016, 2016, 9561058. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, K.; Pei, Y.; Moore, T.L.; Wang, H.; Yu, X.P.; Geiser, A.G.; Chandrasekhar, S. Regulation of the human adamts-4 promoter by transcription factors and cytokines. Biochem. Biophys. Res. Commun. 2006, 345, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.L.; Hui Mingalone, C.K.; Foote, A.T.; Uchimura, T.; Zhang, M.; Zeng, L. Wnt7a inhibits il-1beta induced catabolic gene expression and prevents articular cartilage damage in experimental osteoarthritis. Sci. Rep. 2017, 7, 41823. [Google Scholar] [CrossRef] [PubMed]
- Nalesso, G.; Sherwood, J.; Bertrand, J.; Pap, T.; Ramachandran, M.; De Bari, C.; Pitzalis, C.; Dell’accio, F. Wnt-3a modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J. Cell. Biol. 2011, 193, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; van Blitterswijk, C.A.; Karperien, M. A Wnt/beta-catenin negative feedback loop inhibits interleukin-1-induced matrix metalloproteinase expression in human articular chondrocytes. Arthritis Rheumtol. 2012, 64, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Snelling, S.J.; Davidson, R.K.; Swingler, T.E.; Le, L.T.; Barter, M.J.; Culley, K.L.; Price, A.; Carr, A.J.; Clark, I.M. Dickkopf-3 is upregulated in osteoarthritis and has a chondroprotective role. Osteoarthr. Cartil. 2016, 24, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijten, J.C.; Bos, S.D.; Landman, E.B.; Georgi, N.; Jahr, H.; Meulenbelt, I.; Post, J.N.; van Blitterswijk, C.A.; Karperien, M. GREM1, FRZB and DKK1 mRNA levels correlate with osteoarthritis and are regulated by osteoarthritis-associated factors. Arthritis Res. Ther. 2013, 15, R126. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res. 2009, 24, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Min, J.L.; Meulenbelt, I.; Riyazi, N.; Kloppenburg, M.; Houwing-Duistermaat, J.J.; Seymour, A.B.; Pols, H.A.; van Duijn, C.M.; Slagboom, P.E. Association of the frizzled-related protein gene with symptomatic osteoarthritis at multiple sites. Arthritis Rheumtol. 2005, 52, 1077–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loughlin, J.; Dowling, B.; Chapman, K.; Marcelline, L.; Mustafa, Z.; Southam, L.; Ferreira, A.; Ciesielski, C.; Carson, D.A.; Corr, M. Functional variants within the secreted frizzled-related protein 3 gene are associated with hip osteoarthritis in females. Proc. Natl. Acad. Sci. USA 2004, 101, 9757–9762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdes, A.M.; Loughlin, J.; Oene, M.V.; Chapman, K.; Surdulescu, G.L.; Doherty, M.; Spector, T.D. Sex and ethnic differences in the association of ASPN, CALM1, COL2A1, COMP, and FRZB with genetic susceptibility to osteoarthritis of the knee. Arthritis Rheumtol. 2007, 56, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Oldefest, M.; Dusterhoft, S.; Desel, C.; Thysen, S.; Fink, C.; Rabe, B.; Lories, R.; Grotzinger, J.; Lorenzen, I. Secreted frizzled-related protein 3 (sFRP3)-mediated suppression of interleukin-6 receptor release by a disintegrin and metalloprotease 17 (ADAM17) is abrogated in the osteoarthritis-associated rare double variant of sFRP3. Biochem. J. 2015, 468, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Minina, E.; Wenzel, H.M.; Kreschel, C.; Karp, S.; Gaffield, W.; McMahon, A.P.; Vortkamp, A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development 2001, 128, 4523–4534. [Google Scholar] [PubMed]
- Ohba, S. Hedgehog signaling in endochondral ossification. J. Dev. Biol. 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. PTHrP and skeletal development. Ann. N. Y. Acad. Sci. 2006, 1068, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhou, J.; Wei, X.; Zhang, J.; Fleming, B.C.; Terek, R.; Pei, M.; Chen, Q.; Liu, T.; Wei, L. Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of mmp-13 in human osteoarthritic cartilage. Osteoarthr. Cartil. 2012, 20, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wei, X.; Chen, C.; Cao, K.; Li, Y.; Jiao, Q.; Ding, J.; Zhou, J.; Fleming, B.C.; Chen, Q.; et al. Indian hedgehog in synovial fluid is a novel marker for early cartilage lesions in human knee joint. Int. J. Mol. Sci. 2014, 15, 7250–7265. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, S.A.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med. 2009, 15, 1421–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.L.; Patel, R.; Kelly, T.A.; Wann, A.K.; Hung, C.T.; Chapple, J.P.; Knight, M.M. Hedgehog signalling does not stimulate cartilage catabolism and is inhibited by interleukin-1beta. Arthritis Res. Ther. 2015, 17, 373. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; van den Berg, W.B. Bone morphogenetic proteins and articular cartilage: To serve and protect or a wolf in sheep clothing’s? Osteoarthr. Cartil. 2010, 18, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Fukui, N.; Zhu, Y.; Maloney, W.J.; Clohisy, J.; Sandell, L.J. Stimulation of BMP-2 expression by pro-inflammatory cytokines IL-1 and TNF-alpha in normal and osteoarthritic chondrocytes. J. Bone Jt. Surg. Am. 2003, 85-A (Suppl. 3), 59–66. [Google Scholar] [CrossRef]
- Nakase, T.; Miyaji, T.; Tomita, T.; Kaneko, M.; Kuriyama, K.; Myoui, A.; Sugamoto, K.; Ochi, T.; Yoshikawa, H. Localization of bone morphogenetic protein-2 in human osteoarthritic cartilage and osteophyte. Osteoarthr. Cartil. 2003, 11, 278–284. [Google Scholar] [CrossRef]
- Papathanasiou, I.; Malizos, K.N.; Tsezou, A. Bone morphogenetic protein-2-induced Wnt/beta-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res. Ther. 2012, 14, R82. [Google Scholar] [CrossRef] [PubMed]
