Factors Associated with Heritable Pulmonary Arterial Hypertension Exert Convergent Actions on the miR-130/301-Vascular Matrix Feedback Loop


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
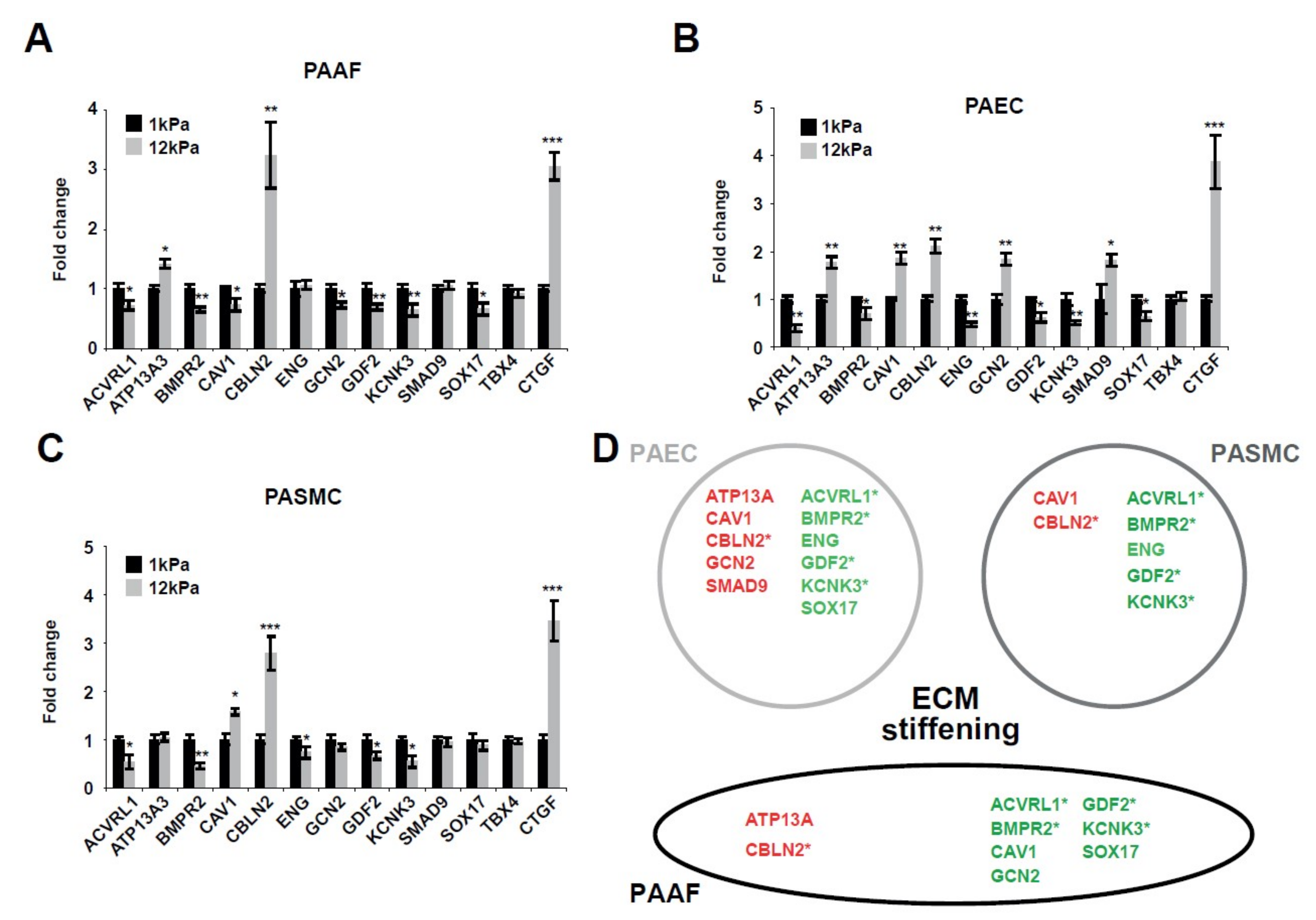
2.1. Matrix Stiffening Modulates the Expression of Several Factors Genetically Associated with PAH
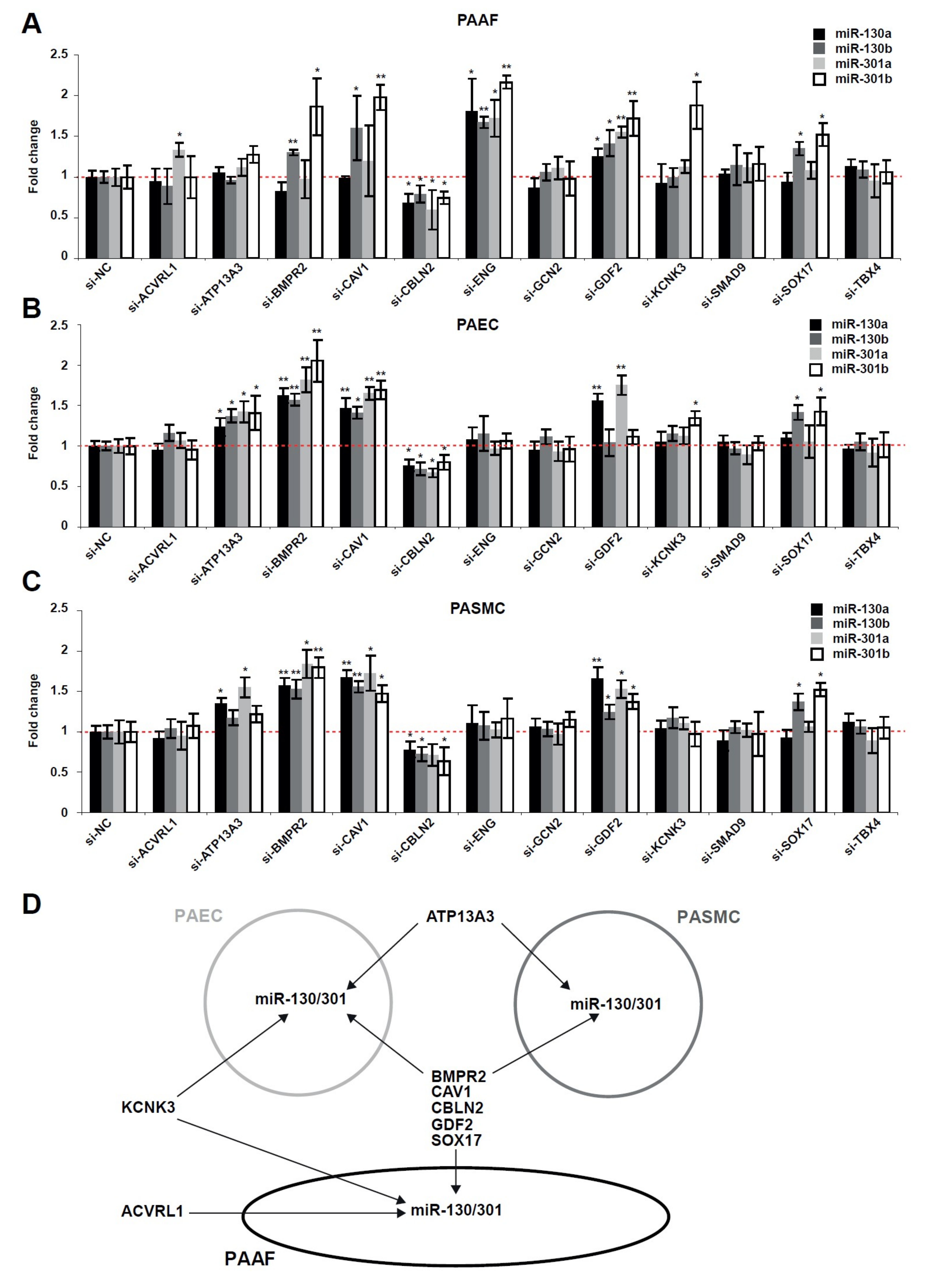
2.2. The miR-130/301 Family is Modulated by Several Factors Genetically Associated with PAH
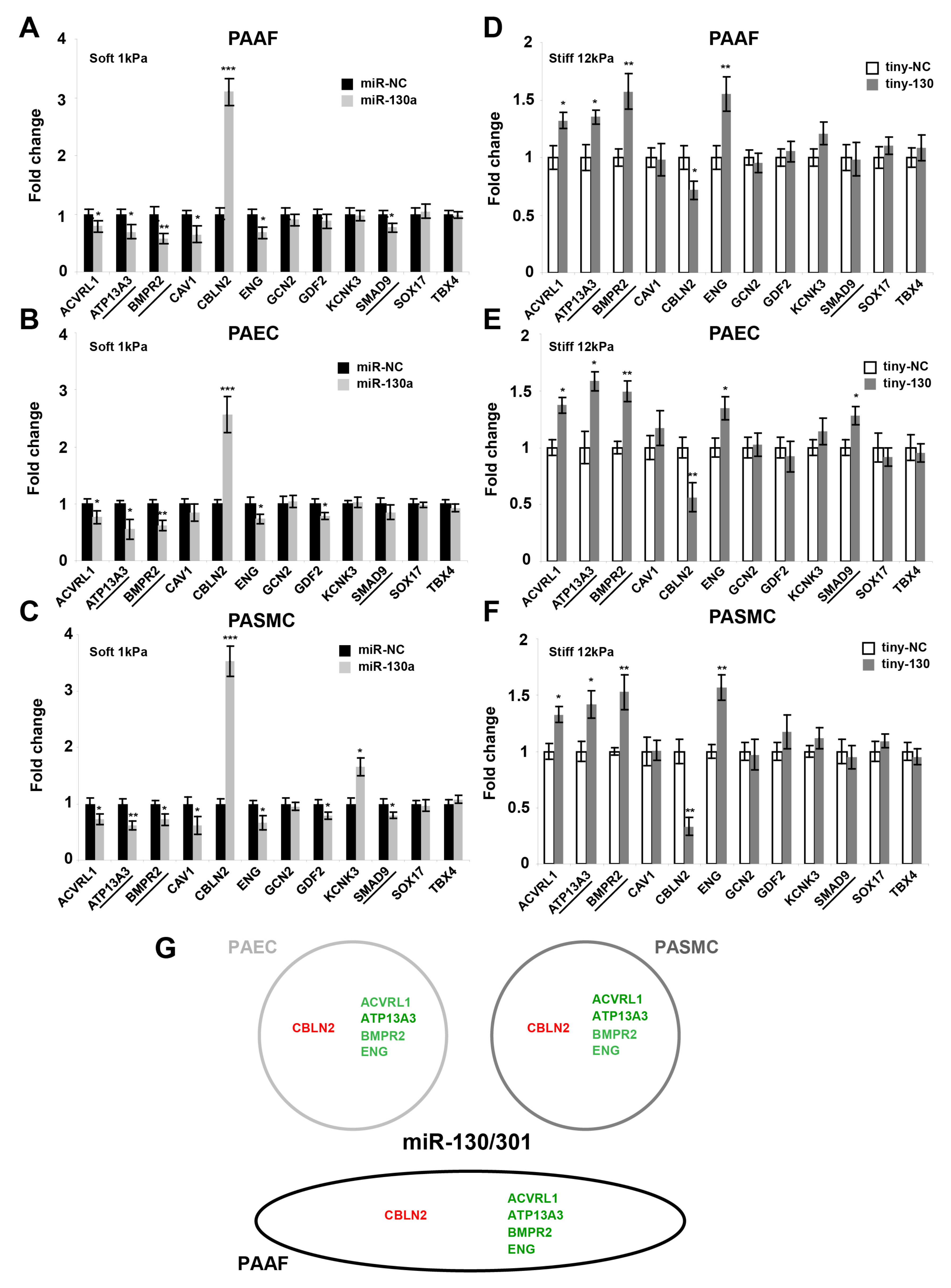
2.3. The miR-130/301 Family Controls the Vascular Expression of Factors Associated with Hereditary PAH
2.4. Factors Genetically Associated with PAH Broadly Impact the PPARγ-ApoE-LRP8-Matrix Remodeling Axis
2.5. A Computational Network Biology Approach Predicts Additional Molecular Pathways Linking Genes of Heritable PAH to the miR-130/301-ECM Axis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Culture Reagents
4.2. Oligonucleotides and Transfection
4.3. Messenger RNA and miRNA Extraction
4.4. Quantitative RT-PCR of Mature miRNAs
4.5. Quantitative RT-PCR of Messenger RNAs
4.6. Computational Gene Network Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
References
- Farber, H.; Loscalzo, J. Pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ. Res. 2014, 115, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Girerd, B.; Weatherald, J.; Montani, D.; Humbert, M. Heritable pulmonary hypertension: From bench to bedside. Eur. Respir. Rev. 2017, 26, 170037. [Google Scholar] [CrossRef] [PubMed]
- Graf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.K.; Sampson, K.S.; Kass, R.S. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 2162. [Google Scholar] [CrossRef] [PubMed]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.; Roofthooft, M.T.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyries, M.; Montani, D.; Girerd, B.; Perret, C.; Leroy, A.; Lonjou, C.; Chelghoum, N.; Coulet, F.; Bonnet, D.; Dorfmuller, P.; et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat. Genet. 2014, 46, 65. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Eyries, M.; Montani, D.; Poirier, O.; Girerd, B.; Dorfmuller, P.; Coulet, F.; Nadaud, S.; Maugenre, S.; Guignabert, C.; et al. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat. Genet. 