Hyaluronic Acid-Decorated Chitosan Nanoparticles for CD44-Targeted Delivery of Everolimus



 ,
, 

Abstract
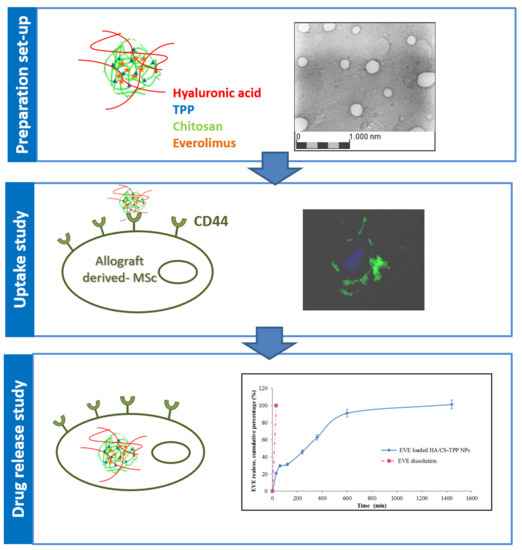
:
1. Introduction
2. Results and Discussion
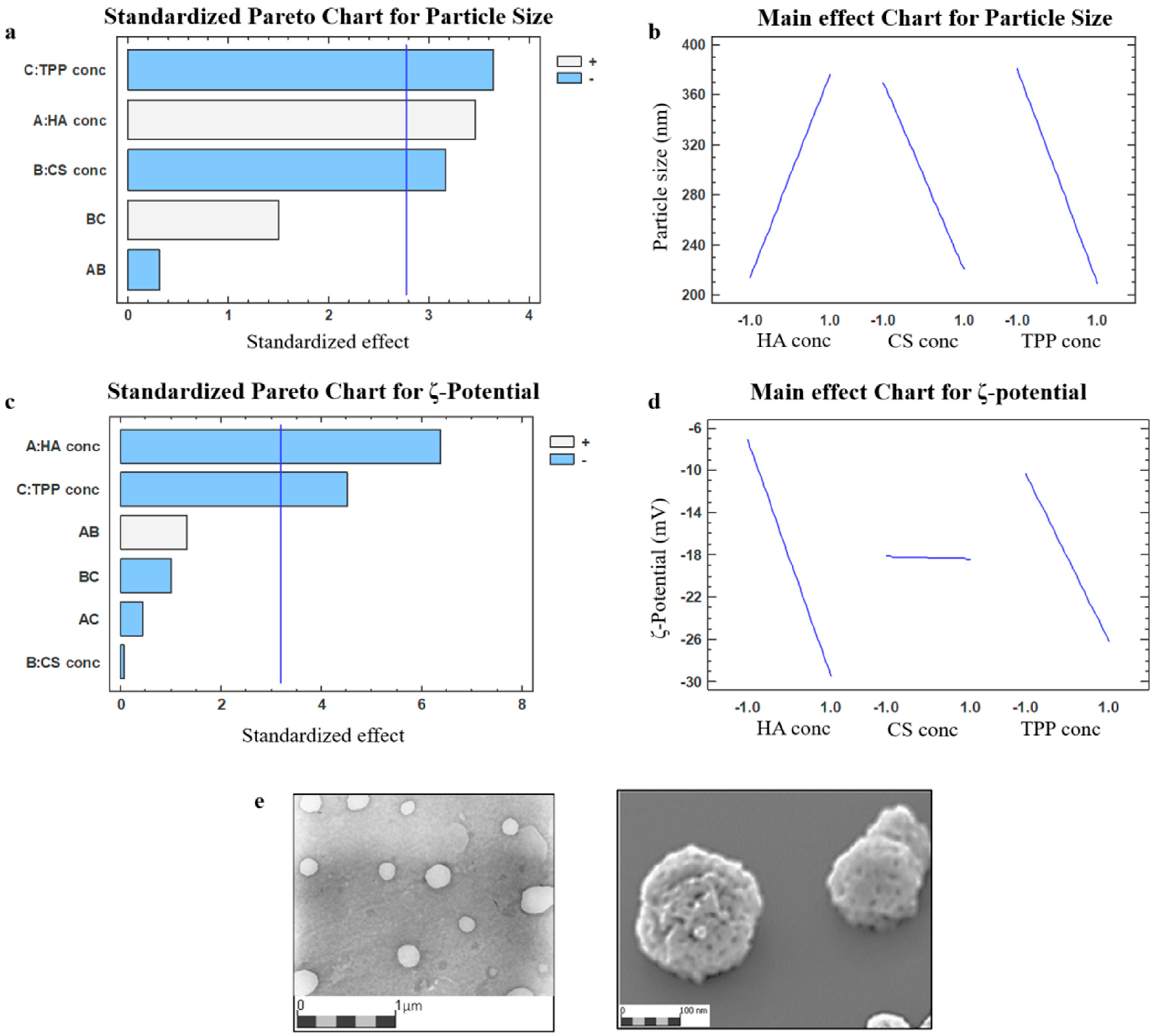
2.1. Nanoparticles Preparation Method Set Up
− 7.425 × [HA] × [CS] + 35.275 × [CS] × [TPP]
+ −0.765 × [HA] × [TPP] − 1.7475 × [CS] × [TPP]
2.2. Nanoparticles Characterization
2.3. EVE Loaded HA/CS-TPP Nanoparticles Preparation
2.4. “Ex Vivo” Cellular Tests
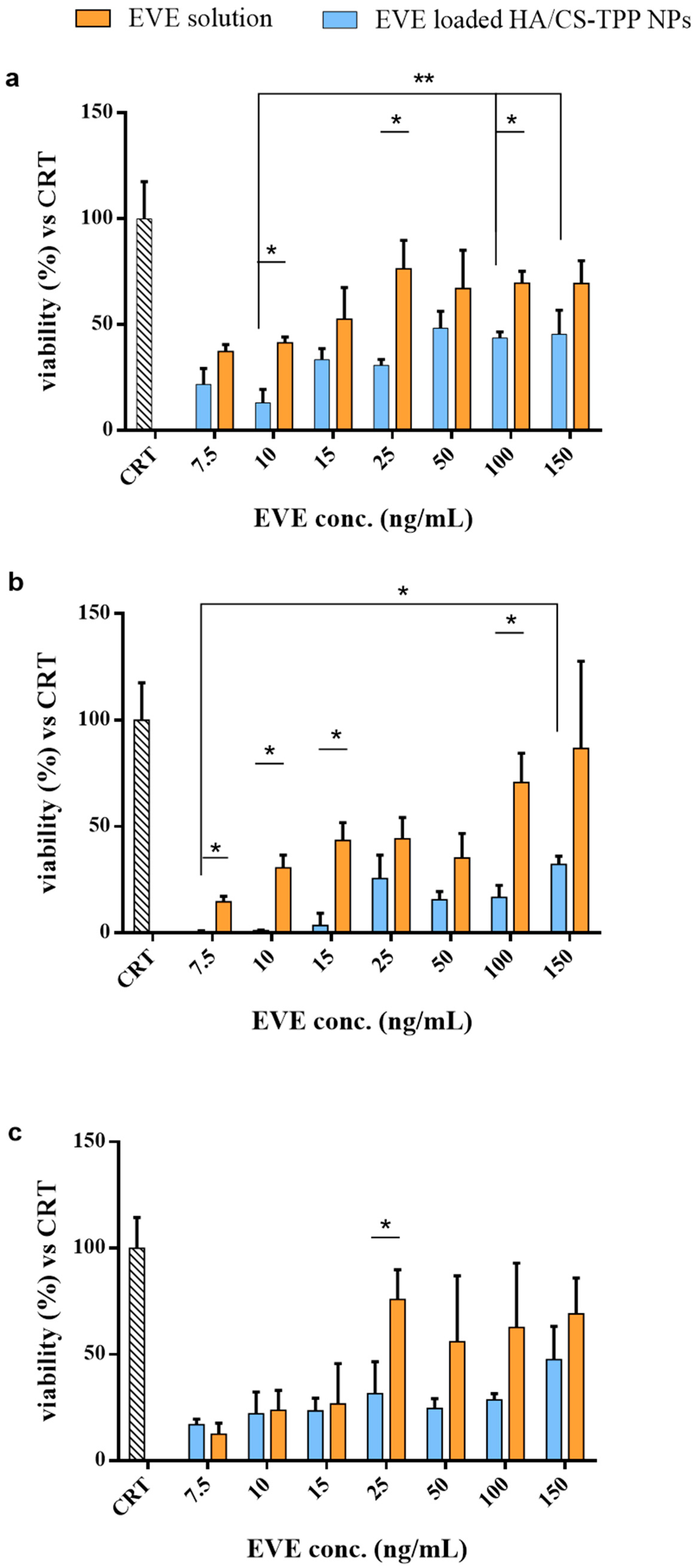
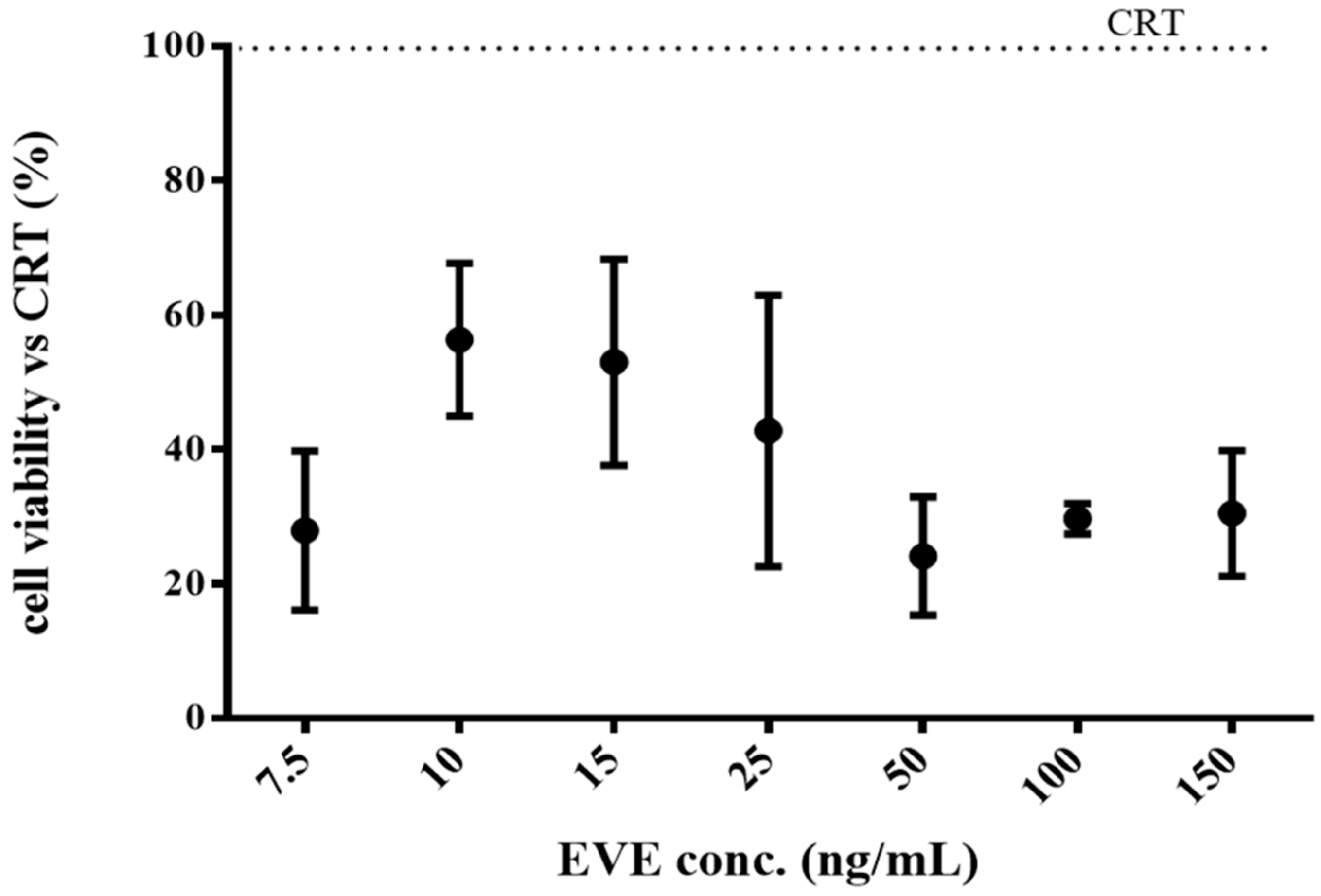
2.4.1. Cytotoxicity
2.4.2. Effect of EVE on NHDFs Growth
2.4.3. Effect of EVE on NHDFs Proliferation
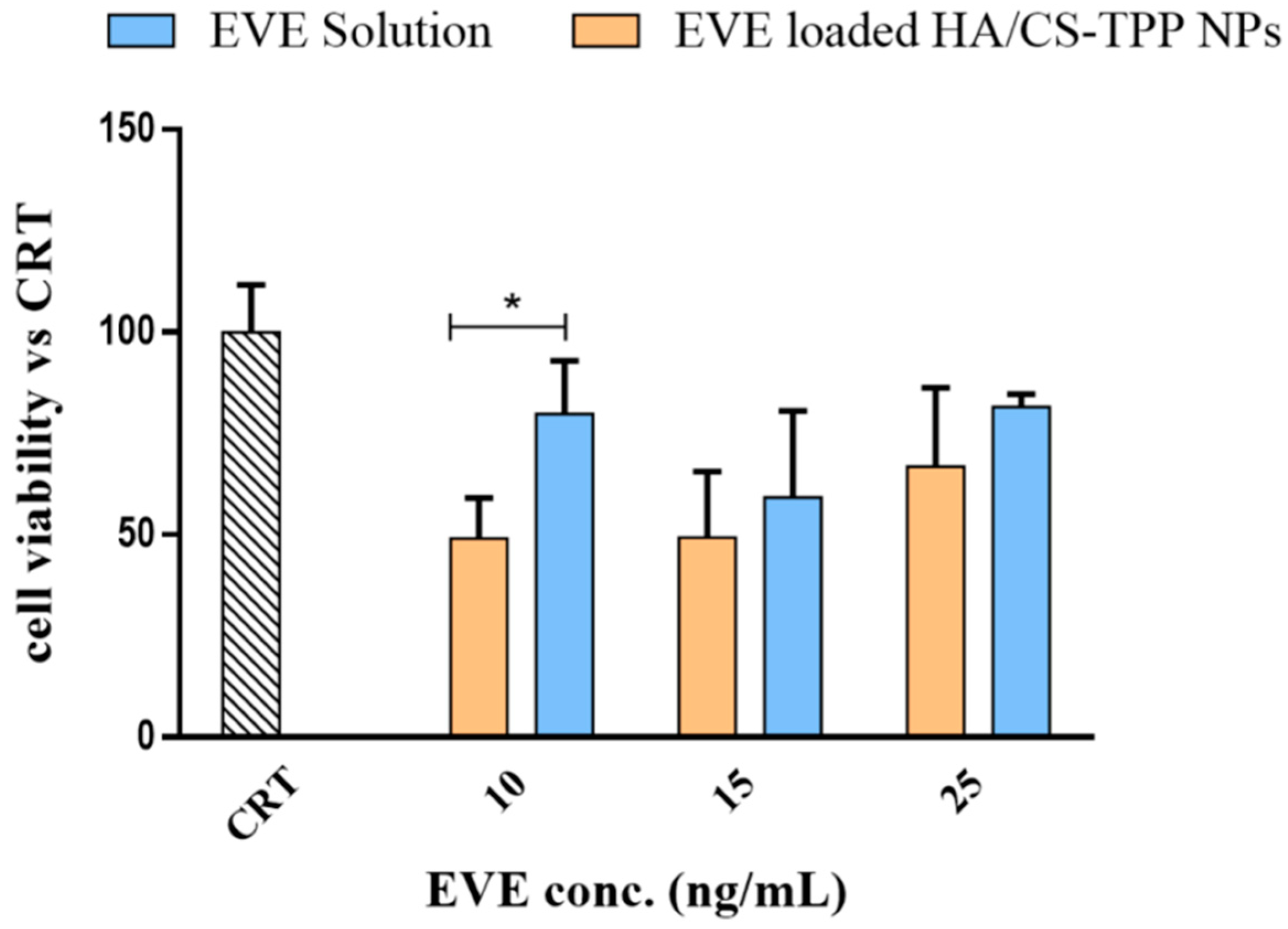
2.4.4. Effect of EVE on MSc Growth
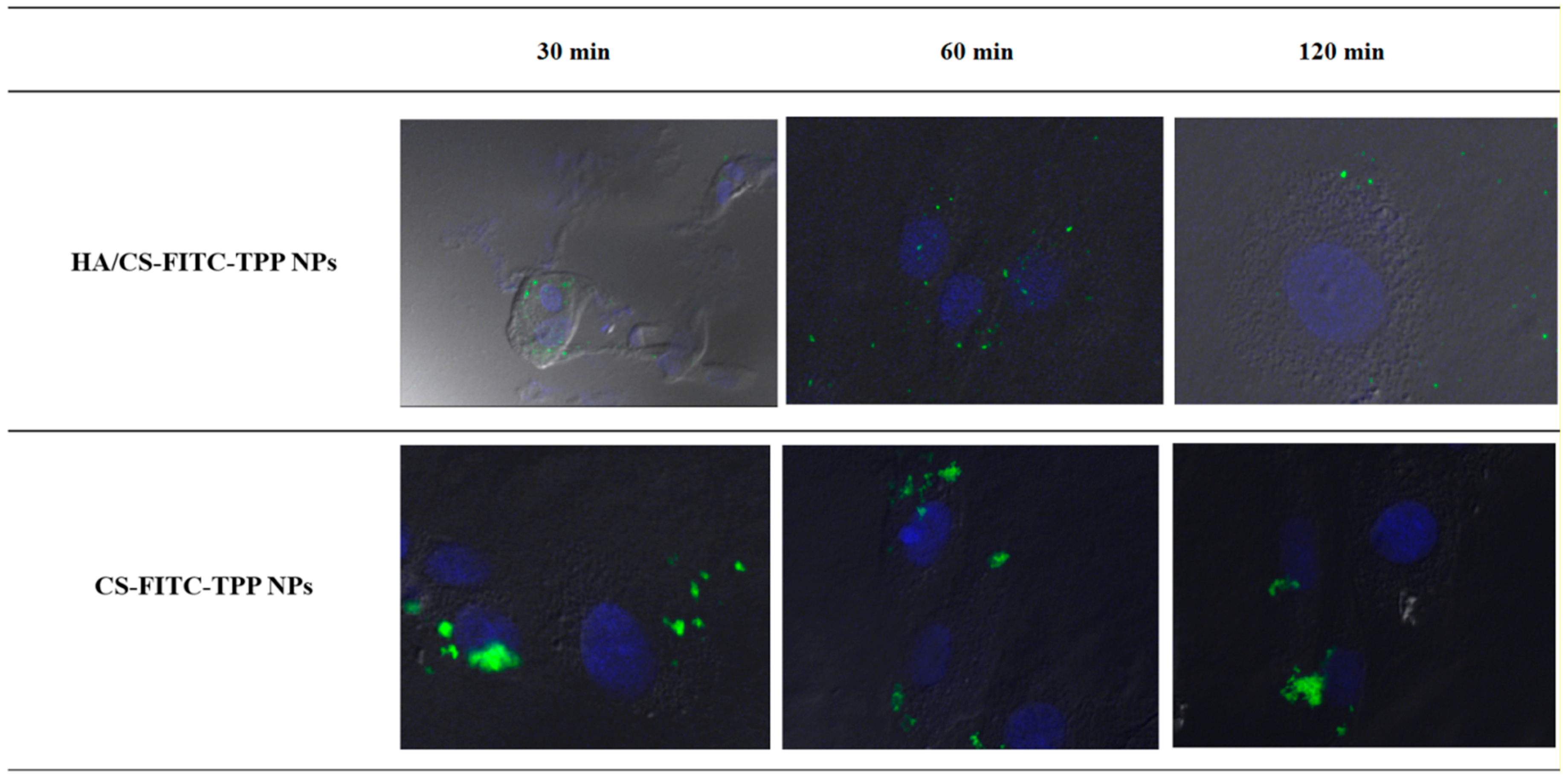
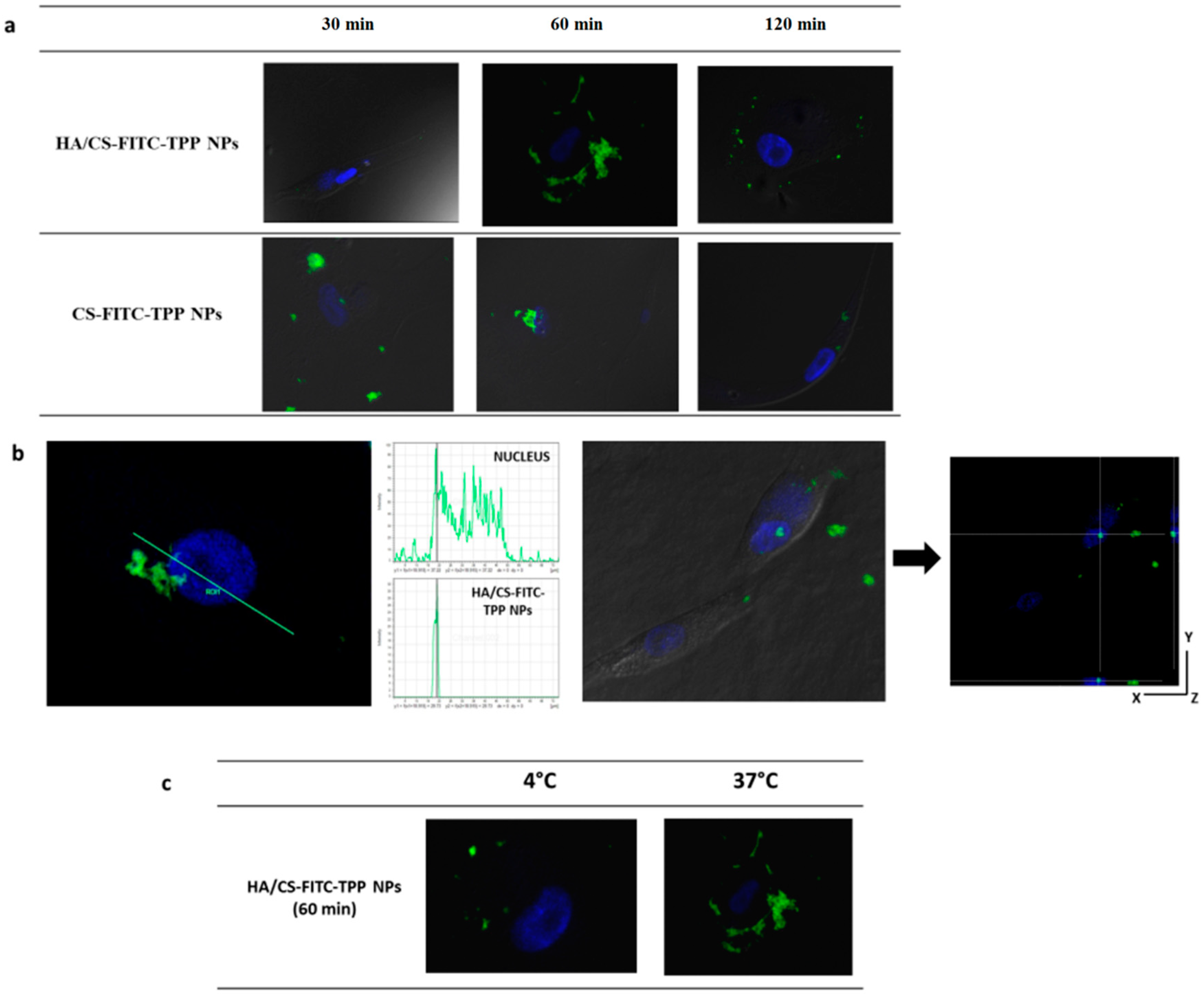
2.4.5. NPs Cellular Uptake Evaluation
3. Materials and Methods
3.1. Materials
3.2. HA/CS-TPP Nanoparticles Preparation Method Set-Up
3.2.1. Screening Design of Experiment (DoE)
3.3. Nanoparticles Characterization
3.3.1. Nanoparticles Dimensional and Morphological Characterization
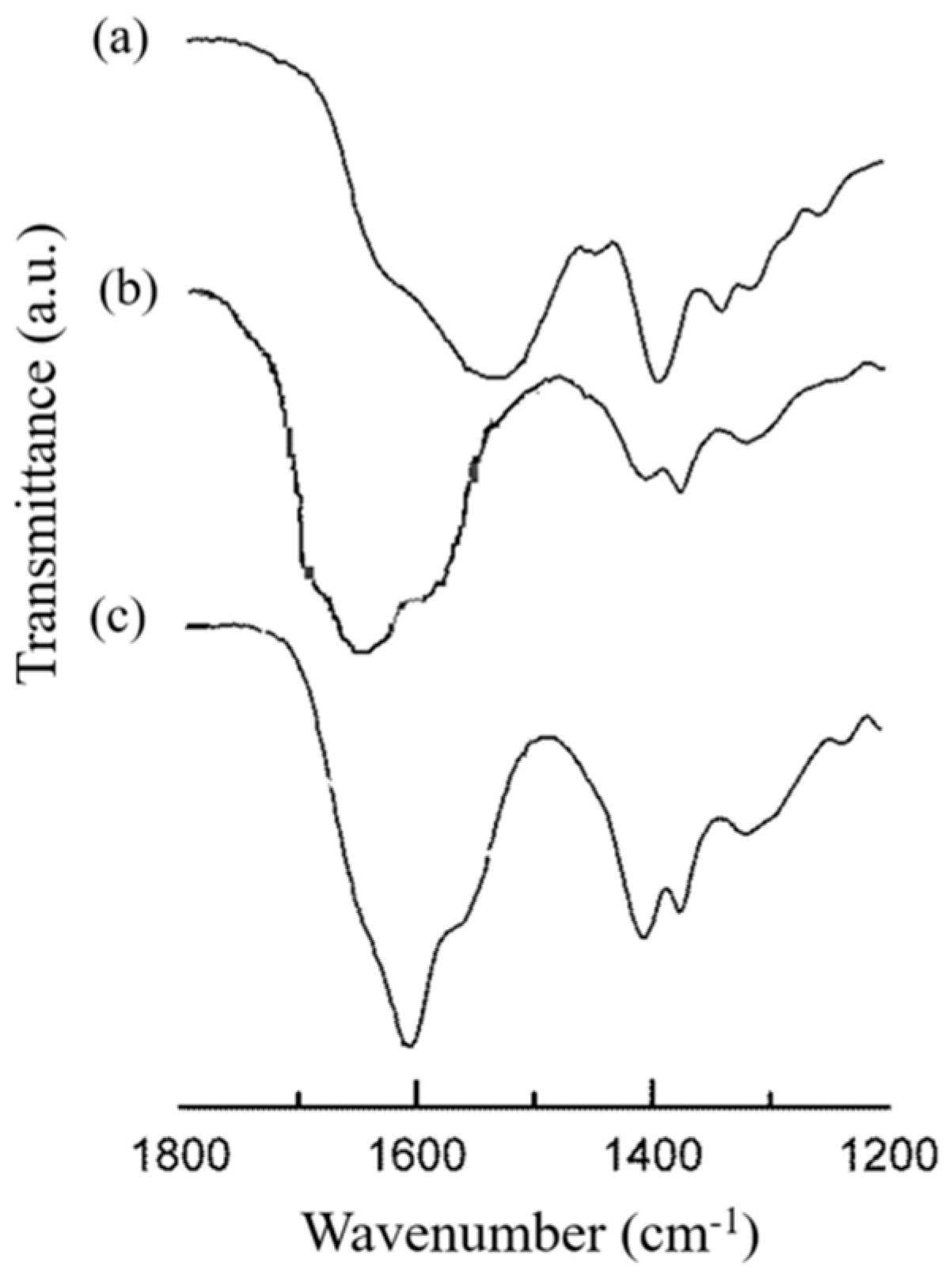
3.3.2. HA/CS-TPP Nanoparticles Polymeric Composition
Hyaluronic Acid Quantification
Chitosan Quantification
Fourier Transform Infrared (FTIR) Spectroscopy
3.4. EVE Loaded HA/CS-TPP Nanoparticles Preparation
3.5. Drug Loading and In Vitro Release of EVE Loaded HA/CS-TPP Nanoparticles
3.5.1. EVE Loading
3.5.2. In Vitro EVE Release
3.6. Ex Vivo Cellular Tests
3.6.1. Cells
3.6.2. HA/CS-TPP Nanoparticles Cytotoxicity
3.6.3. Effect of EVE Loaded HA/CS-TPP Nanoparticles on NHDFs Growth and Proliferation
3.6.4. Effect of EVE Loaded HA/CS-TPP Nanoparticles on MSc Growth
3.6.5. Nanoparticles Cellular Uptake Determination
CS-FITC Synthesis
3.7. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mabasa, V.H.; Ensom, M.H. The role of therapeutic monitoring of everolimus in solid organ transplantation. Ther. Drug Monit. 2005, 27, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Ter Heine, R.; van Erp, N.P.; Guchelaar, H.J.; de Fijter, J.W.; Reinders, M.E.J.; van Herpen, C.M.; Burger, D.M.; Moes, D.J.A.R. A pharmacological rationale for improved everolimus dosing in oncology and transplant patients. Br. J. Clin. Pharmacol. 2018, 84, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Jeng, L.-B.; Lee, S.G.; Soin, A.S.; Lee, W.-C.; Suh, K.-S.; Joo, D.J.; Uemoto, S.; Joh, J.; Yoshizumi, T.; Yang, H.-R.; et al. Efficacy and safety of everolimus with reduced tacrolimus in living- donor liver transplant recipients: 12- month results of a randomized multicenter study. Am. J. Transplant. 2018, 18, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, A.K.; Andersson, B.; Gustafsson, F.; Eiskjær, H.; Rådegran, G.; Gude, E.; Jansson, K.; Solbu, D.; Karason, K.; Arora, S.; et al. Everolimus Initiation with Early Calcineurin Inhibitor Withdrawal in De Novo Heart Transplant Recipients: Three-Year Results from the Randomized SCHEDULE Study. Am. J. Transplant. 