ATP as a Pathophysiologic Mediator of Bacteria-Host Crosstalk in the Gastrointestinal Tract

Abstract
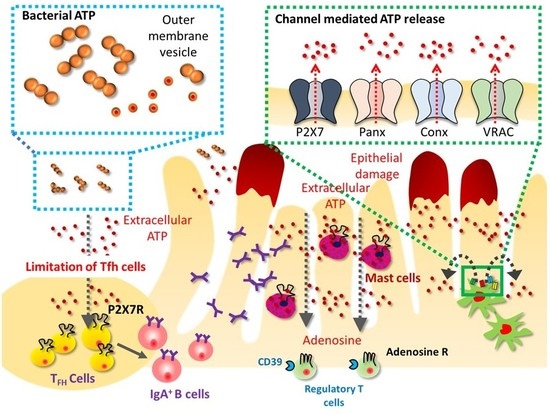
:
{kind=link}
{kind=link}
1. ATP as an Inter-Species Messenger
2. eATP as an Inflammatory Mediator in Intestinal Inflammation
3. Resolution of Inflammation
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| LP | lamina propria |
| OMV | outer membrane vesicle |
| R | receptor |
| ROS | reactive oxygen species |
| Tfh cells | follicular helper T cells |
References
- Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007, 87, 659–797. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.H. The biological significance of the linkages in adenosine triphosphoric acid. J. Physiol. 1934, 80, 345–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar] [PubMed]
- Atarashi, K.; Nishimura, J.; Shima, T.; Umesaki, Y.; Yamamoto, M.; Onoue, M.; Yagita, H.; Ishii, N.; Evans, R.; Honda, K.; et al. ATP drives lamina propria TH17 cell differentiation. Nature 2008, 455, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Perruzza, L.; Gargari, G.; Proietti, M.; Fosso, B.; D’Erchia, A.M.; Faliti, C.E.; Rezzonico-Jost, T.; Scribano, D.; Mauri, L.; Colombo, D.; et al. T follicular helper cells promote a beneficial gut ecosystem for host metabolic homeostasis by sensing microbiota-derived extracellular ATP. Cell Rep. 2017, 18, 2566–2575. [Google Scholar] [CrossRef] [PubMed]
- Lazarowski, E.R. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012, 8, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taruno, A. ATP release channels. Int. J. Mol. Sci. 2018, 19, 808. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, V.; Bidula, S.; Campwala, H.; Katikaneni, D.; Fountain, S.J. Constitutive lysosome exocytosis releases ATP and engages P2Y receptors in human monocytes. J. Cell Sci. 2012, 125, 4567–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodin, P.; Burnstock, G. Purinergic signalling: ATP release. Neurochem. Res. 2001, 26, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T.; Mager, A.; Kuper, N.; Karcher, C.; Weissmuller, T.; Boengler, K.; Schulz, R.; Robson, S.C.; Colgan, S.P. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 2006, 99, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Tsai, S.H.; Kinoshita, M.; Fujimoto, K.; Okumura, R.; Umemoto, E.; Kurashima, Y.; Kiyono, H.; Kayama, H.; Takeda, K. E-NPP3 controls plasmacytoid dendritic cell numbers in the small intestine. PLoS ONE 2017, 12, e0172509. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006, 314, 1792–1795. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yao, Y.; Sumi, Y.; Li, A.; To, U.K.; Elkhal, A.; Inoue, Y.; Woehrle, T.; Zhang, Q.; Hauser, C.; et al. Purinergic signaling: A fundamental mechanism in neutrophil activation. Sci. Signal. 2010, 3, ra45. [Google Scholar] [CrossRef] [PubMed]
- Kronlage, M.; Song, J.; Sorokin, L.; Isfort, K.; Schwerdtle, T.; Leipziger, J.; Robaye, B.; Conley, P.B.; Kim, H.C.; Sargin, S.; et al. Autocrine purinergic receptor signaling is essential for macrophage chemotaxis. Sci. Signal. 2010, 3, ra55. [Google Scholar] [CrossRef] [PubMed]
- Schenk, U.; Westendorf, A.M.; Radaelli, E.; Casati, A.; Ferro, M.; Fumagalli, M.; Verderio, C.; Buer, J.; Scanziani, E.; Grassi, F. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci. Signal. 2008, 1, ra6. [Google Scholar] [CrossRef] [PubMed]
- Woehrle, T.; Yip, L.; Elkhal, A.; Sumi, Y.; Chen, Y.; Yao, Y.; Insel, P.A.; Junger, W.G. Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood 2010, 116, 3475–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurashima, Y.; Amiya, T.; Nochi, T.; Fujisawa, K.; Haraguchi, T.; Iba, H.; Tsutsui, H.; Sato, S.; Nakajima, S.; Iijima, H.; et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat. Commun. 2012, 3, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, H.D.; Schneider, D.A.; Gourse, R.L. Control of rRNA expression by small molecules is dynamic and nonredundant. Mol. Cell 2003, 12, 125–134. [Google Scholar] [CrossRef]
