Autologous Mesenchymal Stroma Cells Are Superior to Allogeneic Ones in Bone Defect Regeneration

 , and
, and
Abstract
:1. Introduction
2. Results
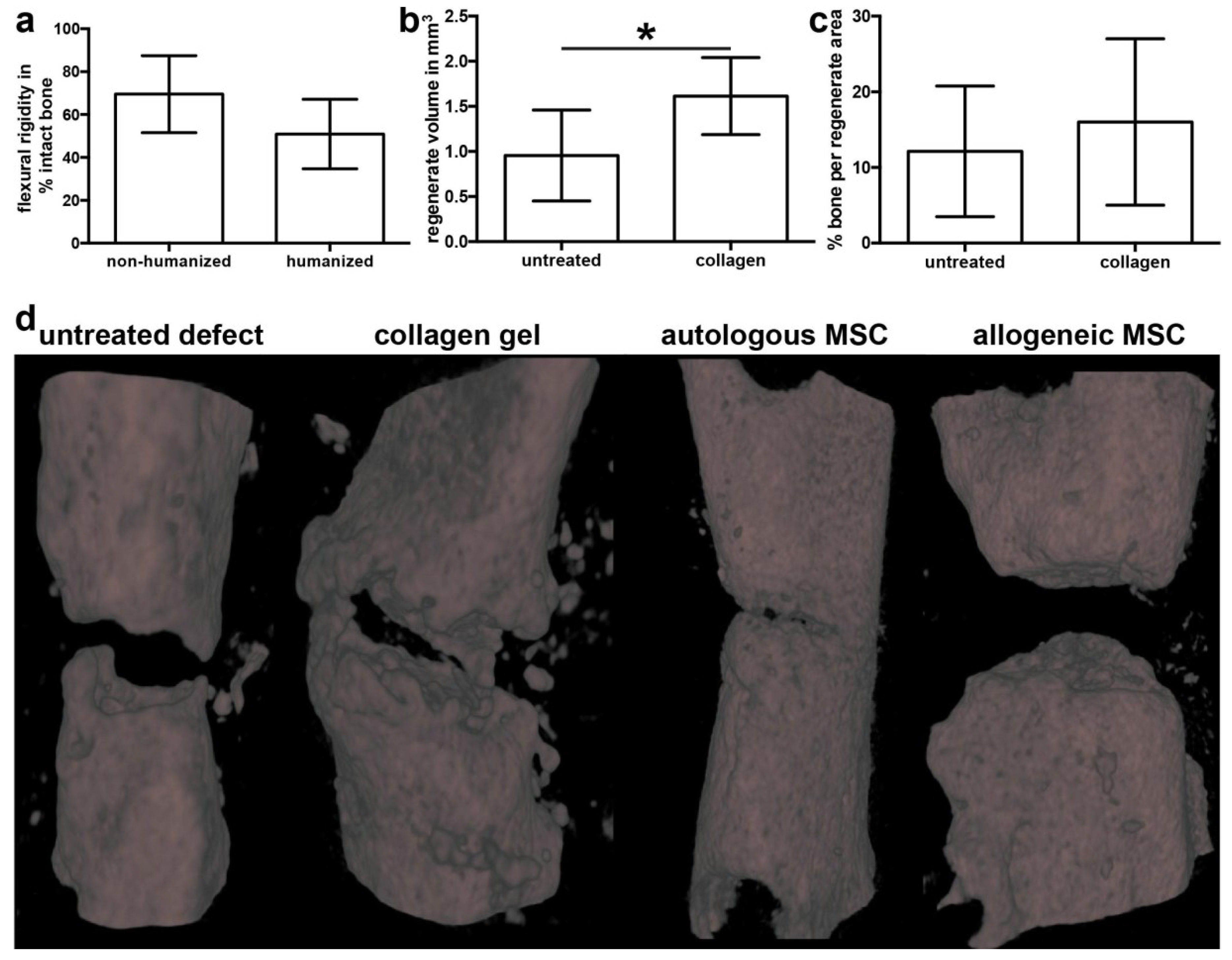
2.1. The Bone-Healing Capacity of Humanized Mice Is Not Significantly Affected by the Humanization Procedure
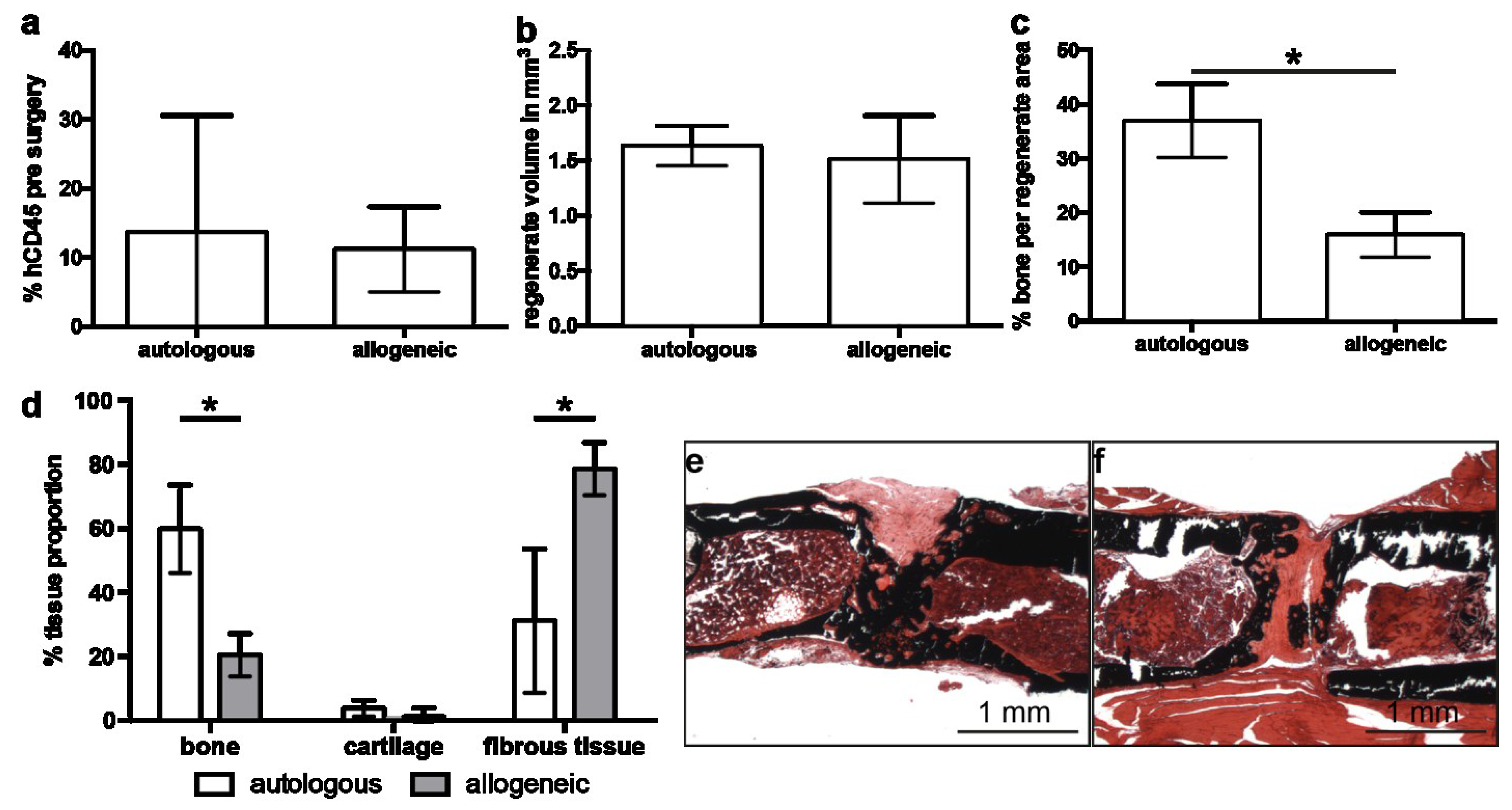
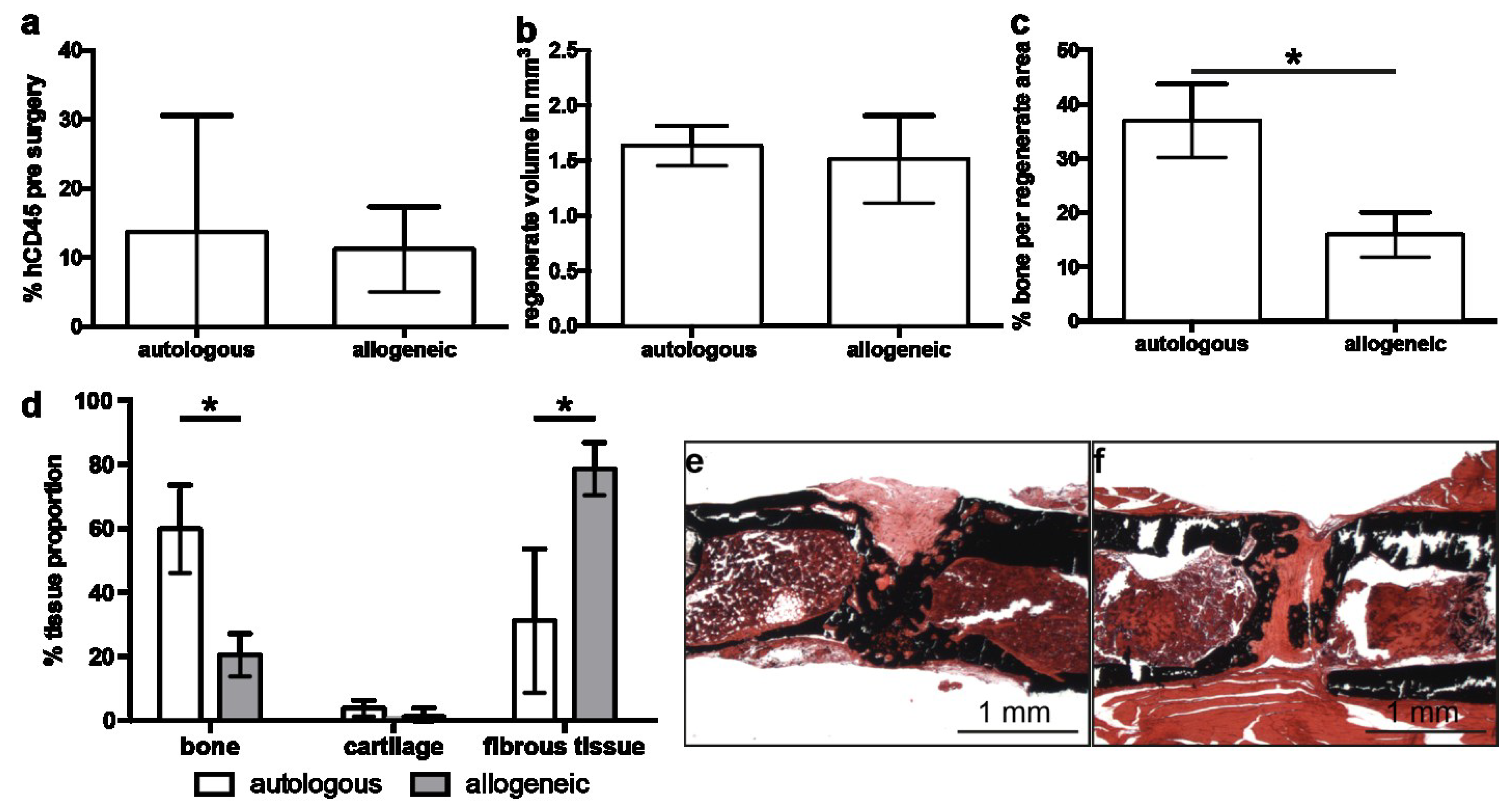
2.2. Implantation of Autologous hMSC Results in Significantly More Bone Formation Compared to Allogeneic hMSC
