Application of Fluorescence Lifetime Imaging Microscopy of DNA Binding Dyes to Assess Radiation-Induced Chromatin Compaction Changes

Abstract
:1. Introduction
2. Results
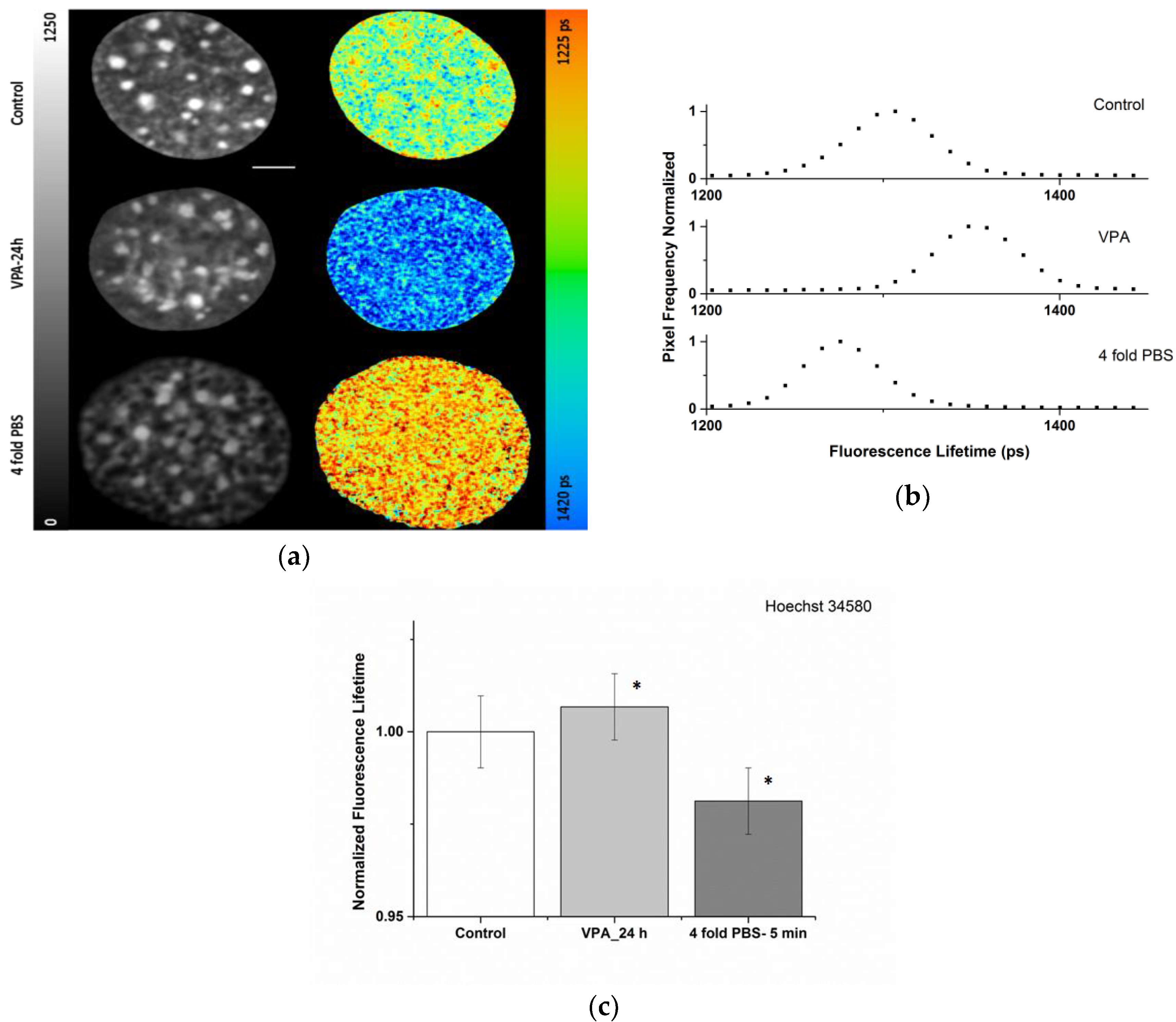
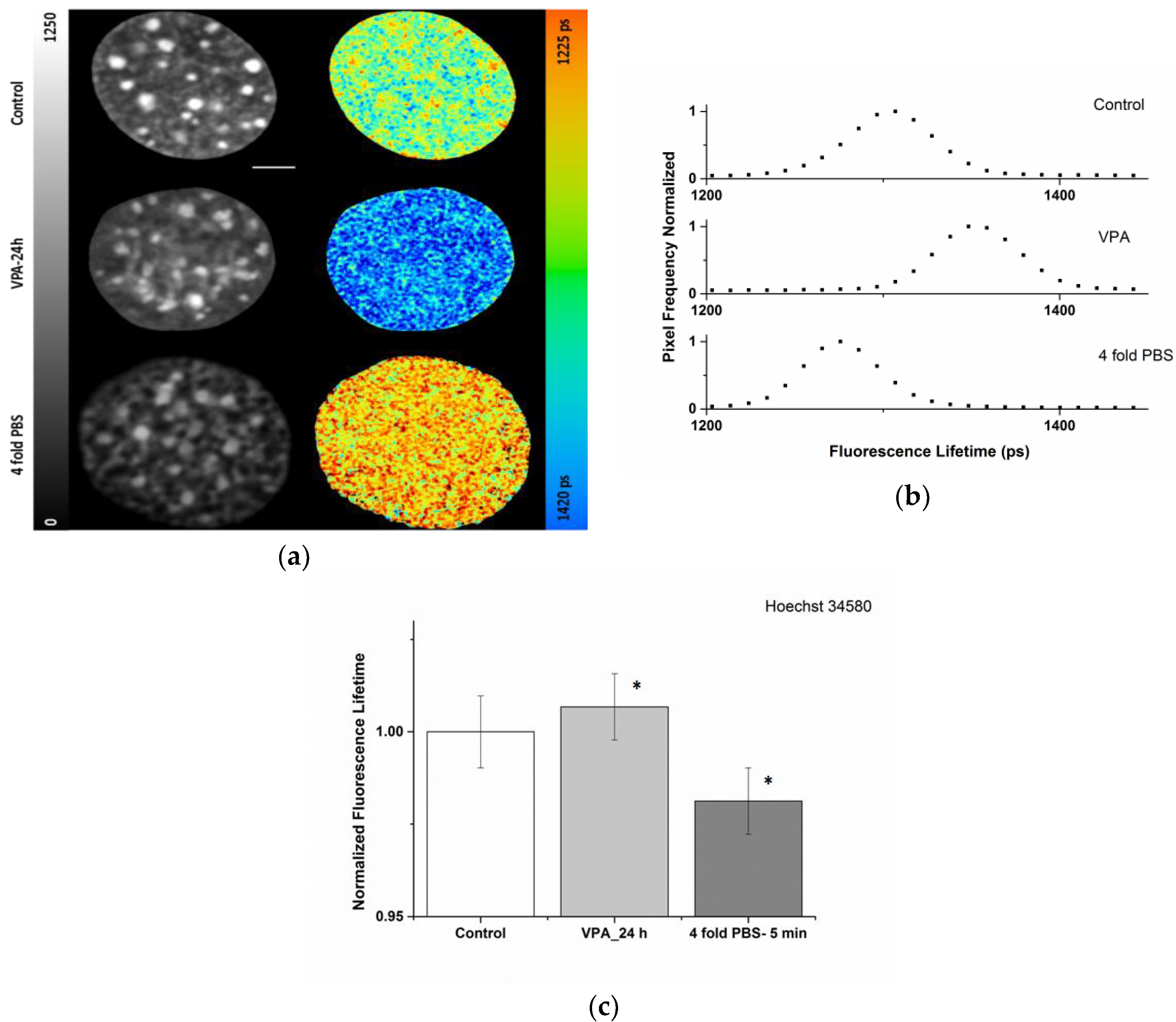
2.1. Sensitivity of the Organic Dye Sensors upon Chromatin Modulation
2.2. Impact of Pile-up Effect and Counting Loss on Fluorescence Lifetime Readout
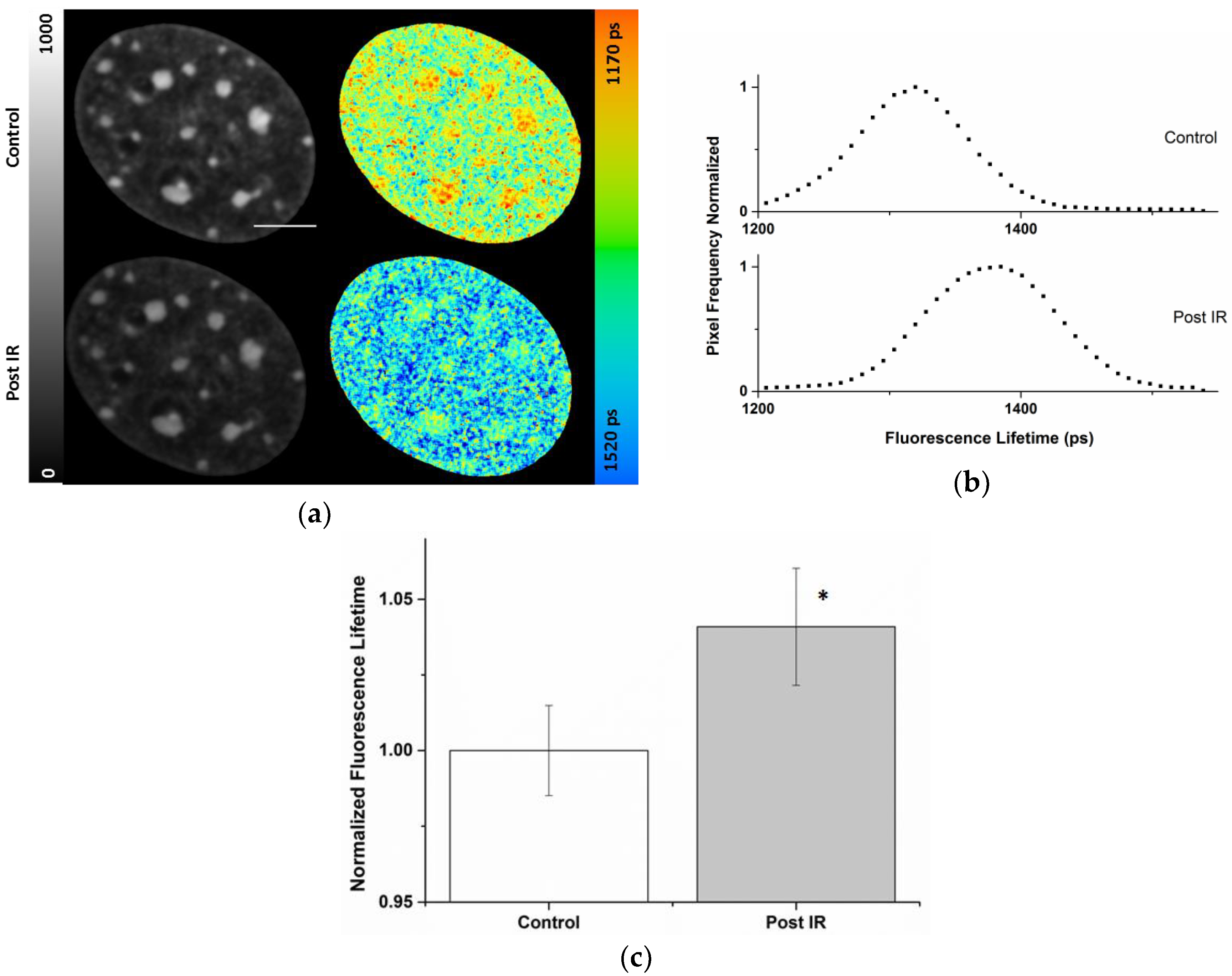
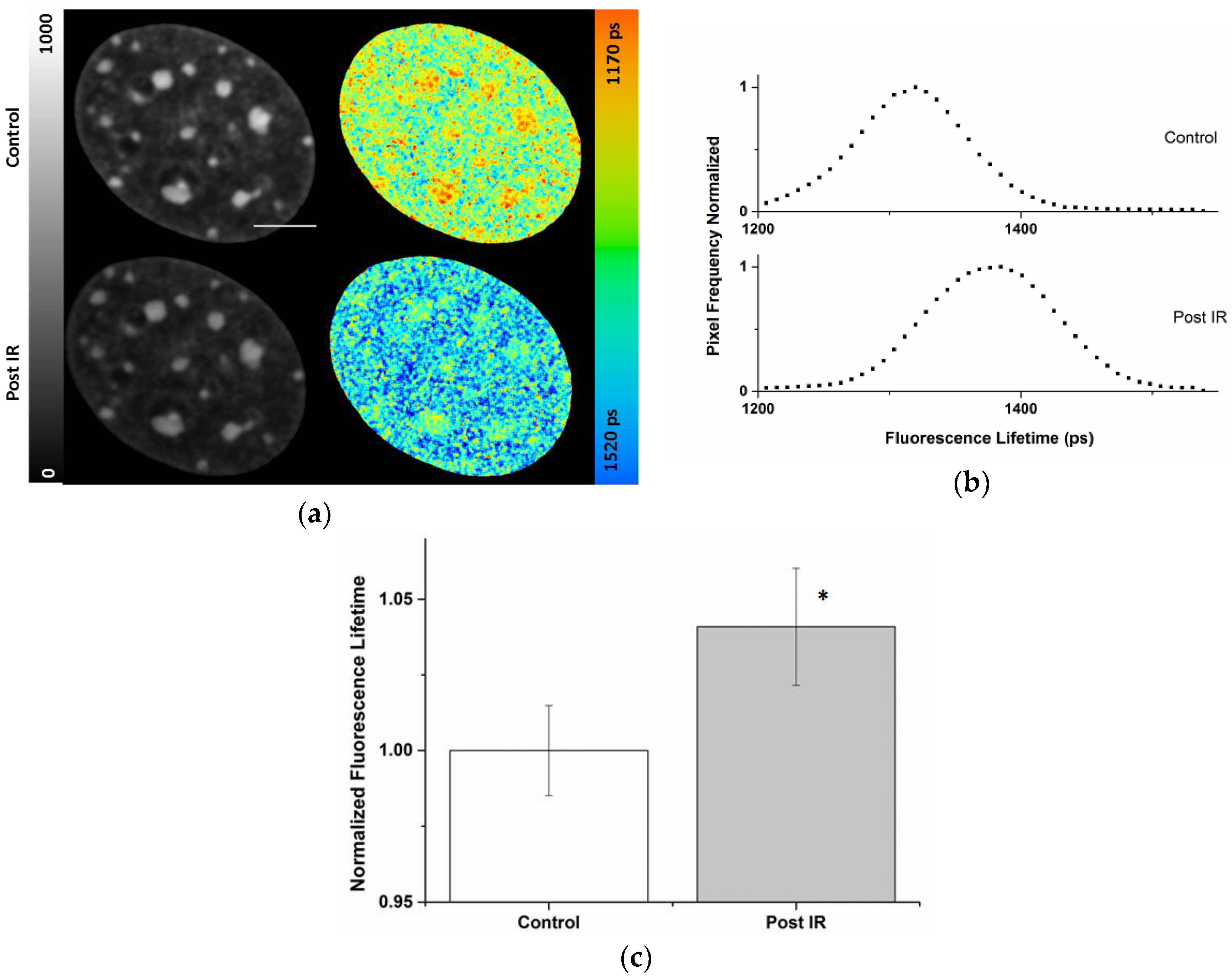
2.3. Radiation-Induced Global Chromatin Decompaction Imaged by FLIM in Living Cells after X-ray Irradiation
3. Discussion
4. Materials and Methods
4.1. Sample Preparation and DNA Staining
4.1.1. Histone Deacetylation Inhibitors, Valproic Acid
4.1.2. Hypertonic Treatment
4.2. Microscopy, Irradiation and Image Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FLIM | Fluorescence Lifetime Imaging Microscopy |
| TCSPC | Time Correlated Single Photon Counting |
| FRET | Förster Resonance Energy Transfer |
| LET | Linear Energy Transfer |
| DSB | Double Strand Break |
| HDACi | Histone Deacetylase Inhibitor |
| VPA | Valproic Acid |
| PBS | Phosphate Buffer Saline |
| FP | Fluorescence Protein |
| LUT | Lookup Table |
| GLCM | Gray Level Co-occurrence Matrix |
References
- Lanctot, C.; Cheutin, T.; Cremer, M.; Cavalli, G.; Cremer, T. Dynamic genome architecture in the nuclear space: Regulation of gene expression in three dimensions. Nat. Rev. Genet. 2007, 8, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Misteli, T. Higher-order genome organization in human disease. Cold Spring Harb. Perspect. Biol. 2010, 2, a00794. [Google Scholar] [CrossRef] [PubMed]