- Mempin, R.; Tran, H.; Chen, C.; Gong, H.; Kim Ho, K.; Lu, S. Release of extracellular ATP by bacteria during growth. BMC Microbiol. 2013, 13, 301. [Google Scholar] [CrossRef] [PubMed]
- Iwase, T.; Shinji, H.; Tajima, A.; Sato, F.; Tamura, T.; Iwamoto, T.; Yoneda, M.; Mizunoe, Y. Isolation and identification of ATP-secreting bacteria from mice and humans. J. Clin. Microbiol. 2010, 48, 1949–1951. [Google Scholar] [CrossRef] [PubMed]
- Hironaka, I.; Iwase, T.; Sugimoto, S.; Okuda, K.; Tajima, A.; Yanaga, K.; Mizunoe, Y. Glucose triggers ATP secretion from bacteria in a growth-phase-dependent manner. Appl. Environ. Microbiol. 2013, 79, 2328–2335. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.L.; Corradi, G.; Lauri, N.; Marginedas-Freixa, I.; Leal Denis, M.F.; Enrique, N.; Mate, S.M.; Milesi, V.; Ostuni, M.A.; Herlax, V.; et al. Dynamic regulation of extracellular ATP in Escherichia coli. Biochem. J. 2017, 474, 1395–1416. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Huang, H.I.; Benzatti, F.P.; Karlsson, A.B.; Zhang, J.J.; Youssef, N.; Ma, A.; Hale, L.P.; Hammer, G.E. Inflammatory Th1 and Th17 in the intestine are each driven by functionally specialized dendritic cells with distinct requirements for MyD88. Cell Rep. 2016, 17, 1330–1343. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, F.; Wu, W.; Cao, A.T.; Xue, X.; Yao, S.; Evans-Marin, H.L.; Li, Y.Q.; Cong, Y. TLR5 mediates CD172α+ intestinal lamina propria dendritic cell induction of Th17 cells. Sci. Rep. 2016, 6, 22040. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Hammad, H.; van Nimwegen, M.; Kool, M.; Willart, M.A.; Muskens, F.; Hoogsteden, H.C.; Luttmann, W.; Ferrari, D.; Di Virgilio, F.; et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat. Med. 2007, 13, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Junger, W.G. Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 2011, 11, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Do some nerve cells release more than one transmitter? Neuroscience 1976, 1, 239–248. [Google Scholar] [CrossRef]
- Van Calker, D.; Muller, M.; Hamprecht, B. Adenosine regulates via two different types of receptors, the accumulation of cyclic amp in cultured brain cells. J. Neurochem. 1979, 33, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic nerves and receptors. Prog. Biochem. Pharmacol. 1980, 16, 141–154. [Google Scholar] [PubMed]
- Schwartzman, M.; Pinkas, R.; Raz, A. Evidence for different purinergic receptors for ATP and ADP in rabbit kidney and heart. Eur. J. Pharmacol. 1981, 74, 167–173. [Google Scholar] [CrossRef]
- Londos, C.; Cooper, D.M.; Wolff, J. Subclasses of external adenosine receptors. Proc. Natl. Acad. Sci. USA 1980, 77, 2551–2554. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Kennedy, C. Is there a basis for distinguishing two types of P2-purinoceptor? Gen. Pharmacol. 1985, 16, 433–440. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G. Purinoceptors: Are there families of P2X and P2Y purinoceptors? Pharmacol. Ther. 1994, 64, 445–475. [Google Scholar] [CrossRef]
- Communi, D.; Gonzalez, N.S.; Detheux, M.; Brezillon, S.; Lannoy, V.; Parmentier, M.; Boeynaems, J.M. Identification of a novel human ADP receptor coupled to Gi. J. Biol. Chem. 2001, 276, 41479–41485. [Google Scholar] [CrossRef] [PubMed]
- Abbracchio, M.P.; Boeynaems, J.M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Miras-Portugal, M.T.; King, B.F.; Gachet, C.; Jacobson, K.A.; Weisman, G.A.; et al. Characterization of the udp-glucose receptor (re-named here the P2Y14 receptor) adds diversity to the P2Y receptor family. Trends Pharmacol. Sci. 2003, 24, 52–55. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef] [PubMed]