- Schmal, H.; Pilz, I.H.; Mehlhorn, A.T.; Dovi-Akue, D.; Kirchhoff, C.; Sudkamp, N.P.; Gerlach, U.; Niemeyer, P. Expression of BMP-receptor type 1A correlates with progress of osteoarthritis in human knee joints with focal cartilage lesions. Cytotherapy 2012, 14, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Bobacz, K.; Gruber, R.; Soleiman, A.; Erlacher, L.; Smolen, J.S.; Graninger, W.B. Expression of bone morphogenetic protein 6 in healthy and osteoarthritic human articular chondrocytes and stimulation of matrix synthesis in vitro. Arthritis Rheumtol. 2003, 48, 2501–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.L.; Fang, C.; Liu, C.; Leslie, M.P.; Chang, E.; Di Cesare, P.E. Expression of bone morphogenetic proteins, receptors, and tissue inhibitors in human fetal, adult, and osteoarthritic articular cartilage. J. Orthop. Res. 2004, 22, 1188–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobinac, D.; Spanjol, J.; Marinovic, M.; Zoricic Cvek, S.; Maric, I.; Cicvaric, T.; Fuckar, D.; Markic, D.; Vojnikovic, B. Expression of bone morphogenetic proteins, cartilage-derived morphogenetic proteins and related receptors in normal and osteoarthritic human articular cartilage. Coll. Antropol. 2008, 32 (Suppl. 2), 83–87. [Google Scholar]
- Lafont, J.E.; Poujade, F.A.; Pasdeloup, M.; Neyret, P.; Mallein-Gerin, F. Hypoxia potentiates the bmp-2 driven COL2A1 stimulation in human articular chondrocytes via p38 MAPK. Osteoarthr. Cartil. 2016, 24, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Lindsey, D.P.; Dhulipala, L.; Harris, A.H.; Goodman, S.B.; Maloney, W.J. Effects of intermittent hydrostatic pressure and BMP-2 on osteoarthritic human chondrocyte metabolism in vitro. J. Orthop. Res. 2011, 29, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Chubinskaya, S.; Segalite, D.; Pikovsky, D.; Hakimiyan, A.A.; Rueger, D.C. Effects induced by BMPs in cultures of human articular chondrocytes: Comparative studies. Growth Factors 2008, 26, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.K.; Huey, D.J.; Hu, J.C.; Athanasiou, K.A. TGF-beta1, GDF-5, and BMP-2 stimulation induces chondrogenesis in expanded human articular chondrocytes and marrow-derived stromal cells. Stem Cells 2015, 33, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Chubinskaya, S.; Rueger, D.C.; Bau, B.; Haag, J.; Aigner, T. Regulation of anabolic and catabolic gene expression in normal and osteoarthritic adult human articular chondrocytes by osteogenic protein-1. Clin. Exp. Rheumatol. 2004, 22, 103–106. [Google Scholar] [PubMed]
- Varas, A.; Valencia, J.; Lavocat, F.; Martinez, V.G.; Thiam, N.N.; Hidalgo, L.; FeRNAndez-Sevilla, L.M.; Sacedon, R.; Vicente, A.; Miossec, P. Blockade of bone morphogenetic protein signaling potentiates the pro-inflammatory phenotype induced by interleukin-17 and tumor necrosis factor-alpha combination in rheumatoid synoviocytes. Arthritis Res. Ther. 2015, 17, 192. [Google Scholar] [CrossRef] [PubMed]
- Mahjoub, M.; Sassi, N.; Driss, M.; Laadhar, L.; Allouche, M.; Hamdoun, M.; Romdhane, K.B.; Sellami, S.; Makni, S. Expression patterns of notch receptors and their ligands in human osteoarthritic and healthy articular cartilage. Tissue Cell 2012, 44, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.; Brantsing, C.; Egell, S.; Lindahl, A. Notch1, Jagged1, and HES5 are abundantly expressed in osteoarthritis. Cells Tissues Organs 2008, 188, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Grogan, S.P.; Miyaki, S.; Asahara, H.; D’Lima, D.D.; Lotz, M.K. Mesenchymal progenitor cell markers in human articular cartilage: Normal distribution and changes in osteoarthritis. Arthritis Res. Ther. 2009, 11, R85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, N.Y.; Distler, A.; Beyer, C.; Philipi-Schobinger, A.; Breda, S.; Dees, C.; Stock, M.; Tomcik, M.; Niemeier, A.; Dell’Accio, F.; et al. Inhibition of notch1 promotes hedgehog signalling in a HES1-dependent manner in chondrocytes and exacerbates experimental osteoarthritis. Ann. Rheum. Dis. 2016, 75, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Schneiderman, R.; Rosenberg, N.; Hiss, J.; Lee, P.; Liu, F.; Hintz, R.L.; Maroudas, A. Concentration and size distribution of insulin-like growth factor-I in human normal and osteoarthritic synovial fluid and cartilage. Arch. Biochem. Biophys. 1995, 324, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Tavera, C.; Abribat, T.; Reboul, P.; Dore, S.; Brazeau, P.; Pelletier, J.P.; Martel-Pelletier, J. IGF and IGF-binding protein system in the synovial fluid of osteoarthritic and rheumatoid arthritic patients. Osteoarthr. Cartil. 1996, 4, 263–274. [Google Scholar] [CrossRef]
- Starkman, B.G.; Cravero, J.D.; Delcarlo, M.; Loeser, R.F. IGF-I stimulation of proteoglycan synthesis by chondrocytes requires activation of the PI 3-kinase pathway but not ERK MAPK. Biochem. J. 2005, 389, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Weimer, A.; Madry, H.; Venkatesan, J.K.; Schmitt, G.; Frisch, J.; Wezel, A.; Jung, J.; Kohn, D.; Terwilliger, E.F.; Trippel, S.B.; et al. Benefits of recombinant adeno-associated virus (rAAV)-mediated insulinlike growth factor I (IGF-I) overexpression for the long-term reconstruction of human osteoarthritic cartilage by modulation of the IGF-I axis. Mol. Med. 2012, 18, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Chubinskaya, S.; Pacione, C.; Im, H.J. Basic fibroblast growth factor inhibits the anabolic activity of insulin-like growth factor 1 and osteogenic protein 1 in adult human articular chondrocytes. Arthritis Rheumtol. 