- Gospodinov, A.; Herceg, Z. Chromatin structure in double strand break repair. DNA Repair 2013, 12, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.A. The heterochromatic barrier to DNA double strand break repair: How to get the entry visa. Int. J. Mol. Sci. 2012, 13, 11844–11860. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Kruhlak, M.; Dotiwala, F.; Nussenzweig, A.; Haber, J.A. Heterochromatin is refractory to γ-H2AX modification in yeast and mammals. JCB 2007, 178, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Nikjoo, H.; Uehara, S.; Wilson, W.E.; Hoshi, M.; Goodhead, D.T. Track structure in radiation biology: Theory and applications. Int. J. Radiat. Biol. 1998, 73, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Scholz, M.; Taucher-Scholz, G. Biological imaging of heavy charged-particle tracks. Radiat. Res. 2003, 159, 676–684. [Google Scholar] [CrossRef]
- Hada, M.; Georgaklias, A.G. Formation of clustered DNA damage after high-LET irradiation: A review. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef]
- Lorat, Y.; Brunner, C.U.; Schanz, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Nanoscale analysis of clustered DNA damage after high-LET irradiation by quantitative electron microscopy—The heavy burden to repair. DNA Repair 2015, 28, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Guppy, B.J.; Sawchuk, L.; Davie, J.R.; McManus, K.J. Regulation of chromatin structure via histone post translational modification and link to carcinogenesis. Cancer Metast. Rev. 2013, 32, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Sishc, B.J.; Davis, A.J. The role of the core non-homologous end joining factors in carcinogenesis and cancer. Cancer 2017, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Rudolph, H.; Gueven, N.; Lavin, M.F.; Taucher-Scholz, G. Live cell imaging of heavy-ion-induced radiation responses by beamline microscopy. Radiat. Res. 2005, 163, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Jakob, B.; Splinter, J.; Durante, M.; Taucher-Scholz, G. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc. Natl. Acad. Sci. USA 2009, 106, 3172–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, B.; Taucher-Scholz, G. Live Cell Imaging to Study Real-Time ATM-Mediated Recruitment of DNA Repair Complexes to Sites of Ionizing Radiation-Induced DNA Damage; Humana Press: New York, NY, USA, 2017; Volume 1599, pp. 287–302. [Google Scholar]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiolo, I.; Minoda, A.; Polyzos, C.S.A.; Costes, S.; Karpen, G. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Merk, B.; Voss, K.; Averbeck, N.; Jakob, B.; Durante, M.; Taucher-Scholz, G. Species conserved DNA damage response at the inactive human X chromosome. Mutat. Res. 2013, 756, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Waters, J. Accuracy and precision in quantitative fluorescence microscopy. JCB 2009, 185, 1135–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakowicz, J. Principle of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006. [Google Scholar]
- Becker, W. The bh TCSPC Handbook; Becker & Hickl: Berlin, Germany, 2017. [Google Scholar]
- Van Muster, E.; Gadella, T. Fluorescence lifetime imaging microscopy (FLIM). In Microscopy Techniques. Advances in Biochemical Engineering in Microscopy Techniques; Rietdorf, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 143–175. [Google Scholar]
- Borst, J.; Visser, A. Fluorescence lifetime imaging microscopy in life sciences. Meas. Sci. Technol. 2010, 21, 102002. [Google Scholar] [CrossRef]
- Llères, D.; James, J.; Swift, S.; Norman, D.; Lamond, A. Quantitative analysis of chromatin compaction in living cells using FLIM-FRET. JCB 2009, 187, 481–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visvanathan, A.; Ahmed, K.; Even-Faitelson, L.; Llѐres, D.; Bazett-Polyamines, D.; Lamond, A. Modulation of higher order chromatin conformation in mammalian cell nuclei can be mediated by polyamines and divalent cations. PLoS ONE 2013, 8, e67689. [Google Scholar] [CrossRef] [PubMed]