2013, 45, 518–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, T.; Cotrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Haeger, C.M.; Dieffenbach, P.B.; Sicard, D.; Chrobak, I.; Coronata, A.M.; Suarez Velandia, M.M.; Vitali, S.; Colas, R.A.; Norris, P.C.; et al. Distal vessel stiffening is an early and pivotal mechanobiological regulator of vascular remodeling and pulmonary hypertension. JCI Insight 2016, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.; Nishimura, R.A.; Sorajja, P.; Cha, S.; McGoon, M.D. Relationship of pulmonary arterial capacitance and mortality in idiopathic pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2006, 47, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Gan, C.T.; Lankhaar, J.W.; Westerhof, N.; Marcus, J.T.; Becker, A.; Twisk, J.W.; Boonstra, A.; Postmus, P.E.; Vonk-Noordegraaf, A. Noninvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest 2007, 132, 1906–1912. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.S.; Lee, P.F.; Lanning, C.J.; Ivy, D.D.; Kirby, K.S.; Claussen, L.R.; Chan, K.C.; Shandas, R. Pulmonary vascular input impedance is a combined measure of pulmonary vascular resistance and stiffness and predicts clinical outcomes better than pulmonary vascular resistance alone in pediatric patients with pulmonary hypertension. Am. Heart J. 2008, 155, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Tabima, D.M.; Roldan-Alzate, A.; Wang, Z.; Hacker, T.A.; Molthen, R.C.; Chesler, N.C. Persistent vascular collagen accumulation alters hemodynamic recovery from chronic hypoxia. J. Biomech. 2012, 45, 799–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, E.M.; Iyer, N.; Ilsar, R.; Bailey, B.P.; Adams, M.R.; Celermajer, D.S. Abnormal pulmonary artery stiffness in pulmonary arterial hypertension: In vivo study with intravascular ultrasound. PLoS ONE 2012, 7, e33331. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Cottrill, K.; Krauszman, A.; Lu, Y.; Annis, S.; Hale, A.; Bhat, B.; Waxman, A.B.; Chau, B.N.; Kuebler, W.M.; et al. The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J. Biol. Chem. 2014, 290, 2069–2085. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Lu, Y.; Annis, S.; Hale, A.; Bhat, B.; Saggar, R.; Saggar, R.; Wallace, W.D.; Ross, D.J.; Vargas, S.O.; et al. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J. Clin. Invest. 2014, 124, 3514–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Invest. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negi, V.; Chan, S.Y. Discerning functional hierarchies of microRNAs in pulmonary hypertension. JCI Insight 2017, 2, e91327. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Cottrill, K.A.; Annis, S.; Bhat, B.; Gochuico, B.R.; Osorio, J.C.; Rosas, I.; Haley, K.J.; Corey, K.E.; Chung, R.T.; et al. A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci. Rep. 2015, 5, 18277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Austin, E.D.; Hemnes, A.R.; Loyd, J.E.; Zhao, Z. An evidence-based knowledgebase of pulmonary arterial hypertension to identify genes and pathways relevant to pathogenesis. Mol. Biosyst. 2014, 10, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Oughtred, R.; Boucher, L.; Heinicke, S.; Chen, D.; Stark, C.; Breitkreutz, A.; Kolas, N.; O’Donnell, L.; et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res. 2015, 43, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Waegele, B.; Lechner, M.; Brauner, B.; Dunger-Kaltenbach, I.; Fobo, G.; Frishman, G.; Montrone, C.; Mewes, H.W. CORUM: The comprehensive resource of mammalian protein complexes 2009. Nucleic Acids Res. 2010, 38, D497–D501. [Google Scholar] [CrossRef] [PubMed]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond—Recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Launay, G.; Salza, R.; Multedo, D.; Thierry-Mieg, N.; Ricard-Blum, S. MatrixDB, the extracellular matrix interaction database: Updated content, a new navigator and expanded functionalities. Nucleic Acids Res. 2015, 43, D321–D327. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.; Mukherjee, S.; Ebert, B.; Gillette, M.; Paulovich, A.; Pomeroy, S.; Golub, T.; Lander, E.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Meloche, J.; Paulin, R.; Boucherat, O.; Provencher, S.; Bonnet, S. DNA Damage and Pulmonary Hypertension. Int. J. Mol. Sci. 2016, 17, 990. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Loscalzo, J. Pathogenic mechanisms of pulmonary arterial hypertension. J. Mol. Cell. Cardiol. 2008, 44, 14–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pousada, G.; Baloira, A.; Valverde, D. Complex inheritance in Pulmonary Arterial Hypertension patients with several mutations. Sci. Rep. 2016, 6, 33570. [Google Scholar] [CrossRef] [PubMed]