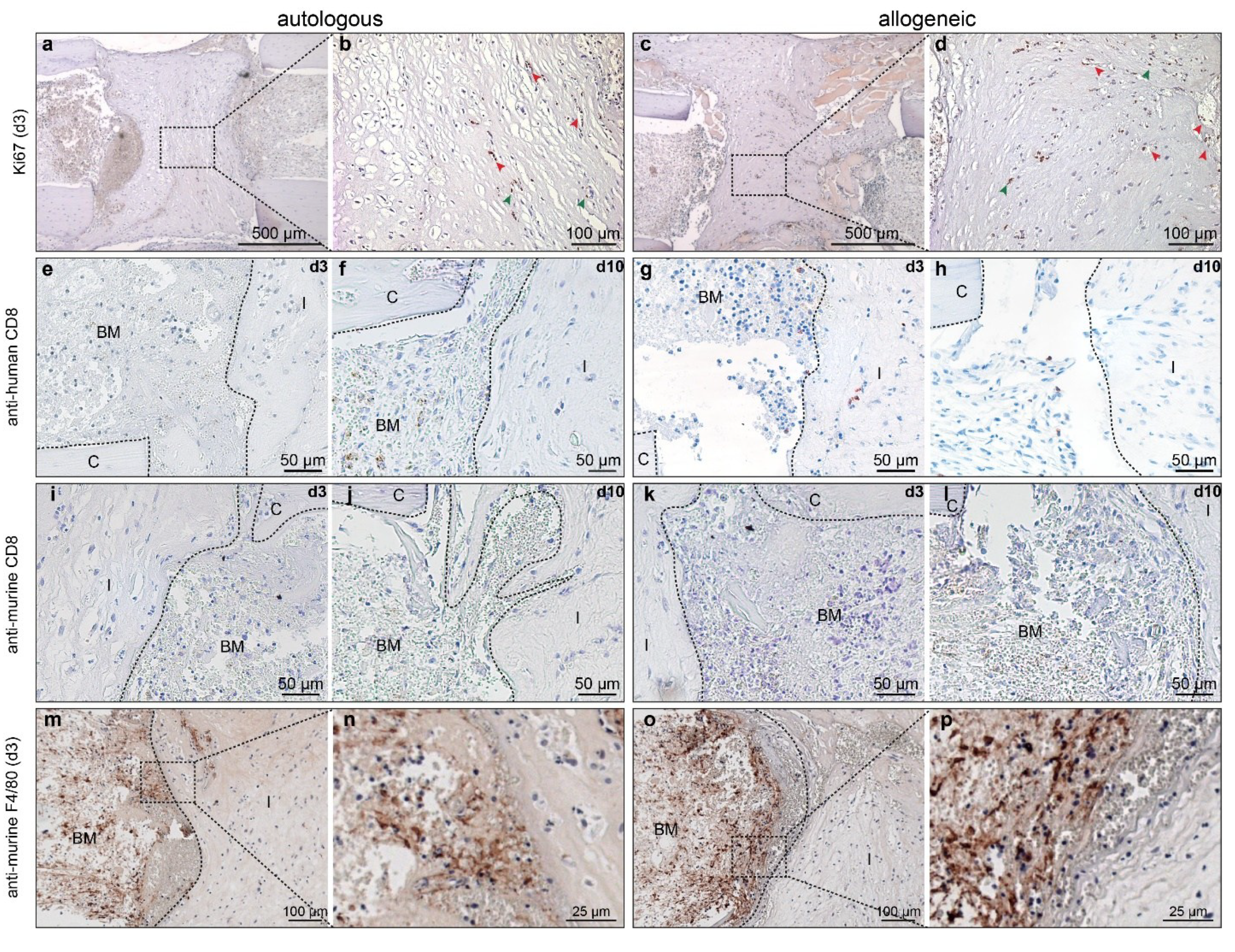
2.3. Allogeneic hMSCs Elicit a Mild Cellular Immune Response
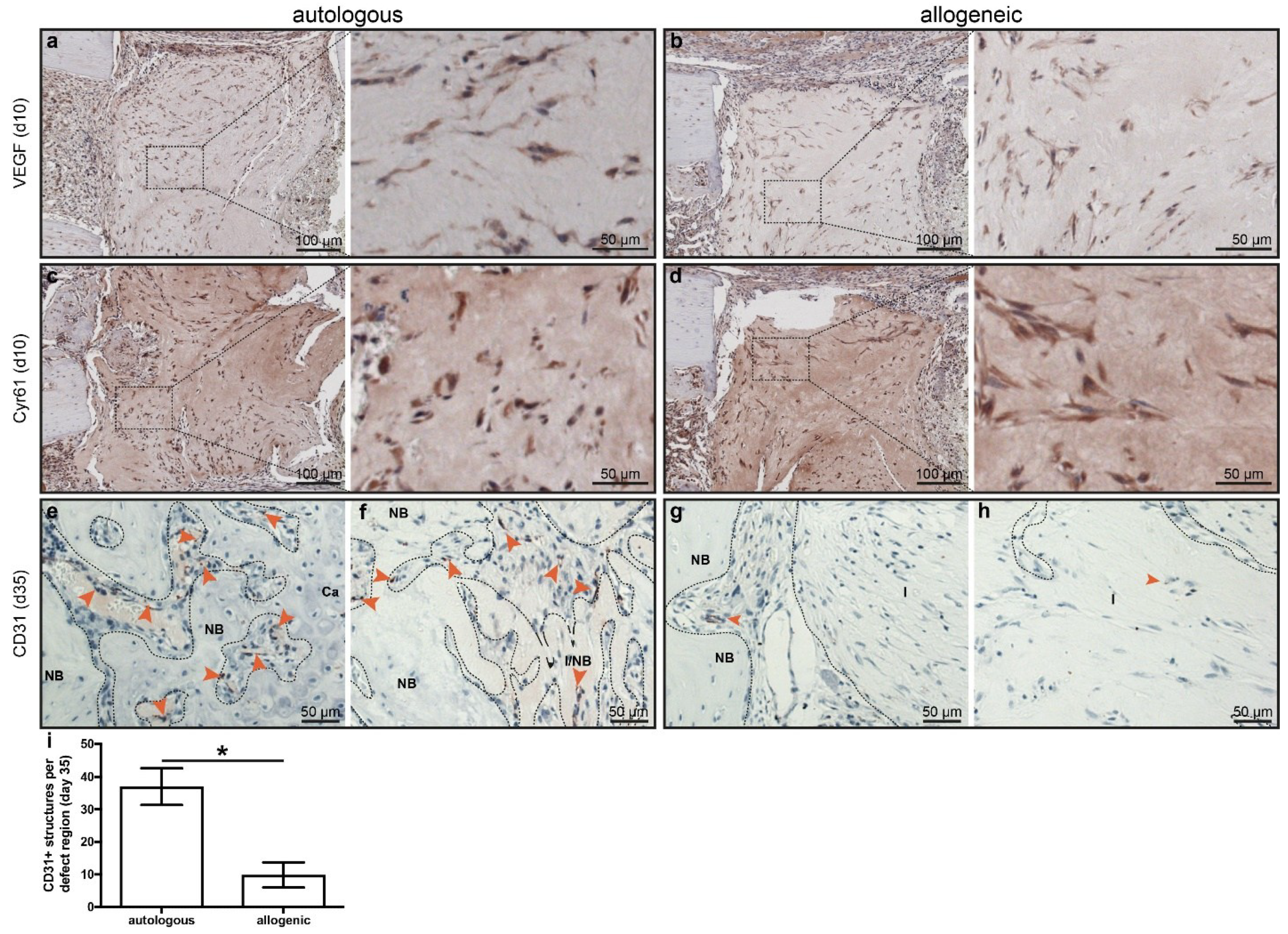
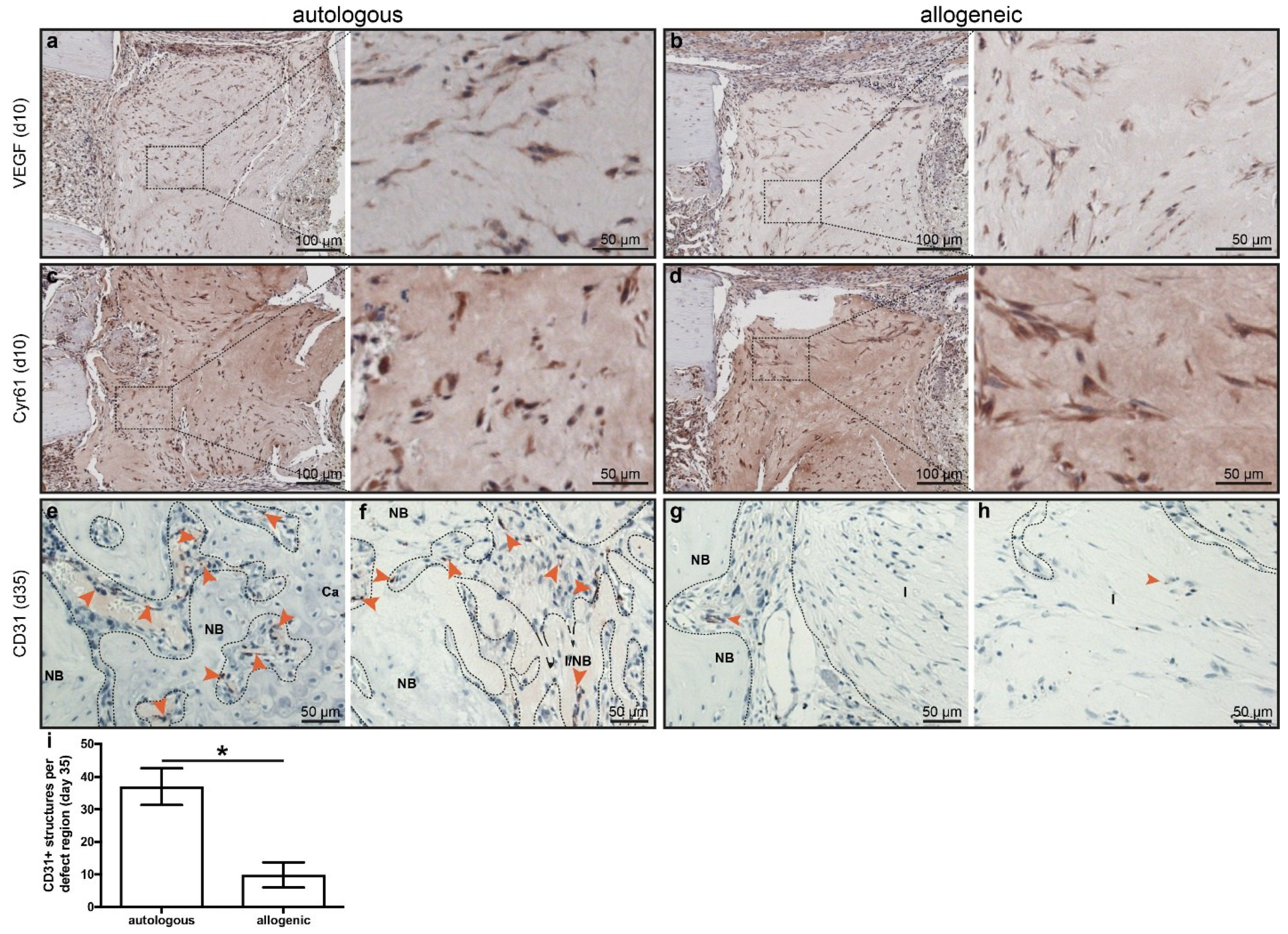
2.4. Early Angiogenesis Was Not Different between Mice Treated with Autologous or Allogeneic hMSC
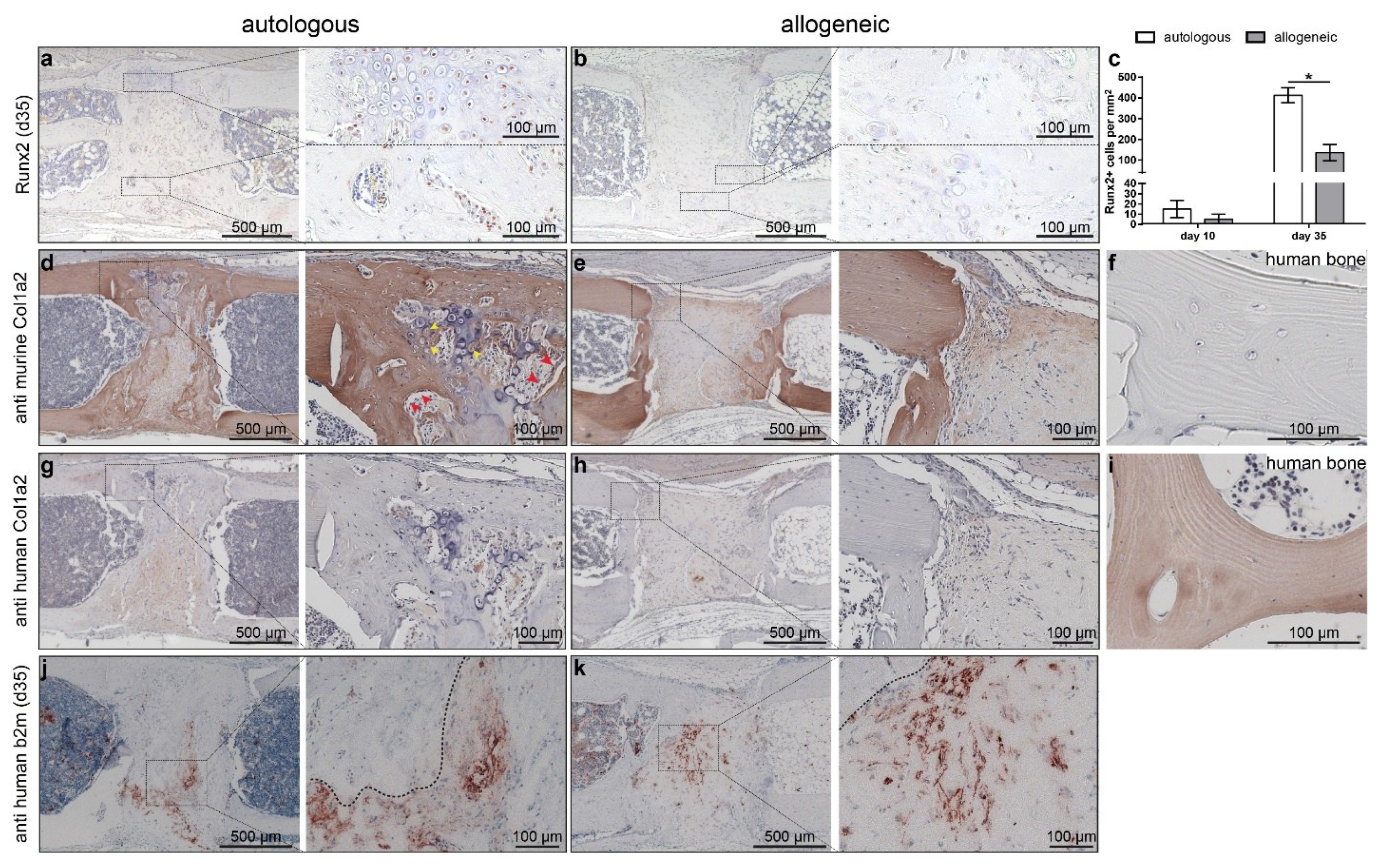
2.5. Human Cells Are Present Up to 35 Days in Defects Treated with hMSC, but These Do Not Synthesize Bone Matrix
3. Discussion
4. Materials and Methods
4.1. Animal Model and Husbandry