- Spagnol, S.; Dahl, K. Active cytoskeletal force and chromatin condensation independently modulate intranuclear network fluctuations. Integr. Biol. 2014, 6, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Estandarte, A.; Botchway, S.; Lynch, C.; Yusuf, M.; Robinson, I. The use of DAPI fluorescence lifetime imaging for investigating chromatin condensation in human chromosome. Nat. Sci. Rep. 2016, 6, 31417. [Google Scholar] [CrossRef] [PubMed]
- Spagnol, S.; Dahl, K. Spatiallly resolved quantification of chromatin condensation through differential local Rheology in cell nuclei fluorescence lifetime imaging. PLoS ONE 2016, 11, e0146244. [Google Scholar]
- Llères, D.; Bailly, A.; Perrin, A.; Norman, D.; Xirodimas, D.; Feil, R. Quantitative FLIM-FRET microscopy to monitor nanoscale chromatin compaction In Vivo reveals structural roles of condensin complexes. Cell Rep. 2017, 18, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, E.; Taucher-Scholz, G.; Jakob, B. Upgrading the GSI beamline microscope with a confocal fluorescence lifetime scanner to monitor charged particle induced chromatin decondensation in living cells. NIMB 2015, 365, 626–630. [Google Scholar] [CrossRef]
- Becker, W. Advanced Time Correlated Single Photon Counting Applications. In Introduction to Multi-Dimensional TCSPC; Springer: Cham, Switzerland, 2015; pp. 1–55. [Google Scholar]
- Sherrard, A.; Bishop, P.; Panagi, M.; Villagomez, M.; Alibhai, D.; Kaidi, A. Streamlined histone-based fluorescence lifetime imaging microscopy (FLIM) for studying chromatin organisation. Biol. Open 2018, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Felisbino, M.; Viccari Gatti, M.; Mello, M. Changes in chromatin structure in NIH 3T3 cells induced by Valproic acid and Trichostatin A. J. Cell. Biochem. 