- Hadinnapola, C.; Bleda, M.; Haimel, M.; Screaton, N.; Swift, A.J.; Dorfmuller, P.; Preston, S.D.; Southwood, M.; Hernandez-Sanchez, J.; Martin, J.; et al. Phenotypic Characterisation of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically with Pulmonary Arterial Hypertension. Circulation 2017, 136, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Madan, M.; Patel, A.; Skruber, K.; Geerts, D.; Altomare, D.A.; Iv, O.P. ATP13A3 and caveolin-1 as potential biomarkers for difluoromethylornithine-based therapies in pancreatic cancers. Am. J. Cancer Res. 2016, 6, 1231–1252. [Google Scholar] [PubMed]
- Michelakis, E.D. Spatio-temporal diversity of apoptosis within the vascular wall in pulmonary arterial hypertension: Heterogeneous BMP signaling may have therapeutic implications. Circ. Res. 2006, 98, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Kanai-Azuma, M.; Hara, K.; Matoba, S.; Hiramatsu, R.; Kawakami, H.; Kurohmaru, M.; Koopman, P.; Kanai, Y. Redundant roles of Sox17 and Sox18 in postnatal angiogenesis in mice. J. Cell Sci. 2006, 119, 3513–3526. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Orsenigo, F.; Morini, M.F.; Pitulescu, M.E.; Bhat, G.; Nyqvist, D.; Breviario, F.; Conti, V.; Briot, A.; Iruela-Arispe, M.L.; et al. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat. Commun. 2013, 4, 2609. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.W.; Haitchi, H.M.; LeCras, T.D.; Sridharan, A.; Xu, Y.; Wert, S.E.; James, J.; Udell, N.; Thurner, P.J.; Whitsett, J.A. Sox17 is required for normal pulmonary vascular morphogenesis. Dev. Biol. 2014, 387, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Yiangou, Y.; Burnet, P.; Nikou, G.; Chrysanthou, B.J.; Bloom, S.R. Purification and characterisation of cerebellins from human and porcine cerebellum. J. Neurochem. 1989, 53, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Metzger, R.J.; Papaioannou, V.E. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet. 2012, 8, e1002866. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Liang, J.; Liu, N.; Huan, C.; Zhang, Y.; Liu, W.; Kumar, M.; Xiao, R.; D’Armiento, J.; Metzger, D.; et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J. Clin. Invest. 