2005, 52, 3910–3917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.; Yudoh, K.; Masuko, K. The potential role of vascular endothelial growth factor (VEGF) in cartilage: How the angiogenic factor could be involved in the pathogenesis of osteoarthritis? Osteoarthr. Cartil. 2008, 16, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.; Varoga, D.; Wruck, C.J.; Kurz, B.; Goldring, M.B.; Pufe, T. Reactive oxygen species induce expression of vascular endothelial growth factor in chondrocytes and human articular cartilage explants. Arthritis Res. Ther. 2006, 8, R189. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Inoki, I.; Komiya, K.; Shiomi, T.; Ikeda, E.; Obata, K.; Matsumoto, H.; Toyama, Y.; Okada, Y. Vascular endothelial growth factor isoforms and their receptors are expressed in human osteoarthritic cartilage. Am. J. Pathol. 2003, 162, 171–181. [Google Scholar] [CrossRef]
- Pufe, T.; Petersen, W.; Tillmann, B.; Mentlein, R. The splice variants VEGF121 and VEGF189 of the angiogenic peptide vascular endothelial growth factor are expressed in osteoarthritic cartilage. Arthritis Rheumtol. 2001, 44, 1082–1088. [Google Scholar] [CrossRef]
- Pfander, D.; Kortje, D.; Zimmermann, R.; Weseloh, G.; Kirsch, T.; Gesslein, M.; Cramer, T.; Swoboda, B. Vascular endothelial growth factor in articular cartilage of healthy and osteoarthritic human knee joints. Ann. Rheum. Dis. 2001, 60, 1070–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Xie, W.; Shang, Q.; Su, B. The long noncoding RNA MEG3 is downregulated and inversely associated with VEGF levels in osteoarthritis. Biomed. Res. Int. 2015, 2015, 356893. [Google Scholar] [CrossRef] [PubMed]
- Pulsatelli, L.; Dolzani, P.; Silvestri, T.; Frizziero, L.; Facchini, A.; Meliconi, R. Vascular endothelial growth factor activities on osteoarthritic chondrocytes. Clin. Exp. Rheumatol. 2005, 23, 487–493. [Google Scholar] [PubMed]
- Shakibaei, M.; Schulze-Tanzil, G.; Mobasheri, A.; Beichler, T.; Dressler, J.; Schwab, W. Expression of the VEGF receptor-3 in osteoarthritic chondrocytes: Stimulation by interleukin-1 beta and association with beta 1-integrins. Histochem. Cell Biol. 2003, 120, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. (Lond.) 2005, 109, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthr. Cartil. 2013, 21, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Sandy, J.D.; Chan, D.D.; Trevino, R.L.; Wimmer, M.A.; Plaas, A. Human genome-wide expression analysis reorients the study of inflammatory mediators and biomechanics in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.; Saal, J.G.; Schaudt, K.; Zacher, J.; Fritz, P.; Pawelec, G. Determination of cytokines in synovial fluids: Correlation with diagnosis and histomorphological characteristics of synovial tissue. Ann. Rheum. Dis. 1992, 51, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; McCollum, R.; DiBattista, J.; Faure, M.P.; Chin, J.A.; Fournier, S.; Sarfati, M.; Pelletier, J.P. The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis Rheumtol. 1992, 35, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Tan, J.; Yuan, Z.; Meng, G.; Bi, L.; Liu, J. Expression profile of cytokines and chemokines in osteoarthritis patients: Proinflammatory roles for CXCL8 and CXCL11 to chondrocytes. Int. Immunopharmacol. 2016, 40, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.; Manthey, A.; Tyler, J. Insulin-like growth factor (IGF) receptor, IGF-i, interleukin-1 beta (IL-1 beta), and IL-6 mRNA expression in osteoarthritic and normal human cartilage. J. Histochem. Cytochem. 1996, 44, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Towle, C.A.; Hung, H.H.; Bonassar, L.J.; Treadwell, B.V.; Mangham, D.C. Detection of interleukin-1 in the cartilage of patients with osteoarthritis: A possible autocrine/paracrine role in pathogenesis. Osteoarthr. Cartil. 1997, 5, 293–300. [Google Scholar] [CrossRef]
- Aigner, T.; McKenna, L.; Zien, A.; Fan, Z.; Gebhard, P.M.; Zimmer, R. Gene expression profiling of serum- and interleukin-1 beta-stimulated primary human adult articular chondrocytes—A molecular analysis based on chondrocytes isolated from one donor. Cytokine 2005, 31, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Honorati, M.C.; Bovara, M.; Cattini, L.; Piacentini, A.; Facchini, A. Contribution of interleukin 17 to human cartilage degradation and synovial inflammation in osteoarthritis. Osteoarthr. Cartil. 2002, 10, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Barak, T.; Quach, J.; Lotz, M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J. Biol. Chem. 1998, 273, 27467–27473. [Google Scholar] [CrossRef] [PubMed]
- Haseeb, A.; Ansari, M.Y.; Haqqi, T.M. Harpagoside suppresses IL-6 expression in primary human osteoarthritis chondrocytes. J. Orthop. Res. 2017, 35, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M.; Plumb, D.A.; Dragomir, C.; Favero, M.; El Hachem, K.; Hashimoto, K.; Roach, H.I.; Olivotto, E.; Borzi, R.M.; et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: Signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur. Cells Mater. 2011, 21, 202–220. [Google Scholar] [CrossRef]
- Otero, M.; Plumb, D.A.; Tsuchimochi, K.; Dragomir, C.L.; Hashimoto, K.; Peng, H.; Olivotto, E.; Bevilacqua, M.; Tan, L.; Yang, Z.; et al. E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (mmp13) transcriptional control in articular chondrocytes under proinflammatory stress. J. Biol. Chem. 2012, 287, 3559–3572. [Google Scholar] [CrossRef] [PubMed]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Zafarullah, M. Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1) and nuclear factor kappa b (NF-kappa b) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol. 2002, 21, 251–262. [Google Scholar] [CrossRef]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Huang, W.; Dehnade, F.; Ahmad, M.; Zafarullah, M. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by map kinases, AP-1, and NF-kappab transcription factors in articular chondrocytes. Exp. Cell Res. 2003, 288, 208–217. [Google Scholar] [CrossRef]