2016, 16, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Bemelman, F.J.; de Fijter, J.W.; Kers, J.; Meyer, C.; Peters-Sengers, H.; de Maar, E.F.; van der Pant, K.A.M.I.; de Vries, A.P.J.; Sanders, J.-S.; Zwinderman, A.; et al. Early Conversion to Prednisolone/Everolimus as an Alternative Weaning Regimen Associates with Beneficial Renal Transplant Histology and Function: The Randomized-Controlled MECANO Trial. Am. J. Transplant. 2017, 17, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Pape, L.; Ganschow, R.; Ahlenstiel, T. Everolimus in pediatric transplantation. Curr. Opin. Organ Transplant. 2012, 17, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Parada, M.T.; Alba, A.; Sepulveda, C.; Melo, J. Long-term use of everolimus in lung transplant patients. Transplant. Proc. 2011, 43, 2313–2315. [Google Scholar] [CrossRef] [PubMed]
- Azzola, A.; Havryk, A.; Chhajed, P.; Hostettler, K.; Black, J.; Johnson, P.; Roth, M.; Glanville, A.; Tamm, M. Everolimus and mycophenolate mofetil are potent inhibitors of fibroblast proliferation after lung transplantation. Transplantation 2004, 77, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Belperio, J.A.; Weigt, S.S.; Fishbein, M.C.; Lynch, J.P., III. Chronic lung allograft rejection: Mechanisms and therapy. Proc. Am. Thor. Soc. 2009, 6, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.M.; Nunley, D.R.; Rojas, M.; Roman, J. Activation of Tissue Remodeling Precedes Obliterative Bronchiolitis in Lung Transplant Recipients. Biomarker Insights 2008, 3, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Strueber, M.; Warnecke, G.; Fuge, J.; Simon, A.R.; Zhang, R.; Welte, T.; Haverich, A.; Gottlieb, J. Everolimus Versus Mycophenolate Mofetil De Novo After Lung Transplantation: A Prospective, Randomized, Open-Label Trial. Am. J. Transplant. 2016, 16, 3171–3180. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.I.; Sharples, L.D.; Stewart, S. Bronchiolitis obliterans syndrome: Risk factors and therapeutic strategies. Drugs 2005, 65, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Weigt, S.S.; DerHovanessian, A.; Wallace, W.D.; Lynch, J.P., III; Belperio, J.A. Bronchiolitis obliterans syndrome: The Achilles’ heel of lung transplantation. Semin. Respir. Crit. Care Med. 2013, 34, 336–351. [Google Scholar] [PubMed]
- Budde, K.; Becker, T.; Arns, W.; Sommerer, C.; Reinke, P.; Eisenberger, U.; Kramer, S.; Fischer, W.; Gschaidmeier, H.; Pietruck, F.; et al. Everolimus-based, calcineurin-inhibitor-free regimen in recipients of de-novo kidney transplants: An open-label, randomised, controlled trial. Lancet 2011, 377, 837–847. [Google Scholar] [CrossRef]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Paplomata, E.; Zelnak, A.; O’Regan, R. Everolimus: Side effect profile and management of toxicities in breast cancer. Breast Cancer Res. Treat. 2013, 140, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Cova, E.; Colombo, M.; Inghilleri, S.; Morosini, M.; Miserere, S.; Penaranda-Avila, J.; Santini, B.; Piloni, D.; Magni, S.; Gramatica, F.; et al. Antibody-engineered nanoparticles selectively inhibit mesenchymal cells isolated from patients with chronic lung allograft dysfunction. Nanomedicine 2015, 10, 9–23. [Google Scholar] [CrossRef] [PubMed]