2016, 126, 3063–3079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Shao, N.Y.; Sa, S.; Li, D.; Termglinchan, V.; Ameen, M.; Karakikes, I.; Sosa, G.; Grubert, F.; Lee, J.; et al. Patient-Specific iPSC-Derived Endothelial Cells Uncover Pathways that Protect against Pulmonary Hypertension in BMPR2 Mutation Carriers. Cell Stem Cell 2017, 20, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 2009, 33, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, G.; Rabinovitch, M. The protective role of adiponectin in pulmonary vascular disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L1–L2. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Bigorgne, A.; Hautefort, A.; Girerd, B.; Sitbon, O.; Montani, D.; Humbert, M.; Tcherakian, C.; Perros, F. Gut-Lung Connection in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2017, 56, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids. Res. 2016, 44, D481–D487. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D. BioCarta. Bio. Softw. Int. Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertero, T.; Handen, A.L.; Chan, S.Y. Factors Associated with Heritable Pulmonary Arterial Hypertension Exert Convergent Actions on the miR-130/301-Vascular Matrix Feedback Loop. Int. J. Mol. Sci. 2018, 19, 2289. https://doi.org/10.3390/ijms19082289
Bertero T, Handen AL, Chan SY. Factors Associated with Heritable Pulmonary Arterial Hypertension Exert Convergent Actions on the miR-130/301-Vascular Matrix Feedback Loop. International Journal of Molecular Sciences. 2018; 19(8):2289. https://doi.org/10.3390/ijms19082289
Chicago/Turabian StyleBertero, Thomas, Adam L. Handen, and Stephen Y. Chan. 2018. "Factors Associated with Heritable Pulmonary Arterial Hypertension Exert Convergent Actions on the miR-130/301-Vascular Matrix Feedback Loop" International Journal of Molecular Sciences 19, no. 8: 2289. https://doi.org/10.3390/ijms19082289
APA StyleBertero, T., Handen, A. L., & Chan, S. Y. (2018). Factors Associated with Heritable Pulmonary Arterial Hypertension Exert Convergent Actions on the miR-130/301-Vascular Matrix Feedback Loop. International Journal of Molecular Sciences, 19(8), 2289. https://doi.org/10.3390/ijms19082289