- Benderdour, M.; Tardif, G.; Pelletier, J.P.; Di Battista, J.A.; Reboul, P.; Ranger, P.; Martel-Pelletier, J. Interleukin 17 (IL-17) induces collagenase-3 production in human osteoarthritic chondrocytes via AP-1 dependent activation: Differential activation of AP-1 members by IL-17 and IL-1beta. J. Rheumatol. 2002, 29, 1262–1272. [Google Scholar] [PubMed]
- Wang, J.; Elewaut, D.; Veys, E.M.; Verbruggen, G. Insulin-like growth factor 1-induced interleukin-1 receptor ii overrides the activity of interleukin-1 and controls the homeostasis of the extracellular matrix of cartilage. Arthritis Rheumtol. 2003, 48, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.Y.; Huang, C.Y.; Tsai, C.H.; Wang, S.W.; Lin, Y.M.; Tang, C.H. Interleukin-1beta induces fibroblast growth factor 2 expression and subsequently promotes endothelial progenitor cell angiogenesis in chondrocytes. Clin. Sci. (Lond.) 2016, 130, 667–681. [Google Scholar] [CrossRef] [PubMed]
- De Andres, M.C.; Takahashi, A.; Oreffo, R.O. Demethylation of an NF-kappaB enhancer element orchestrates inos induction in osteoarthritis and is associated with altered chondrocyte cell cycle. Osteoarthr. Cartil. 2016, 24, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Jikko, A.; Wakisaka, T.; Iwamoto, M.; Hiranuma, H.; Kato, Y.; Maeda, T.; Fujishita, M.; Fuchihata, H. Effects of interleukin-6 on proliferation and proteoglycan metabolism in articular chondrocyte cultures. Cell Biol. Int. 1998, 22, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Quintavalla, J.; Kumar, C.; Daouti, S.; Slosberg, E.; Uziel-Fusi, S. Chondrocyte cluster formation in agarose cultures as a functional assay to identify genes expressed in osteoarthritis. J. Cell. Physiol. 2005, 204, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Makki, M.S.; Haseeb, A.; Haqqi, T.M. MicroRNA-9 promotion of interleukin-6 expression by inhibiting monocyte chemoattractant protein-induced protein 1 expression in interleukin-1beta-stimulated human chondrocytes. Arthritis Rheumatol. 2015, 67, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Kopanska, M.; Szala, D.; Czech, J.; Gablo, N.; Gargasz, K.; Trzeciak, M.; Zawlik, I.; Snela, S. MiRNA expression in the cartilage of patients with osteoarthritis. J. Orthop. Surg. Res. 2017, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Watkins, G.; Le Good, N.; Roberts, S.; Murphy, C.L.; Brockbank, S.M.; Needham, M.R.; Read, S.J.; Newham, P. The identification of differentially expressed microRNA in osteoarthritic tissue that modulate the production of TNF-alpha and MMP13. Osteoarthr. Cartil. 2009, 17, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Liu, N.; Luo, S.; Huang, W.; Zha, Z.; Yang, J. MicroRNA-9 regulates the development of knee osteoarthritis through the NF-kappaB1 pathway in chondrocytes. Medicine (Baltimore) 2016, 95, e4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Chen, B.; Fan, X.; Li, G.; Dong, P.; Zheng, J. Epigenetically-regulated microRNA-9-5p suppresses the activation of hepatic stellate cells via TGFBR1 and TGFBR2. Cell. Physiol. Biochem. 2017, 43, 2242–2252. [Google Scholar] [CrossRef] [PubMed]
- Borgonio Cuadra, V.M.; Gonzalez-Huerta, N.C.; Romero-Cordoba, S.; Hidalgo-Miranda, A.; Miranda-Duarte, A. Altered expression of circulating microRNA in plasma of patients with primary osteoarthritis and in silico analysis of their pathways. PLoS ONE 2014, 9, e97690. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Malizos, K.N.; Oikonomou, P.; Tsezou, A. Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE 2008, 3, e3740. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jia, J.; Liu, X.; Yang, S.; Ye, S.; Yang, W.; Zhang, Y. MicroRNA-16-5p controls development of osteoarthritis by targeting SMAD3 in chondrocytes. Curr. Pharm. Des. 2015, 21, 5160–5167. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Ren, X.; Chen, J.; Li, Y.; Tang, X.; Wen, X.; Yang, X.; Zhang, J.; Wang, Y.; Ma, J.; et al. miR-16 targets fibroblast growth factor 2 to inhibit NPC cell proliferation and invasion via PI3K/AKT and MAPK signaling pathways. Oncotarget 2016, 7, 3047–3058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jia, J.; Yang, S.; Liu, X.; Ye, S.; Tian, H. MicroRNA-21 controls the development of osteoarthritis by targeting GDF-5 in chondrocytes. Exp. Mol. Med. 2014, 46, e79. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ni, S.; Cao, Y.; Zhang, T.; Wu, T.; Yin, X.; Lang, Y.; Lu, H. The angiogenic effect of microRNA-21 targeting TIMP3 through the regulation of MMP2 and MMP9. PLoS ONE 2016, 11, e0149537. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Harries, L.W. MicroRNAs as mediators of the ageing process. Genes (Basel) 2014, 5, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.; Yang, C.; Song, Y.; Liu, W.; Wang, K.; Li, S.; Zhang, Y. MicroRNA-23a-3p promotes the development of osteoarthritis by directly targeting smad3 in chondrocytes. Biochem. Biophys. Res. Commun. 2016, 478, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Tavallaee, G.; Tokar, T.; Nakamura, A.; Sundararajan, K.; Weston, A.; Sharma, A.; Mahomed, N.N.; Gandhi, R.; Jurisica, I.; et al. Identification of synovial fluid microRNA signature in knee osteoarthritis: Differentiating early- and late-stage knee osteoarthritis. Osteoarthr. Cartil. 