2014, 115, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Isbaner, S.; Karedla, N.; Ruhlandt, D.; Stein, S.C.; Chizhik, A.; Gregor, I.; Enderlein, J. Dead-time correction of fluorescence lifetime imaging. Opt. Express 2016, 24, 9429–9445. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.C.; King, T.A. Correction methods for photon pile-up in lifetime determination by single-photon counting. J. Phys. A Gen. Phys. 1970, 3, 101–109. [Google Scholar] [CrossRef]
- Coates, P.B. Pile-up corrections in the measurements of lifetimes. J. Phys. E Sci. Instrum. 1972, 5, 148–150. [Google Scholar] [CrossRef]
- Ferraro, A. Altered primary chromatin structures and their implications in cancer development. Cell. Oncol. 2016, 39, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M. Studies in heterochromatin DNA: Accessibility of late replicating heterochromatin DNA in chromatin to micrococcal nuclease digestion. Chromosoma 1979, 70, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Imran Khan, M.; Mishra, A.; Das, P.; Sinha, K. HAT2 mediates histone H4K4 acetylation and affects micrococal nuclease sensitivity of chromatin in Leishmania donovani. PLoS ONE 2017, 12, e0177372. [Google Scholar] [CrossRef] [PubMed]
- Dobrucki, J.; Darzynkiewicz, Z. Chromatin condensation and sensitivity of DNA in situ to denaturation during cell cycle and apoptosis-a confocal microscopy study. Micron 2001, 32, 645–652. [Google Scholar] [CrossRef]
- Ou, H.; Phan, S.; Deerinck, T.; Thor, A.; Ellisman, M.; O’Shea, C. ChromEMT: Visualizing 3D chromatin structure and compaction n interphase and mitotic cells. Science 2017, 357, eaag0025. [Google Scholar] [CrossRef] [PubMed]