- North, R.A.; Surprenant, A. Pharmacology of cloned P2X receptors. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic receptors as future targets for treatment of functional Gi disorders. Gut 2008, 57, 1193–1194. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Debetto, P.; Giusti, P. The P2X7 purinergic receptor: From physiology to neurological disorders. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, A.; Brest, P.; Hofman, V.; Hebuterne, X.; Wildman, S.; Ferrua, B.; Marchetti, S.; Doglio, A.; Vouret-Craviari, V.; Galland, F.; et al. Amplification loop of the inflammatory process is induced by P2X7R activation in intestinal epithelial cells in response to neutrophil transepithelial migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G32–G42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.A.; Vial, C.; Digby, H.R.; Agboh, K.C.; Wen, H.; Atterbury-Thomas, A.; Evans, R.J. Molecular properties of P2X receptors. Pflugers Arch. Eur. J. Physiol. 2006, 452, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Denlinger, L.C.; Fisette, P.L.; Sommer, J.A.; Watters, J.J.; Prabhu, U.; Dubyak, G.R.; Proctor, R.A.; Bertics, P.J. Cutting edge: The nucleotide receptor P2X7 contains multiple protein- and lipid–interaction motifs including a potential binding site for bacterial lipopolysaccharide. J. Immunol. 2001, 167, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jiang, L.H.; Wilson, H.L.; North, R.A.; Surprenant, A. Proteomic and functional evidence for a P2X7 receptor signalling complex. EMBO J. 2001, 20, 6347–6358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, B.J.; Rathsam, C.; Stokes, L.; McGeachie, A.B.; Wiley, J.S. Extracellular ATP dissociates nonmuscle myosin from P2X7 complex: This dissociation regulates P2X7 pore formation. Am. J. Physiol. Cell Physiol. 2009, 297, C430–C439. [Google Scholar] [CrossRef] [PubMed]
- Donnelly-Roberts, D.L.; Namovic, M.T.; Faltynek, C.R.; Jarvis, M.F. Mitogen-activated protein kinase and caspase signaling pathways are required for P2X7 receptor (P2X7R)-induced pore formation in human THP-1 cells. J. Pharmacol. Exp. Ther. 2004, 308, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Korcok, J.; Raimundo, L.N.; Ke, H.Z.; Sims, S.M.; Dixon, S.J. Extracellular nucleotides act through P2X7 receptors to activate NF-κB in osteoclasts. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2004, 19, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Sandilos, J.K.; Chiu, Y.H.; Chekeni, F.B.; Armstrong, A.J.; Walk, S.F.; Ravichandran, K.S.; Bayliss, D.A. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated c-terminal autoinhibitory region. J. Biol. Chem. 2012, 287, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic signaling during inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, K.; Ganesan, J.; Muller, T.; Durr, C.; Grimm, M.; Beilhack, A.; Krempl, C.D.; Sorichter, S.; Gerlach, U.V.; Juttner, E.; et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 2010, 16, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Takemura, N.; Kurashima, Y.; Mori, Y.; Okada, K.; Ogino, T.; Osawa, H.; Matsuno, H.; Aayam, L.; Kaneto, S.; Park, E.J.; et al. Eosinophil depletion suppresses radiation-induced small intestinal fibrosis. Sci. Transl. Med. 2018, 10, eaan0333. [Google Scholar] [CrossRef] [PubMed]
- Somers, G.; Hammet, F.M.A.; Trute, L.; Southey, M.; Venter, D. Expression of the P2Y6 purinergic receptor in human T cells infiltrating inflammatory bowel disease. Lab. Investig. J. Tech. Methods Pathol. 1998, 78, 1375–1383. [Google Scholar]