2016, 24, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, Z.; Pan, Y.; Ma, J.; Miao, X.; Qi, X.; Zhou, H.; Jia, L. miR-26a and mir-26b mediate osteoarthritis progression by targeting FUT4 via NF-kappaB signaling pathway. Int. J. Biochem. Cell Biol. 2018, 94, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wang, J.Q.; Yan, S.Y. Reduced miR26a and miR26b expression contributes to the pathogenesis of osteoarthritis via the promotion of p65 translocation. Mol. Med. Rep. 2017, 15, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wei, M.; Kang, X.; Liu, D.; Quan, Y.; Pan, X.; Liu, X.; Liao, D.; Liu, J.; Zhang, B. Reciprocal inhibition between miR-26a and NF-kappaB regulates obesity-related chronic inflammation in chondrocytes. Biosci. Rep. 2015, 35, e00204. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chang, J.T.; Ho, Y.F.; Shyu, A.B. miR-26 down-regulates TNF-alpha/NF-kappaB signalling and IL-6 expression by silencing HMGA1 and MALT1. Nucleic Acids Res. 2016, 44, 3772–3787. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wang, R.; Zhou, L.; Zhu, Y.; Gong, J.; Zhuang, S.M. MicroRNA-26b suppresses the nf-kappab signaling and enhances the chemosensitivity of hepatocellular carcinoma cells by targeting TAK1 and TAB3. Mol. Cancer 2014, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Chun, H.J.; Azuma, J.; Maegdefessel, L.; Kundu, R.K.; Quertermous, T.; Tsao, P.S.; Spin, J.M. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J. Cell. Physiol. 2011, 226, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Kim, I.K.; Kumar, S.; Jayasinghe, S.; Hong, N.; Castoldi, G.; Catalucci, D.; Jones, W.K.; Gupta, S. NF-kappaB mediated mir-26a regulation in cardiac fibrosis. J. Cell. Physiol. 2013, 228, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Li, S.H.; Liu, Y.; Luo, Y.T. Long noncoding RNA CIR promotes chondrocyte extracellular matrix degradation in osteoarthritis by acting as a sponge for mir-27b. Cell. Physiol. Biochem. 2017, 43, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Cheleschi, S.; De Palma, A.; Pecorelli, A.; Pascarelli, N.A.; Valacchi, G.; Belmonte, G.; Carta, S.; Galeazzi, M.; Fioravanti, A. Hydrostatic pressure regulates microRNA expression levels in osteoarthritic chondrocyte cultures via the wnt/beta-catenin pathway. Int. J. Mol. Sci. 2017, 18, 133. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chang, A.C.; Chen, H.T.; Wang, S.W.; Lo, Y.S.; Tang, C.H. Adiponectin promotes VEGF-C-dependent lymphangiogenesis by inhibiting mir-27b through a caMKII/AMPK/p38 signaling pathway in human chondrosarcoma cells. Clin. Sci. (Lond.) 2016, 130, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, A.J.; Liu, Y.; Fang, Y.; Ding, X.; Liang, M. The mir-29 family: Genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol. Genom. 2012, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Le, L.T.; Swingler, T.E.; Crowe, N.; Vincent, T.L.; Barter, M.J.; Donell, S.T.; Delany, A.M.; Dalmay, T.; Young, D.A.; Clark, I.M. The microRNA-29 family in cartilage homeostasis and osteoarthritis. J. Mol. Med. (Berl.) 2016, 94, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Q.; Xu, X.; Xu, Y.; Fan, Z.; Kang, L.; Li, L.; Liang, Y.; Guo, J.; Hong, T.; Li, Z.; et al. miR-105/Runx2 axis mediates FGF2-induced ADAMTS expression in osteoarthritis cartilage. J. Mol. Med. (Berl.) 2016, 94, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Santini, P.; Politi, L.; Vedova, P.D.; Scandurra, R.; Scotto d’Abusco, A. The inflammatory circuitry of mir-149 as a pathological mechanism in osteoarthritis. Rheumatol. Int. 2014, 34, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, Y.; Zhao, M.; Wu, C.; Zhang, P.; Tang, L.; Zhang, H.; Chen, X.; Yang, Y.; Liu, G. miRNA-29c suppresses lung cancer cell adhesion to extracellular matrix and metastasis by targeting integrin beta1 and matrix metalloproteinase2 (MMP2). PLoS ONE 2013, 8, e70192. [Google Scholar]
- Liu, Y.; Taylor, N.E.; Lu, L.; Usa, K.; Cowley, A.W., Jr.; Ferreri, N.R.; Yeo, N.C.; Liang, M. Renal medullary microRNAs in dahl salt-sensitive rats: Mir-29b regulates several collagens and related genes. Hypertension 2010, 55, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Moulin, D.; Salone, V.; Koufany, M.; Clement, T.; Behm-Ansmant, I.; Branlant, C.; Charpentier, B.; Jouzeau, J.Y. MicroRNA-29b contributes to collagens imbalance in human osteoarthritic and dedifferentiated articular chondrocytes. BioMed Res. Int. 2017, 2017, 9792512. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Xie, J.; Li, H.; Li, D.; Liu, P.; Hu, Y. MicroRNA-30a promotes extracellular matrix degradation in articular cartilage via downregulation of Sox9. Cell Prolif. 2016, 49, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, C.; Liu, X.; Yang, S.; Ye, S.; Jia, J.; Liu, W.; Zhang, Y. Elevated expression of microRNA-30b in osteoarthritis and its role in erg regulation of chondrocyte. Biomed. Pharmacother. 2015, 76, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Xu, X.; Zhang, Q.; Kang, L.; Xu, Y.; Zhang, K.; Li, L.; Liang, Y.; Hong, T.; Ye, Q.; et al. The IL-1beta/AP-1/miR-30a/ADAMTS-5 axis regulates cartilage matrix degradation in human osteoarthritis. J. Mol. Med. (Berl.) 2016, 94, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, F.; Malizos, K.N.; Papathanasiou, I.; Tsezou, A. MicroRNA-33a regulates cholesterol synthesis and cholesterol efflux-related genes in osteoarthritic chondrocytes. Arthritis Res. Ther. 2015, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Ni, J.; Long, H.; Huang, J.; Yang, C.; Huang, X. IL-1beta-induced miR-34a up-regulation inhibits cyr61 to modulate osteoarthritis chondrocyte proliferation through ADAMTS-4. J. Cell. Biochem. 2017. [Google Scholar] [CrossRef]