- Krufczik, M.; Sievers, A.; Hausmann, A.; Lee, J.; Hildenbrand, G.; Schaufler, W.; Hausmann, M. Combining low temperature fluorescence DNA-hybridization, immunostaining, and super-resolution localization microscopy for nano-structure analysis of ALU elements and their influence on chromatin structure. Int. J. Mol. Sci. 2017, 18, 1005. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Roberti, M.; Hériché, J.; Huet, S.; Alexander, M.; Ellenberg, J. Correlative live and super-resolution imaging reveals the dynamic structure of replication domains. JCB 2018, 217, 1973–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczurek, A.; Klewes, L.; Xing, J.; Gourram, A.; Birk, U.; Knecht, H.; Dobrucki, J.W.; Mai, S.; Cremer, C. Imaging chromatin nanostructure with binding-activated localization microscopy based on DNA structure fluctuations. Nucleic Acids Res. 2017, 45, e56. [Google Scholar] [CrossRef] [PubMed]
- Szczurek, A.; Birk, U.; Knecht, H.; Dobrucki, J.W.; Mai, S.; Cremer, C. Super-resolution binding activated localization microscopy through reversible change of DNA conformation. Nucleus 2018, 9, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Cardoso, M. Chromatin condensation modulates access and binding of nuclear proteins. FASEB J. 2010, 24, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, M.; Bradly, M. Influence of chromatin structure on the induction of DNA double strand breaks by ionizing radiation. Cancer Res. 1992, 52, 1580–1586. [Google Scholar] [PubMed]
- Waters, R.; Lyons, B. Variation in radiation-induced formation of DNA double strand breaks as a function of chromatin structure. Radiat. Res. 1992, 130, 309–318. [Google Scholar] [CrossRef]