- Grbic, D.M.; Degagne, E.; Langlois, C.; Dupuis, A.A.; Gendron, F.P. Intestinal inflammation increases the expression of the P2Y6 receptor on epithelial cells and the release of CXC chemokine ligand 8 by UDP. J. Immunol. 2008, 180, 2659–2668. [Google Scholar] [CrossRef] [PubMed]
- Layhadi, J.A.; Turner, J.; Crossman, D.; Fountain, S.J. ATP evokes Ca2+ responses and CXCL5 secretion via P2X4 receptor activation in human monocyte-derived macrophages. J. Immunol. 2018, 200, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Prudic, K.; Pippel, A.; Klapperstuck, M.; Braam, U.; Muller, C.E.; Schmalzing, G.; Markwardt, F. Interaction of purinergic P2X4 and P2X7 receptor subunits. Front. Pharmacol. 2017, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- Suurvali, J.; Boudinot, P.; Kanellopoulos, J.; Ruutel Boudinot, S. P2X4: A fast and sensitive purinergic receptor. Biomed. J. 2017, 40, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Surprenant, A. Functional properties of native and cloned P2X receptors. Ciba Found. Symp. 1996, 198, 208–222. [Google Scholar] [PubMed]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, P.; Barroso-Gutierrez, C.; Surprenant, A. P2X7 receptor differentially couples to distinct release pathways for IL-1β in mouse macrophage. J. Immunol. 2008, 180, 7147–7157. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Kanneganti, T.D.; Dubyak, G.R.; Nunez, G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 2007, 282, 18810–18818. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Munoz-Planillo, R.; Reimer, T.; Eigenbrod, T.; Nunez, G. Inflammasomes as microbial sensors. Eur. J. Immunol. 2010, 40, 611–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gicquel, T.; Robert, S.; Loyer, P.; Victoni, T.; Bodin, A.; Ribault, C.; Gleonnec, F.; Couillin, I.; Boichot, E.; Lagente, V. IL-1β production is dependent on the activation of purinergic receptors and NLRP3 pathway in human macrophages. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 4162–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Franchi, L.; Nunez, G. TLR agonists stimulate NLRP3-dependent il-1beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J. Immunol. 2013, 190, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, T.; Izawa, K.; Isobe, M.; Takahashi, M.; Maehara, A.; Yamanishi, Y.; Kaitani, A.; Okumura, K.; Teshima, T.; Kitamura, T.; et al. Ceramide-CD300F binding suppresses experimental colitis by inhibiting ATP-mediated mast cell activation. Gut 2016, 65, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Ohbori, K.; Fujiwara, M.; Ohishi, A.; Nishida, K.; Uozumi, Y.; Nagasawa, K. Prophylactic oral administration of magnesium ameliorates dextran sulfate sodium-induced colitis in mice through a decrease of colonic accumulation of P2X7 receptor-expressing mast cells. Biol. Pharm. Bull. 2017, 40, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Figliuolo, V.R.; Savio, L.E.B.; Safya, H.; Nanini, H.; Bernardazzi, C.; Abalo, A.; de Souza, H.S.P.; Kanellopoulos, J.; Bobe, P.; Coutinho, C.; et al. P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim. Biophys. Acta 2017, 1863, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.R.; Castelo-Branco, M.T.; Figliuolo, V.R.; Bernardazzi, C.; Buongusto, F.; Yoshimoto, A.; Nanini, H.F.; Coutinho, C.M.; Carneiro, A.J.; Coutinho-Silva, R.; et al. Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.C.; Castelo-Branco, M.T.; Pacheco, R.G.; Buongusto, F.; do Rosario, A., Jr.; Schanaider, A.; Coutinho-Silva, R.; de Souza, H.S. Prophylactic systemic P2X7 receptor blockade prevents experimental colitis. Biochim. Biophys. Acta 2014, 1842, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Jacobson, K.A.; Christofi, F.L. Purinergic drug targets for gastrointestinal disorders. Curr. Opin. Pharmacol. 