- De Backer, L.; Braeckmans, K.; Stuart, M.C.; Demeester, J.; De Smedt, S.C.; Raemdonck, K. Bio-inspired pulmonary surfactant-modified nanogels: A promising siRNA delivery system. J. Control. Release 2015, 206, 177–186. [Google Scholar] [CrossRef] [PubMed]
- De Backer, L.; Cerrada, A.; Pérez-Gil, J.; De Smedt, S.C.; Raemdonck, K. Bio-inspired materials in drug delivery: Exploring the role of pulmonary surfactant in siRNA inhalation therapy. J. Control. Release 2015, 220, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Kuzmov, A.; Minko, T. Nanotechnology approaches for inhalation treatment of lung diseases. J. Control. Release 2015, 219, 500–518. [Google Scholar] [CrossRef] [PubMed]
- Cova, E.; Ceccarelli, G.; Pandolfi, L.; Agozzino, M.; Allevi, R.; Benedetti, L.; Colombo, M.; Inghilleri, S.; Magni, S.; Oggionni, T.; et al. Engineered gold nanoparticles targeted to mesenchymal cells from BOS patients can be safely administered to normal mice by inhalation. Eur. Respir. J. 2015, 46 (Suppl. 59), PA1793. [Google Scholar]
- Ossipov, D.A. Nanostructured hyaluronic acid-based materials for active delivery to cancer. Expert Opin. Drug Deliv. 2010, 7, 681–703. [Google Scholar] [CrossRef] [PubMed]
- Arpicco, S.; Lerda, C.; Dalla Pozza, E.; Costanzo, C.; Tsapis, N.; Stella, B.; Donadelli, M.; Dando, I.; Fattal, E.; Cattel, L.; et al. Hyaluronic acid-coated liposomes for active targeting of gemcitabine. Eur. J. Pharm. Biopharm. 2013, 85, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peer, D.; Margalit, R. Loading mitomycin C inside long circulating hyaluronan targeted nano-liposomes increases its antitumor activity in three mice tumor models. Int. J. Cancer 2004, 108, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Margalit, R. Tumor-targeted hyaluronan nanoliposomes increase the antitumor activity of liposomal Doxorubicin in syngeneic and human xenograft mouse tumor models. Neoplasia 2004, 6, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Almalik, A.; Donno, R.; Cadman, C.J.; Cellesi, F.; Day, P.J.; Tirelli, N. Hyaluronic acid-coated chitosan nanoparticles: Molecular weight-dependent effects on morphology and hyaluronic acid presentation. J. Control. Release 2013, 172, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Almalik, A.; Karimi, S.; Ouasti, S.; Donno, R.; Wandrey, C.; Day, P.J.; Tirelli, N. Hyaluronic acid (HA) presentation as a tool to modulate and control the receptor-mediated uptake of HA-coated nanoparticles. Biomaterials 2013, 34, 5369–5380. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Cao, M.; Zhang, J.; Hu, K.; Yin, Z.; Zhou, Z.; Xiao, X.; Yang, Y.; Sheng, W.; Wu, Y.; et al. Hyaluronic acid-chitosan nanoparticles for co-delivery of MiR-34a and doxorubicin in therapy against triple negative breast cancer. Biomaterials 2014, 35, 4333–4344. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Iyer, A.K.; Morrissey, D.V.; Amiji, M.M. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials 2013, 34, 3489–3502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Mok, H.; Lee, S.; Oh, Y.K.; Park, T.G. Target-specific intracellular delivery of siRNA using degradable hyaluronic acid nanogels. J. Control. Release 2007, 119, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Y.; Wang, L.; Wang, X.; Tu, P. Comparison of hyaluronic acid-based micelles and polyethylene glycol-based micelles on reversal of multidrug resistance and enhanced anticancer efficacy in vitro and in vivo. Drug Del. 2018, 25, 330–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.Y.; Chung, H.; Min, K.H.; Yoon, H.Y.; Kim, K.; Park, J.H.; Kwon, I.C.; Jeong, S.Y. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials 2010, 31, 106–114. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Seijo, B.; Alonso, M.J. Bioadhesive hyaluronan-chitosan nanoparticles can transport genes across the ocular mucosa and transfect ocular tissue. Gene Ther. 2008, 15, 668–676. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Seijo, B.; Alonso, M.J. Novel hyaluronan-based nanocarriers for transmucosal delivery of macromolecules. Macromol. Biosci. 2008, 8, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Lallana, E.; Rios de la Rosa, J.M.; Tirella, A.; Pelliccia, M.; Gennari, A.; Stratford, I.J.; Puri, S.; Ashford, M.; Tirelli, N. Chitosan/Hyaluronic acid nanoparticles: Rational design revisited for RNA delivery. Mol. Pharm. 2017, 14, 2422–2436. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Khor, E.; Lim, L.Y. Implantable applications of chitin and chitosan. Biomaterials 2003, 24, 2339–2349. [Google Scholar] [CrossRef]
- De la Fuente, M.; Seijo, B.; Alonso, M.J. Novel hyaluronic acid-chitosan nanoparticles for ocular gene therapy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2016–2024. [Google Scholar] [CrossRef] [PubMed]
- Colonna, C.; Conti, B.; Perugini, P.; Pavanetto, F.; Modena, T.; Dorati, R.; Iadarola, P.; Genta, I. Ex vivo evaluation of prolidase loaded chitosan nanoparticles for the enzyme replacement therapy. Eur. J. Pharm. Biopharm. 2008, 70, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Mihoub, A.B.; Saidat, B.; Bal, Y.; Frochot, C.; Vanderesse, R.; Acherar, S. Development of new ionic gelation strategy: Towards the preparation of new monodisperse and stable hyaluronic acid/β-ciclodextrin-grafted chitosan nanoparticles as drug delivery carriers for doxorubicin. Front. Mater. Sci. 2018, 12, 83–94. [Google Scholar] [CrossRef]
- Raemdonck, K.; Martens, T.F.; Braeckmans, K.; Demeester, J.; De Smedt, S.C. Polysaccharide-based nucleic acid nanoformulations. Adv. Drug Deliv. Rev. 2013, 65, 1123–1147. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.M.; Nasti, A.; Tirelli, N. Nanocarriers for cytoplasmic delivery: Cellular uptake and intracellular fate of chitosan and hyaluronic acid-coated chitosan nanoparticles in a phagocytic cell model. Macromol. Biosci. 2011, 11, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.Z.; Zhu, K.J. The influence of multivalent phosphate structure on the properties of ionically cross-linked chitosan films for controlled drug release. Eur. J. Pharm. Biopharm. 