- Yan, S.; Wang, M.; Zhao, J.; Zhang, H.; Zhou, C.; Jin, L.; Zhang, Y.; Qiu, X.; Ma, B.; Fan, Q. MicroRNA-34a affects chondrocyte apoptosis and proliferation by targeting the sirt1/p53 signaling pathway during the pathogenesis of osteoarthritis. Int. J. Mol. Med. 2016, 38, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, N.R.; Shalgi, R.; Frankel, L.B.; Leucci, E.; Lees, M.; Klausen, M.; Pilpel, Y.; Nielsen, F.C.; Oren, M.; Lund, A.H. P53-independent upregulation of mir-34a during oncogene-induced senescence represses myc. Cell Death Differ. 2010, 17, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.L.; Sun, B.F.; Huang, L.R.; Yuan, H.B.; Zhang, S.; Chen, J.; Yu, Z.J.; Luo, H. Potent inhibition of mir-34b on migration and invasion in metastatic prostate cancer cells by regulating the TGF-beta pathway. Int. J. Mol. Sci. 2017, 18, 2762. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, H.; Zhu, Z.; Ye, Y.; Mao, H.; Zhang, S. MicroRNA-105 targets SOX9 and inhibits human glioma cell progression. FEBS Lett. 2016, 590, 4329–4342. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, T.; Sakai, T.; Yonezawa, T.; Hiraiwa, H.; Hamada, T.; Nakashima, M.; Ono, Y.; Ishizuka, S.; Nakahara, H.; Lotz, M.K.; et al. MicroRNA-125b regulates the expression of aggrecanase-1 (ADAMTS-4) in human osteoarthritic chondrocytes. Arthritis Res. Ther. 2013, 15, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaskas, P.; Goljanek-Whysall, K.; Clegg, P.; Fang, Y.; Cremers, A.; Emans, P.; Welting, T.; Peffers, M. MicroRNA profiling in cartilage ageing. Int. J. Genom. 2017, 2017, 2713725. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wei, J.; Li, X.; Cheng, Y.; Chen, W.; Cui, Y.; Simoncini, T.; Gu, Z.; Yang, J.; Fu, X. 17beta-estradiol enhances vascular endothelial Ets-1/miR-126-3p expression: The possible mechanism for attenuation of atherosclerosis. J. Clin. Endocrinol. Metab. 2017, 102, 594–603. [Google Scholar] [PubMed]
- Li, Y.; Li, Y.; Ge, P.; Ma, C. Mir-126 regulates the ERK pathway via targeting KRAS to inhibit the glioma cell proliferation and invasion. Mol. Neurobiol. 2017, 54, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Cheon, E.J.; Lee, M.H.; Kim, H.A. MicroRNA-127-5p regulates matrix metalloproteinase 13 expression and interleukin-1beta-induced catabolic effects in human chondrocytes. Arthritis Rheumtol. 2013, 65, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.; Li, Y.; Zeng, C.; Deng, Z.; Gao, S.; Xiao, W.; Luo, W.; Jiang, W.; Li, L.; Lei, G. MicroRNA-127-5p regulates osteopontin expression and osteopontin-mediated proliferation of human chondrocytes. Sci. Rep. 2016, 6, 25032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makki, M.S.; Haqqi, T.M. Mir-139 modulates mcpip1/IL-6 expression and induces apoptosis in human OA chondrocytes. Exp. Mol. Med. 2015, 47, e189. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Nakasa, T.; Otsuki, S.; Grogan, S.P.; Higashiyama, R.; Inoue, A.; Kato, Y.; Sato, T.; Lotz, M.K.; Asahara, H. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheumtol. 2009, 60, 2723–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, H.; Zeng, Y.; Zhou, Z.; Pei, F.; Lu, Y.; Cheng, J.; Shen, B. Expression of miRNA-140 in chondrocytes and synovial fluid of knee joints in patients with osteoarthritis. Chin. Med. Sci. J. 2016, 31, 207–212. [Google Scholar] [CrossRef]
- Liang, Z.J.; Zhuang, H.; Wang, G.X.; Li, Z.; Zhang, H.T.; Yu, T.Q.; Zhang, B.D. MiRNA-140 is a negative feedback regulator of MMP-13 in IL-1beta-stimulated human articular chondrocyte c28/i2 cells. Inflamm. Res. 2012, 61, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Kitajima, S.; Li, F.; Cheng, C.; Takegami, Y.; Kohno, S.; Wan, Y.S.; Hayashi, N.; Muranaka, H.; Nishimoto, Y.; et al. MicroRNA-140 mediates RB tumor suppressor function to control stem cell-like activity through interleukin-6. Oncotarget 2017, 8, 13872–13885. [Google Scholar] [CrossRef] [PubMed]
- Tardif, G.; Pelletier, J.P.; Fahmi, H.; Hum, D.; Zhang, Y.; Kapoor, M.; Martel-Pelletier, J. Nfat3 and TGF-beta/smad3 regulate the expression of mir-140 in osteoarthritis. Arthritis Res. Ther. 2013, 15, R197. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Kang, X.; Xing, Y.; Dou, C.; Kang, F.; Li, J.; Quan, Y.; Dong, S. Effect of microRNA-145 on il-1beta-induced cartilage degradation in human chondrocytes. FEBS Lett. 2014, 588, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Zhao, X.; Wang, C.; Geng, Y.; Zhao, J.; Xu, J.; Zuo, B.; Zhao, C.; Wang, C.; Zhang, X. MicroRNA-145 attenuates TNF-alpha-driven cartilage matrix degradation in osteoarthritis via direct suppression of mkk4. Cell Death Dis. 2017, 8, e3140. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.D.; Zhao, X.W.; Zhang, Y.G.; Kong, Y.; Niu, S.S.; Ma, L.F.; Zhang, Y.M. Effects of mir-145 on the inhibition of chondrocyte proliferation and fibrosis by targeting tnfrsf11b in human osteoarthritis. Mol. Med. Rep. 2017, 15, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sanchez, A.; Dudek, K.A.; Murphy, C.L. Regulation of human chondrocyte function through direct inhibition of cartilage master regulator SOX9 by microRNA-145 (miRNA-145). J. Biol. Chem. 2012, 287, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Lorente-Cebrian, S.; Mejhert, N.; Kulyte, A.; Laurencikiene, J.; Astrom, G.; Heden, P.; Ryden, M.; Arner, P. MicroRNAs regulate human adipocyte lipolysis: Effects of mir-145 are linked to TNF-alpha. PLoS ONE 2014, 9, e86800. [Google Scholar] [CrossRef] [PubMed]