2017, 37, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.X.; Wang, M.X.; Nie, W.; Liu, D.W.; Zhang, Y.; Liu, H.B. P2X7R blockade prevents NLRP3 inflammasome activation and pancreatic fibrosis in a mouse model of chronic pancreatitis. Pancreas 2017, 46, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Fay, S.; Vieira, R.P.; Karmouty-Quintana, H.; Cicko, S.; Ayata, C.K.; Zissel, G.; Goldmann, T.; Lungarella, G.; Ferrari, D.; et al. P2Y6 receptor activation promotes inflammation and tissue remodeling in pulmonary fibrosis. Front. Immunol. 2017, 8, 1028. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.L.; Mediero, A.; Corciulo, C.; Liu, H.; Zhang, J.; Perez-Aso, M.; Picard, L.; Wilder, T.; Cronstein, B. The antiviral drug tenofovir, an inhibitor of pannexin-1-mediated ATP release, prevents liver and skin fibrosis by downregulating adenosine levels in the liver and skin. PLoS ONE 2017, 12, e0188135. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.G.; Gabrich, L.; Rosario, A., Jr.; Takiya, C.M.; Ferreira, M.L.; Chiarini, L.B.; Persechini, P.M.; Coutinho-Silva, R.; Leite, M., Jr. The role of purinergic P2X7 receptors in the inflammation and fibrosis of unilateral ureteral obstruction in mice. Kidney Int. 2006, 70, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Historical review: ATP as a neurotransmitter. Trends Pharmacol. Sci. 2006, 27, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.C.; Robson, S.; Quintana, F.J. Regulation of the T cell response by CD39. Trends Immunol. 2016, 37, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Kunzli, B.M.; YI, A.R.; Sevigny, J.; Berberat, P.O.; Enjyoji, K.; Csizmadia, E.; Friess, H.; Robson, S.C. From the cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16788–16793. [Google Scholar] [CrossRef] [PubMed]
- Louis, N.A.; Robinson, A.M.; MacManus, C.F.; Karhausen, J.; Scully, M.; Colgan, S.P. Control of IFN-α by CD73: Implications for mucosal inflammation. J. Immunol. 2008, 180, 4246–4255. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Kobie, J.J.; Mosmann, T.R. CD73 and LY-6a/e distinguish in vivo primed but uncommitted mouse CD4 T cells from type 1 or type 2 effector cells. J. Immunol. 2005, 175, 6458–6464. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, K.M.; Hanidziar, D.; Putheti, P.; Hill, P.A.; Pommey, S.; McRae, J.L.; Winterhalter, A.; Doherty, G.; Deaglio, S.; Koulmanda, M.; et al. Expression of CD39 by human peripheral blood CD4+ CD25+ T cells denotes a regulatory memory phenotype. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2010, 10, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Borsellino, G.; Kleinewietfeld, M.; Di Mitri, D.; Sternjak, A.; Diamantini, A.; Giometto, R.; Hopner, S.; Centonze, D.; Bernardi, G.; Dell’Acqua, M.L.; et al. Expression of ectonucleotidase CD39 by Foxp3+ treg cells: Hydrolysis of extracellular ATP and immune suppression. Blood 2007, 110, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Beldi, G.; Wu, Y.; Banz, Y.; Nowak, M.; Miller, L.; Enjyoji, K.; Haschemi, A.; Yegutkin, G.G.; Candinas, D.; Exley, M.; et al. Natural killer T cell dysfunction in CD39-null mice protects against concanavalin a-induced hepatitis. Hepatology 2008, 48, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Mandapathil, M.; Hilldorfer, B.; Szczepanski, M.J.; Czystowska, M.; Szajnik, M.; Ren, J.; Lang, S.; Jackson, E.K.; Gorelik, E.; Whiteside, T.L. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25HighFOXP3+ regulatory T cells. J. Biol. Chem. 2010, 285, 7176–7186. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Lonergan, R.; Costelloe, L.; Kinsella, K.; Moran, B.; O’Farrelly, C.; Tubridy, N.; Mills, K.H. CD39+foxp3+ regulatory T cells suppress pathogenic TH17 cells and are impaired in multiple sclerosis. J. Immunol. 2009, 183, 7602–7610. [Google Scholar] [CrossRef] [PubMed]