2002, 54, 235–243. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Alatorre-Meda, M.; Zaghloul, E.M.; Taboada, P.; Remunan-Lopez, C. Chitosan-hyaluronic acid nanoparticles for gene silencing: The role of hyaluronic acid on the nanoparticles’ formation and activity. Colloids Surf. B Biointerfaces 2013, 103, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Oyarzun-Ampuero, F.A.; Goycoolea, F.M.; Torres, D.; Alonso, M.J. A new drug nanocarrier consisting of polyarginine and hyaluronic acid. Eur. J. Pharm. Biopharm. 2011, 79, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Murgia, X.; Pawelzyk, P.; Schaefer, U.F.; Wagner, C.; Willenbacher, N.; Lehr, C.-M. Size-Limited Penetration of Nanoparticles into Porcine Respiratory Mucus after Aerosol Deposition. Biomacromolecules 2016, 17, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, L.; Li, X.; Hu, X.; Han, Y.; Yao, L.; Wang, Z.; Li, Q.; Aldalbahi, A.; Wang, L.; et al. Size-Dependent Regulation of Intracellular Trafficking of Polystyrene Nanoparticle-Based Drug-Delivery Systems. ACS Appl. Mater. Interfaces 2017, 9, 18619–18625. [Google Scholar] [CrossRef] [PubMed]
- Nasti, A.; Zaki, N.M.; de Leonardis, P.; Ungphaiboon, S.; Sansongsak, P.; Rimoli, M.G.; Tirelli, N. Chitosan/TPP and chitosan/TPP-hyaluronic acid nanoparticles: systematic optimisation of the preparative process and preliminary biological evaluation. Pharm. Res. 2009, 26, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Calvo, P.; Remunan-Lopez, C.; Vila-Jato, J.L.; Alonso, M.J. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J. Appl. Pol. Sci. 1997, 63, 125–132. [Google Scholar] [CrossRef]
- Croll, T.I.; O’Connor, A.J.; Stevens, G.W.; Cooper-White, J.J. A blank state? Layer-by-layer deposition of hyaluronic acid and chitosan onto various surfaces. Biomacromolecules 2006, 7, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Richert, L.; Lavalle, P.; Payan, E.; Shu, X.Z.; Prestwich, G.D.; Stoltz, J.F.; Schaaf, P.; Voegel, J.-C.; Picart, C. Layer-by-layer buildup of polysaccharide films: Physical chemistry and cellular adhesion aspects. Langmuir 2004, 20, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Yan, W.; Xu, Z.; Ni, H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf. B Biointerfaces 2012, 90, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Gaumet, M.; Vargas, A.; Gurny, R.; Delie, F. Nanoparticles for drug delivery: The need for precision in reporting particle size parameters. Eur. J. Pharm. Biopharm. 2008, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Arpicco, S.; Milla, P.; Stella, B.; Dosio, F. Hyaluronic acid conjugates as vectors for the active targeting of drugs, genes and nanocomposites in cancer treatment. Molecules 2014, 19, 3193–3230. [Google Scholar] [CrossRef] [PubMed]