- Doberstein, K.; Steinmeyer, N.; Hartmetz, A.K.; Eberhardt, W.; Mittelbronn, M.; Harter, P.N.; Juengel, E.; Blaheta, R.; Pfeilschifter, J.; Gutwein, P. MicroRNA-145 targets the metalloprotease ADAM17 and is suppressed in renal cell carcinoma patients. Neoplasia 2013, 15, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Budd, E.; de Andres, M.C.; Sanchez-Elsner, T.; Oreffo, R.O.C. Mir-146b is down-regulated during the chondrogenic differentiation of human bone marrow derived skeletal stem cells and up-regulated in osteoarthritis. Sci. Rep. 2017, 7, 46704. [Google Scholar] [CrossRef] [PubMed]
- Soyocak, A.; Kurt, H.; Ozgen, M.; Turgut Cosan, D.; Colak, E.; Gunes, H.V. MiRNA-146a, miRNA-155 and jnk expression levels in peripheral blood mononuclear cells according to grade of knee osteoarthritis. Gene 2017, 627, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhao, J.; Jing, W.; Yan, S.; Wang, X.; Xiao, C.; Ma, B. Role of miR-146a in human chondrocyte apoptosis in response to mechanical pressure injury in vitro. Int. J. Mol. Med. 2014, 34, 451–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Gibson, G.; Kim, J.S.; Kroin, J.; Xu, S.; van Wijnen, A.J.; Im, H.J. MicroRNA-146a is linked to pain-related pathophysiology of osteoarthritis. Gene 2011, 480, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Y.; Bai, W.D.; Liu, J.Q.; Zheng, Z.; Guan, H.; Zhou, Q.; Su, L.L.; Xie, S.T.; Wang, Y.C.; Li, J.; et al. Up-regulation of FGFBP1 signaling contributes to mir-146a-induced angiogenesis in human umbilical vein endothelial cells. Sci. Rep. 2016, 6, 25272. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Prado, S.; Cicione, C.; Muinos-Lopez, E.; Hermida-Gomez, T.; Oreiro, N.; FeRNAndez-Lopez, C.; Blanco, F.J. Characterization of microRNA expression profiles in normal and osteoarthritic human chondrocytes. BMC Musculoskelet. Disord. 2012, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- De Palma, A.; Cheleschi, S.; Pascarelli, N.A.; Giannotti, S.; Galeazzi, M.; Fioravanti, A. Hydrostatic pressure as epigenetic modulator in chondrocyte cultures: A study on miRNA-155, miRNA-181a and miRNA-223 expression levels. J. Biomech. 2018, 66, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Zhou, Z.H.; Zou, J. MicroRNA-181 inhibits proliferation and promotes apoptosis of chondrocytes in osteoarthritis by targeting PTEN. Biochem. Cell Biol. 2017, 95, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Gabler, J.; Ruetze, M.; Kynast, K.L.; Grossner, T.; Diederichs, S.; Richter, W. Stage-specific mirs in chondrocyte maturation: Differentiation-dependent and hypertrophy-related miR clusters and the miR-181 family. Tissue Eng. Part A 2015, 21, 2840–2851. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Meng, R.; Li, H.; Li, J.; Jing, L.; Qin, L.; Gao, Y. Mir-181a modulates chondrocyte apoptosis by targeting glycerol-3-phosphate dehydrogenase 1-like protein (GPD1L) in osteoarthritis. Med. Sci. Monit. 2017, 23, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Schwind, S.; Santhanam, R.; Eisfeld, A.K.; Chiang, C.L.; Lankenau, M.; Yu, B.; Hoellerbauer, P.; Jin, Y.; Tarighat, S.S.; et al. Targeting the ras/mapk pathway with mir-181a in acute myeloid leukemia. Oncotarget 2016, 7, 59273–59286. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 activation of miR-21 and mir-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, X.; Chen, H.P.; Li, L.; Xie, W.; Lan, G.; Zhao, Z.W.; Zheng, X.L.; Wang, Z.B.; Tang, C.K. MicroRNA-186 promotes macrophage lipid accumulation and secretion of pro-inflammatory cytokines by targeting cystathionine gamma-lyase in thp-1 macrophages. Atherosclerosis 2016, 250, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jiang, H.; Wang, S.; Chen, B. Dual functional microRNA-186-5p targets both FGF2 and RelA to suppress tumorigenesis of glioblastoma multiforme. Cell. Mol. Neurobiol. 2017, 37, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Meng, D.; Li, G.; Xu, J.; Tian, K.; Li, Y. Overexpression of microRNA-210 promotes chondrocyte proliferation and extracellular matrix deposition by targeting hif-3alpha in osteoarthritis. Mol. Med. Rep. 2016, 13, 2769–2776. [Google Scholar] [CrossRef] [PubMed]
- Phuah, N.H.; Azmi, M.N.; Awang, K.; Nagoor, N.H. Down-regulation of microRNA-210 confers sensitivity towards 1’s-1’-acetoxychavicol acetate (ACA) in cervical cancer cells by targeting SMAD4. Mol. Cells 2017, 40, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Bavelloni, A.; Ramazzotti, G.; Poli, A.; Piazzi, M.; Focaccia, E.; Blalock, W.; Faenza, I. MiRNA-210: A current overview. Anticancer Res. 2017, 37, 6511–6521. [Google Scholar] [PubMed]