- Saldanha-Araujo, F.; Ferreira, F.I.; Palma, P.V.; Araujo, A.G.; Queiroz, R.H.; Covas, D.T.; Zago, M.A.; Panepucci, R.A. Mesenchymal stromal cells up-regulate CD39 and increase adenosine production to suppress activated T-lymphocytes. Stem Cell Res. 2011, 7, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Kuhny, M.; Hochdorfer, T.; Ayata, C.K.; Idzko, M.; Huber, M. CD39 is a negative regulator of P2X7-mediated inflammatory cell death in mast cells. Cell Commun. Signal. CCS 2014, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldi, S.; Marino, A.; Tomita, K.; Corti, F.; Anand, R.; Olson, K.E.; Marcus, A.J.; Levi, R. E-NTPDase1/CD39 modulates renin release from heart mast cells during ischemia/reperfusion: A novel cardioprotective role. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.H.; Kinoshita, M.; Kusu, T.; Kayama, H.; Okumura, R.; Ikeda, K.; Shimada, Y.; Takeda, A.; Yoshikawa, S.; Obata-Ninomiya, K.; et al. The ectoenzyme e-npp3 negatively regulates ATP-dependent chronic allergic responses by basophils and mast cells. Immunity 2015, 42, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Bühring, H.-J.; Streble, A.; Valent, P. The Basophil-Specific Ectoenzyme e-npp3 (CD203C) as a marker for cell activation and allergy diagnosis. Int. Arch. Allergy Immunol. 2004, 133, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Kusu, T.; Kayama, H.; Kinoshita, M.; Jeon, S.G.; Ueda, Y.; Goto, Y.; Okumura, R.; Saiga, H.; Kurakawa, T.; Ikeda, K.; et al. Ecto-nucleoside triphosphate diphosphohydrolase 7 controls TH17 cell responses through regulation of luminal ATP in the small intestine. J. Immunol. 2013, 190, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Linden, J. Molecular approach to adenosine receptors: Receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Resta, R.; Yamashita, Y.; Thompson, L.F. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol. Rev. 1998, 161, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.F.; Eltzschig, H.K.; Ibla, J.C.; van de Wiele, C.J.; Resta, R.; Morote-Garcia, J.C.; Colgan, S.P. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 2004, 200, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, C.; Steinsdoerfer, M.; Offers, M.; Fischer, E.; Schierl, R.; Heseler, K.; Daubener, W.; Seissler, J. Inhibition of T-cell proliferation by murine multipotent mesenchymal stromal cells is mediated by CD39 expression and adenosine generation. Cell Transplant. 2011, 20, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.V.; Ohta, A. The ‘danger’ sensors that stop the immune response: The A2 adenosine receptors? Trends Immunol. 2005, 26, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Sugiyama, H.; Zhang, Q.; Yan, K.; Fang, X.; McCormick, T.S.; Cooper, K.D.; Huang, Q. Phenotypical analysis of ectoenzymes CD39/CD73 and adenosine receptor 2a in CD4+ CD25high foxp3+ regulatory T-cells in psoriasis. Aust. J. Dermatol. 2018, 59, e31–e38. [Google Scholar] [CrossRef] [PubMed]
- Rybaczyk, L.; Rozmiarek, A.; Circle, K.; Grants, I.; Needleman, B.; Wunderlich, J.E.; Huang, K.; Christofi, F.L. New bioinformatics approach to analyze gene expressions and signaling pathways reveals unique purine gene dysregulation profiles that distinguish between CD and UC. Inflamm. Bowel Dis. 2009, 15, 971–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Smyth, M.J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29, 5346–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasko, G.; Cronstein, B.N. Adenosine: An endogenous regulator of innate immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Chern, Y.; Franco, R.; Sitkovsky, M. Aspects of the general biology of adenosine A2A signaling. Prog. Neurobiol. 2007, 83, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Zanin, R.F.; Braganhol, E.; Bergamin, L.S.; Campesato, L.F.; Filho, A.Z.; Moreira, J.C.; Morrone, F.B.; Sevigny, J.; Schetinger, M.R.; de Souza Wyse, A.T.; et al. Differential macrophage activation alters the expression profile of NTPDase and ecto-5′-nucleotidase. PLoS ONE 2012, 7, e31205. [Google Scholar] [CrossRef] [PubMed]