- Colonna, C.; Conti, B.; Perugini, P.; Pavanetto, F.; Modena, T.; Dorati, R.; Genta, I. Chitosan glutamate nanoparticles for protein delivery: Development and effect on prolidase stability. J. Microencapsul. 2007, 24, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Malatesta, M.; Grecchi, S.; Chiesa, E.; Cisterna, B.; Costanzo, M.; Zancanaro, C. Internalized chitosan nanoparticles persist for long time in cultured cells. Eur. J. Histochem. 2015, 59, 2492–2496. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Lapitsky, Y. Formation and dissolution of chitosan/pyrophosphate nanoparticles: Is the ionic crosslinking of chitosan reversible? Colloids Surf. B Biointerfaces 2014, 115, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gao, S.; Asghar, S.; Liu, G.; Song, J.; Wang, X.; Ping, Q.; Zhang, C.; Xiao, Y. Hyaluronic acid/chitosan nanoparticles for delivery of curcuminoid and its in vitro evaluation in glioma cells. Int. J. Biol. Macromol. 2015, 72, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.J. Zeta Potential in Colloid Science—Principles and Applications, 1st ed.; Academic Press: London, UK, 1988; ISBN 0123619602 9780123619600. [Google Scholar]
- Bang, S.; Das, D.; Yu, J.; Noh, I. Evaluation of MC3T3 cells proliferation and drug release study from sodium hyaluronate-1,4-butendiol diglycidyl ether patterned gel. Nanomaterials 2017, 7, 328. [Google Scholar] [CrossRef] [PubMed]
- Mattu, C.; Li, R.; Ciardelli, G. Chitosan nanoparticles as therapeutic protein nanocarriers: The effect of pH on nanoparticle formation and encapsulation efficiency. Polym. Compos. 2013, 34, 1538–1545. [Google Scholar] [CrossRef]
- Lee, S.B.; Lee, Y.M.; Song, K.W.; Park, M.H. Preparation and properties of polyelectrolyte complex sponges composed of hyaluronic acid and chitosan and their biological behaviors. J. Appl. Polym. Sci. 2003, 90, 925–932. [Google Scholar] [CrossRef]
- Janes, K.A.; Calvo, P.; Alonso, M.J. Polysaccharide colloidal particles as delivery systems for macromolecules. Adv. Drug Deliv. Rev. 2001, 47, 83–97. [Google Scholar] [CrossRef]
- Gan, Q.; Wang, T. Chitosan nanoparticle as protein delivery carrier—systematic examination of fabrication conditions for efficient loading and release. Colloids Surf. B Biointerfaces 2007, 59, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zuo, J.; Bou-Chacra, N.; Pinto Tde, J.; Clas, S.D.; Walker, R.B.; Lobenberg, R. In vitro release kinetics of antituberculosis drugs from nanoparticles assessed using a modified dissolution apparatus. BioMed Res. Int. 2013, 2013, 136590–136598. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for the determination of solute release from swellable device. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Cuff, C.A.; Kothapalli, D.; Azonobi, I.; Chun, S.; Zhang, Y.; Belkin, R.; Yeh, C.; Secreto, A.; Assoian, R.K.; Rader, D.J.; et al. The adhesion receptor CD44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. J. Clin. Investig. 2001, 108, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, D.; Zhao, L.; Hawthorne, E.A.; Cheng, Y.; Lee, E.; Pure, E.; Assoian, R.K. Hyaluronan and CD44 antagonize mitogen-dependent cyclin D1 expression in mesenchymal cells. J. Cell Biol. 2007, 176, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Bellich, B.; D’Agostino, I.; Semeraro, S.; Gamini, A.; Cesaro, A. “The Good, the Bad and the Ugly” of Chitosans. Mar. Drugs 2016, 14, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An Update on Potential Biomedical and Pharmaceutical Applications. Mar. Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Lorenzo, C.; Blanco-Fernandez, B.; Puga, A.M.; Concheiro, A. Crosslinked ionic polysaccharides for stimuli-sensitive drug delivery. Adv. Drug Deliv. Rev. 2013, 65, 1148–1171. [Google Scholar] [CrossRef] [PubMed]
- Mizrahy, S.; Raz, S.R.; Hasgaard, M.; Liu, H.; Soffer-Tsur, N.; Cohen, K.; Dvash, R.; Landsman-Milo, D.; Bremer, M.G.; Moghimi, S.M.; et al. Hyaluronan-coated nanoparticles: The influence of the molecular weight on CD44-hyaluronan interactions and on the immune response. J. Control. Release 2011, 156, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Wolny, P.M.; Banerji, S.; Gounou, C.; Brisson, A.R.; Day, A.J.; Jackson, D.G.; Richter, R.P. Analysis of CD44-hyaluronan interactions in an artificial membrane system: Insights into the distinct binding properties of high and low molecular weight hyaluronan. J. Biol. Chem. 2010, 285, 30170–30180. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.L.; Wilson, C.L.; Kidambi, S. Hyaluronic acid-conjugated liposome nanoparticles for targeted delivery to CD44 overexpressing glioblastoma cells. Oncotarget 2016, 7, 34158–34171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Tiruppathi, C.; Minshall, R.C.; Malik, A.B. Size and Dynamics of Caveolae Studied Using Nanoparticles in Living Endothelial Cells. ACS Nano 2008, 3, 4110–4116. [Google Scholar] [CrossRef] [PubMed]
- Colzani, B.; Speranza, G.; Dorati, R.; Conti, B.; Modena, T.; Bruni, G.; Zagato, E.; Vermeulen, L.; Dakwar, G.R.; Braeckmans, K.; et al. Design of smart GE11-PLGA/PEG-PLGA blend nanoparticulate platforms for parenteral administration of hydrophilic macromolecular drugs: Synthesis, preparation and in vitro/ex vivo characterization. Int. J. Pharm. 2016, 511, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Pistone, S.; Qoragllu, D.; Smistad, G.; Hiorth, M. Multivariate analysis for the optimization of polysaccharide-based nanoparticles prepared by self-assembly. Colloids Surf. B Biointerfaces 2016, 146, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Ruckmani, K.; Shaikh, S.Z.; Khalil, P.; Muneera, M.S.; Thusleem, O.A. Determination of sodium hyaluronate in pharmaceutical formulations by HPLC-UV. J. Pharm. Anal. 2013, 3, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Colonna, C.; Conti, B.; Genta, I.; Alpar, O.H. Non-viral dried powders for respiratory gene delivery preparedby cationic and chitosan loaded liposomes. Int. J. Pharm. 2008, 364, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, R.A.A. Colorimetric determination of chitosan. Anal. Biochem. 1998, 260, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Werner, J.P.F.; Moreno-Villoslada, I.; Goycoolea, F.M. New insights into the nature of the Cibacron brilliant red 3B-A—Chitosan interaction. Pure Appl. Chem. 2016, 88, 891–904. [Google Scholar] [CrossRef]