- Nygren, J.; Ljungman, M.; Ahnstörm, M. Chromatin structure and radiation-induced DNA strand breaks in human cells: Soluble scavengers and DNA-bound proteins offer a better protection against single than double strand breaks. Int. J. Radiat. Biol. 1995, 68, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin compaction protects genomic DNA from radiation damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Lukasova, E.; Gabrielova, B.; Ondrej, V.; Kozubek, S. Chromatin dynamics during DSB repair. Biochem. Biophys. Acta 2007, 1773, 1534–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D. Radiation induced chromatin conformation changes analysed by fluorescent localization microscopy, statistical physics, and graph theory. PLoS ONE 2015, 10, e0128555. [Google Scholar] [CrossRef] [PubMed]
- Bancaud, A.; Huet, S.; Daigle, N.; Mozziconacci, J.; Beaudouin, J.; Ellenberg, J. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 2009, 28, 3785–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Żurek-Biesiada, D.; Waligórski, P.; Dobrucki, J.W. UV-induced spectral shift and protonation of DNA fluorescent dye Hoechst 33258. J Fluoresc. 2014, 24, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.A.; Manzo, C.; Garcı’a-Parajo, M.F.; Lakadamyali, M.; Pia Cosma, M. Chromatin fibers are formed by heterogeneous groups of nucleosomes in Vivo. Cell 2015, 160, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, P.M.; Borovsk, T.; Stap, J.; Cijsouw, T.; ten Cate, R.; Medema, J.P.; Kanaar, R.; Franken, N.A.P.; Aten, J.A. Chromatin mobility is increased at sites of DNA double-strand breaks. J. Cell Sci. 2011, 125, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K.; Knoch, T.; Wachsmuth, M.; Frank-Stöhr, M.; Ströhr, M.; Bacher, C.; Müller, G.; Rippe, K. Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J. Cell. Sci. 2004, 117, 4277–4287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halarick, R.M.; Shanmugam, K.; Dinstein, I. Textural Features for Image Classification. IEEE Trans. Syst. Man Cybern. 1973, 3, 610–621. [Google Scholar] [Green Version]