- Liu, S.C.; Chuang, S.M.; Hsu, C.J.; Tsai, C.H.; Wang, S.W.; Tang, C.H. CTGF increases vascular endothelial growth factor-dependent angiogenesis in human synovial fibroblasts by increasing miR-210 expression. Cell Death Dis. 2014, 5, e1485. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhao, F.C.; Pang, Y.; Li, D.Y.; Yao, S.C.; Sun, S.S.; Guo, K.J. Downregulation of miR-221-3p contributes to IL-1beta-induced cartilage degradation by directly targeting the SDF1/CXCR4 signaling pathway. J. Mol. Med. (Berl.) 2017, 95, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, Y.; Deng, M.; Li, J.; Cai, H.; Meng, Q.; Fang, W.; Long, X.; Ke, J. MicroRNA221-3p modulates Ets-1 expression in synovial fibroblasts from patients with osteoarthritis of temporomandibular joint. Osteoarthr. Cartil. 2016, 24, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.W.; Wang, Y.T.; Liao, Y.C.; Chuang, S.C.; Wang, S.N.; Juo, S.H. Decreased microRNA-221 is associated with high levels of TNF-alpha in human adipose tissue-derived mesenchymal stem cells from obese woman. Cell. Physiol. Biochem. 2013, 32, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Guan, Y.; Tian, S.; Wang, Y.; Sun, K.; Chen, Q. Mechanical and il-1beta responsive mir-365 contributes to osteoarthritis development by targeting histone deacetylase 4. Int. J. Mol. Sci. 2016, 17, 436. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Xiao, S.B.; Xu, P.; Xie, Q.; Cao, L.; Wang, D.; Luo, R.; Zhong, Y.; Chen, H.C.; Fang, L.R. miR-365, a novel negative regulator of interleukin-6 gene expression, is cooperatively regulated by Sp1 and NF-kappaB. J. Biol. Chem. 2011, 286, 21401–21412. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, Y.; Zhao, X.; Meng, C.; Ma, L.; Kong, Y. MicroRNA-411 inhibited matrix metalloproteinase 13 expression in human chondrocytes. Am. J. Transl. Res. 2015, 7, 2000–2006. [Google Scholar] [PubMed]
- Wang, H.; Zhang, H.; Sun, Q.; Wang, Y.; Yang, J.; Yang, J.; Zhang, T.; Luo, S.; Wang, L.; Jiang, Y.; et al. Intra-articular delivery of antago-mir-483-5p inhibits osteoarthritis by modulating matrilin 3 and tissue inhibitor of metalloproteinase 2. Mol. Ther. 2017, 25, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Li, J.; Wei, B.; Huo, W.; Wang, L. MicroRNA-483-5p modulates the expression of cartilage-related genes in human chondrocytes through down-regulating tgf-beta1 expression. Tohoku J. Exp. Med. 2017, 243, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kim, D.; Lee, C.H.; Lee, M.S.; Chun, C.H.; Jin, E.J. MicroRNA-488 regulates zinc transporter SLC39A8/ZIP8 during pathogenesis of osteoarthritis. J. Biomed. Sci. 2013, 20, 31. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jeon, J.; Shin, M.; Won, Y.; Lee, M.; Kwak, J.S.; Lee, G.; Rhee, J.; Ryu, J.H.; Chun, C.H.; et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell 2014, 156, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Rolauffs, B.; Rothdiener, M.; Bahrs, C.; Badke, A.; Weise, K.; Kuettner, K.E.; Kurz, B.; Aurich, M.; Grodzinsky, A.J.; Aicher, W.K. Onset of preclinical osteoarthritis: The angular spatial organization permits early diagnosis. Arthritis Rheumtol. 2011, 63, 1637–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.D.; Chubinskaya, S.; Guilak, F.; Martin, J.A.; Oegema, T.R.; Olson, S.A.; Buckwalter, J.A. Post-traumatic osteoarthritis: Improved understanding and opportunities for early intervention. J. Orthop. Res. 2011, 29, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Lieberthal, J.; Sambamurthy, N.; Scanzello, C.R. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Rolauffs, B.; Kurz, B.; Felka, T.; Rothdiener, M.; Uynuk-Ool, T.; Aurich, M.; Frank, E.; Bahrs, C.; Badke, A.; Stockle, U.; et al. Stress-vs-time signals allow the prediction of structurally catastrophic events during fracturing of immature cartilage and predetermine the biomechanical, biochemical, and structural impairment. J. Struct. Biol. 2013, 183, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolauffs, B.; Muehleman, C.; Li, J.; Kurz, B.; Kuettner, K.E.; Frank, E.; Grodzinsky, A.J. Vulnerability of the superficial zone of immature articular cartilage to compressive injury. Arthritis Rheumtol. 2010, 62, 3016–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrendt, P.; Feldheim, M.; Preusse-Prange, A.; Weitkamp, J.T.; Haake, M.; Eglin, D.; Rolauffs, B.; Fay, J.; Seekamp, A.; Grodzinsky, A.J.; et al. Chondrogenic potential of IL-10 in mechanically injured cartilage and cellularized collagen aci grafts. Osteoarthr. Cartil. 2018, 26, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, P.; Preusse-Prange, A.; Kluter, T.; Haake, M.; Rolauffs, B.; Grodzinsky, A.J.; Lippross, S.; Kurz, B. IL-10 reduces apoptosis and extracellular matrix degradation after injurious compression of mature articular cartilage. Osteoarthr. Cartil. 2016, 24, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg, J.; Rolauffs, B.; Grodzinsky, A.J.; Schunke, M.; Kurz, B. Estrogen reduces mechanical injury-related cell death and proteoglycan degradation in mature articular cartilage independent of the presence of the superficial zone tissue. Osteoarthr. Cartil. 2013, 21, 1738–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boehme, K.A.; Rolauffs, B. Onset and Progression of Human Osteoarthritis—Can Growth Factors, Inflammatory Cytokines, or Differential miRNA Expression Concomitantly Induce Proliferation, ECM Degradation, and Inflammation in Articular Cartilage? Int. J. Mol. Sci. 2018, 19, 2282. https://doi.org/10.3390/ijms19082282
Boehme KA, Rolauffs B. Onset and Progression of Human Osteoarthritis—Can Growth Factors, Inflammatory Cytokines, or Differential miRNA Expression Concomitantly Induce Proliferation, ECM Degradation, and Inflammation in Articular Cartilage? International Journal of Molecular Sciences. 2018; 19(8):2282. https://doi.org/10.3390/ijms19082282
Chicago/Turabian StyleBoehme, Karen A., and Bernd Rolauffs. 2018. "Onset and Progression of Human Osteoarthritis—Can Growth Factors, Inflammatory Cytokines, or Differential miRNA Expression Concomitantly Induce Proliferation, ECM Degradation, and Inflammation in Articular Cartilage?" International Journal of Molecular Sciences 19, no. 8: 2282. https://doi.org/10.3390/ijms19082282