- Baldelli, S.; Murgia, S.; Merlini, S.; Zenoni, S.; Perico, N.; Remuzzi, G.; Cattaneo, D. High-performance liquid chromatography with ultraviolet detection for therapeutic drug monitoring of everolimus. J. Chrom. B Anal. Technol. Biomed. Life Sci. 2005, 816, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Khoschsorur, G. Simultaneous measurement of sirolimus and everolimus in whole blood by HPLC with ultraviolet detection. Clin. Chem. 2005, 51, 1721–1724. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Huang, M.; Ma, Z.; Khor, E.; Lim, L.-Y. Uptake of FITC-Chitosan Nanoparticles by A549 Cells. Pharm. Res. 2002, 19, 1488–1494. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DoE Batch # | HA Solution (mg/mL) (Level) | CS Solution (mg/mL) (Level) | TPP Solution (mg/mL) (Level) | Size ± SD (nm) | PDI ± SD | ζ Potential ± SD (mV) |
|---|---|---|---|---|---|---|
| A | 1 (−1) | 0.3 (−1) | 0.25 (−1) | 385.2 ± 90.7 | 0.240 ± 0.096 | −1.1 ± 0.3 |
| B | 1 (−1) | 0.5 (+1) | 0.5 (+1) | 181.9 ± 65.6 | 0.171 ± 0.040 | −30.9 ± 2.5 |
| C | 1 (−1) | 0.5 (+1) | 0.25 (−1) | 195.4 ± 75.2 | 0.421 ± 0.163 | −1.5 ± 0.8 |
| D | 1 (−1) | 0.3 (−1) | 0.5 (+1) | 210.8 ± 94.9 | 0.202 ± 0.086 | −10.8 ± 2.8 |
| E | 2 (+1) | 0.5 (+1) | 0.5 (+1) | 242.5 ± 197.9 | 0.334 ± 0.103 | −38.5 ± 5.2 |
| F | 2 (+1) | 0.3 (−1) | 0.5 (+1) | 320.9 ± 322.4 | 0.382 ± 0.087 | −40.5 ± 8.3 |
| G | 2 (+1) | 0.3 (−1) | 0.25 (−1) | 630.5 ± 343.4 | 0.275 ± 0.069 | −25.5 ± 4.6 |
| H | 2 (+1) | 0.5 (+1) | 0.25 (−1) | 310.8 ± 162.2 | 0.272 ±0.023 | −18.7 ± 3.9 |
| I | 1.5 (0) | 0.4 (0) | 0.375 (0) | 230.3 ± 144.6 | 0.393 ± 0.042 | −13.5 ± 1.6 |
| L | 1.5 (0) | 0.4 (0) | 0.375 (0) | 223.1 ± 114.7 | 0.264 ± 0.062 | −12.8 ± 3.2 |
| Input | Level | ||
|---|---|---|---|
| −1 | 0 | +1 | |
| HA solution (mg/mL) | 1 | 1.5 | 2 |
| CS solution (mg/mL) | 0.3 | 0.4 | 0.5 |
| TPP solution (mg/mL) | 0.25 | 0.375 | 0.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiesa, E.; Dorati, R.; Conti, B.; Modena, T.; Cova, E.; Meloni, F.; Genta, I. Hyaluronic Acid-Decorated Chitosan Nanoparticles for CD44-Targeted Delivery of Everolimus. Int. J. Mol. Sci. 2018, 19, 2310. https://doi.org/10.3390/ijms19082310
Chiesa E, Dorati R, Conti B, Modena T, Cova E, Meloni F, Genta I. Hyaluronic Acid-Decorated Chitosan Nanoparticles for CD44-Targeted Delivery of Everolimus. International Journal of Molecular Sciences. 2018; 19(8):2310. https://doi.org/10.3390/ijms19082310
Chicago/Turabian StyleChiesa, Enrica, Rossella Dorati, Bice Conti, Tiziana Modena, Emanuela Cova, Federica Meloni, and Ida Genta. 2018. "Hyaluronic Acid-Decorated Chitosan Nanoparticles for CD44-Targeted Delivery of Everolimus" International Journal of Molecular Sciences 19, no. 8: 2310. https://doi.org/10.3390/ijms19082310
APA StyleChiesa, E., Dorati, R., Conti, B., Modena, T., Cova, E., Meloni, F., & Genta, I. (2018). Hyaluronic Acid-Decorated Chitosan Nanoparticles for CD44-Targeted Delivery of Everolimus. International Journal of Molecular Sciences, 19(8), 2310. https://doi.org/10.3390/ijms19082310