- Kruhlak, M.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Müller, W.; McNally, J.; Bazett-Jones, D.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double strand breaks. JCB 2006, 172, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Strickfaden, H.; McDonald, D.; Kruhlak, M.; Haince, J.; Th’ng, J.; Rouleau, M.; Ishibashi, T.; Corry, G.; Ausio, J.; Underhill, D.; et al. Poly(ADP-ribosyl)ation-dependent Transient Chromatin Decondensation and Histone Displacement following Laser Microirradiation. J. Biol. Chem. 2016, 291, 1789–1802. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Condition | ti (ps) |
|---|---|
| Control | 1330 ± 12 |
| VPA-24 h | 1342 ± 12 |
| 4-fold PBS | 1308 ± 12 |
| Cell Compartment | ti (ps) Pre-Correction | ti (ps) Post-Correction | Photon Pre-Correction | Photon Post-Correction |
|---|---|---|---|---|
| Nucleus | 1370 ± 26 | 1372 ± 24 | 293 ± 77 | 419 ± 186 |
| Chromocenters | 1306 ± 26 | 1340 ± 21 | 480 ± 68 | 885 ± 227 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdollahi, E.; Taucher-Scholz, G.; Jakob, B. Application of Fluorescence Lifetime Imaging Microscopy of DNA Binding Dyes to Assess Radiation-Induced Chromatin Compaction Changes. Int. J. Mol. Sci. 2018, 19, 2399. https://doi.org/10.3390/ijms19082399
Abdollahi E, Taucher-Scholz G, Jakob B. Application of Fluorescence Lifetime Imaging Microscopy of DNA Binding Dyes to Assess Radiation-Induced Chromatin Compaction Changes. International Journal of Molecular Sciences. 2018; 19(8):2399. https://doi.org/10.3390/ijms19082399
Chicago/Turabian StyleAbdollahi, Elham, Gisela Taucher-Scholz, and Burkhard Jakob. 2018. "Application of Fluorescence Lifetime Imaging Microscopy of DNA Binding Dyes to Assess Radiation-Induced Chromatin Compaction Changes" International Journal of Molecular Sciences 19, no. 8: 2399. https://doi.org/10.3390/ijms19082399