- Hamidzadeh, K.; Mosser, D.M. Purinergic signaling to terminate TLR responses in macrophages. Front. Immunol. 2016, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Khoa, N.D.; Montesinos, M.C.; Reiss, A.B.; Delano, D.; Awadallah, N.; Cronstein, B.N. Inflammatory cytokines regulate function and expression of adenosine a(2a) receptors in human monocytic THP-1 cells. J. Immunol. 2001, 167, 4026–4032. [Google Scholar] [CrossRef] [PubMed]
- Xaus, J.; Mirabet, M.; Lloberas, J.; Soler, C.; Lluis, C.; Franco, R.; Celada, A. IFN-γ up-regulates the A2B adenosine receptor expression in macrophages: A mechanism of macrophage deactivation. J. Immunol. 1999, 162, 3607–3614. [Google Scholar] [PubMed]
- Bruns, R.F.; Lu, G.H.; Pugsley, T.A. Characterization of the A2 adenosine receptor labeled by [3h]neca in rat striatal membranes. Mol. Pharmacol. 1986, 29, 331–346. [Google Scholar] [PubMed]
- Aherne, C.M.; Kewley, E.M.; Eltzschig, H.K. The resurgence of A2B adenosine receptor signaling. Biochim. Biophys. Acta 2011, 1808, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Shepherd, R.K.; Duling, B.R.; Linden, J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J. Clin. Investig. 1997, 100, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Gomez, G.; Sitkovsky, M.V. Differential requirement for A2A and A3 adenosine receptors for the protective effect of inosine in vivo. Blood 2003, 102, 4472–4478. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Hoang, T.K.; Wang, T.; Ferris, M.; Taylor, C.M.; Tian, X.; Luo, M.; Tran, D.Q.; Zhou, J.; Tatevian, N.; et al. Resetting microbiota by lactobacillus reuteri inhibits T REG deficiency-induced autoimmunity via adenosine A2A receptors. J. Exp. Med. 2017, 214, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Kolachala, V.; Asamoah, V.; Wang, L.; Obertone, T.S.; Ziegler, T.R.; Merlin, D.; Sitaraman, S.V. TNF-α upregulates adenosine 2B (A2B) receptor expression and signaling in intestinal epithelial cells: A basis for A2Br overexpression in colitis. Cell. Mol. Life Sci. CMLS 2005, 62, 2647–2657. [Google Scholar] [CrossRef] [PubMed]
- Aherne, C.M.; Saeedi, B.; Collins, C.B.; Masterson, J.C.; McNamee, E.N.; Perrenoud, L.; Rapp, C.R.; Curtis, V.F.; Bayless, A.; Fletcher, A.; et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal Immunol. 2015, 8, 1324–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.M.; Kurtz, C.C.; Black, S.G.; Ross, W.G.; Alam, M.S.; Linden, J.; Ernst, P.B. The A2B adenosine receptor promotes TH17 differentiation via stimulation of dendritic cell il-6. J. Immunol. 2011, 186, 6746–6752. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inami, A.; Kiyono, H.; Kurashima, Y. ATP as a Pathophysiologic Mediator of Bacteria-Host Crosstalk in the Gastrointestinal Tract. Int. J. Mol. Sci. 2018, 19, 2371. https://doi.org/10.3390/ijms19082371
Inami A, Kiyono H, Kurashima Y. ATP as a Pathophysiologic Mediator of Bacteria-Host Crosstalk in the Gastrointestinal Tract. International Journal of Molecular Sciences. 2018; 19(8):2371. https://doi.org/10.3390/ijms19082371
Chicago/Turabian StyleInami, Akie, Hiroshi Kiyono, and Yosuke Kurashima. 2018. "ATP as a Pathophysiologic Mediator of Bacteria-Host Crosstalk in the Gastrointestinal Tract" International Journal of Molecular Sciences 19, no. 8: 2371. https://doi.org/10.3390/ijms19082371