4.2. Generation of Humanized Mice
4.3. Cell Culture and Preparation for Local Delivery
4.4. Generation of Critically Sized Bone Defects
4.5. Sample Collection and Processing
4.6. µCT Analyses
4.7. Multiplex Cytokine Analyses
4.8. Immunohistochemical Staining
4.9. Histomorphometric Analysis
4.10. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Connolly, J.F.; Guse, R.; Tiedeman, J.; Dehne, R. Autologous marrow injection for delayed unions of the tibia: A preliminary report. J. Orthop. Trauma 1989, 3, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.F.; Guse, R.; Tiedeman, J.; Dehne, R. Autologous marrow injection as a substitute for operative grafting of tibial nonunions. Clin. Orthop. Relat. Res. 1991, 266, 259–270. [Google Scholar] [CrossRef]
- Hernigou, P.; Poignard, A.; Beaujean, F.; Rouard, H. Percutaneous Autologous Bone-Marrow Grafting for Nonunions. Influence of the Number and Concentration of Progenitor Cells. JBJS 2005, 87, 1430–1437. [Google Scholar]
- Gomez-Barrena, E.; Rosset, P.; Gebhard, F.; Hernigou, P.; Baldini, N.; Rouard, H.; Sensebé, L.; Gonzalo-Daganzo, R.M.; Giordano, R.; Padilla-Eguiluz, N. Feasibility and safety of treating non-unions in tibia, femur and humerus with autologous, expanded, bone marrow-derived mesenchymal stromal cells associated with biphasic calcium phosphate biomaterials in a multicentric, non-comparative trial. Biomaterials 2018, in press. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Girolamo, L.; Arrigoni, E.; Stanco, D.; Lopa, S.; Di Giancamillo, A.; Addis, A.; Borgonovo, S.; Dellavia, C.; Domeneghini, C.; Brini, A.T. Role of Autologous Rabbit Adipose-Derived Stem Cells in the Early Phases of the Repairing Process of Critical Bone Defects. J. Orthop. Res. 2011, 29, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Coathup, M.J.; Kalia, P.; Konan, S.; Mirza, K.; Blunn, G.W. A comparison of allogeneic and autologous mesenchymal stromal cells and osteoprogenitor cells in augmenting bone formation around massive bone tumor prostheses. J. Biomed. Mater. Res. Part A 2013, 101, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Bunnell, B.A.; Betancourt, A.M.; Sullivan, D.E. New concepts on the immune modulation mediated by mesenchymal stem cells. Stem Cell Res. Ther. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.M.; Gordon, P.L.; Koo, W.K.; Marx, J.C.; Neel, M.D.; McNall, R.Y.; Muul, L.; Hofmann, T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc. Natl. Acad. Sci. USA 2002, 99, 8932–8937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, E.M.; Prockop, D.J.; Fitzpatrick, L.A.; Koo, W.W.; Gordon, P.L.; Neel, M.; Sussman, M.; Orchard, P.; Marx, J.C.; Pyeritz, R.E.; et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat. Med. 1999, 5, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.M.; Prockop, D.J.; Gordon, P.L.; Koo, W.W.; Fitzpatrick, L.A.; Neel, M.D.; McCarville, M.E.; Orchard, P.J.; Pyeritz, R.E.; Brenner, M.K. Clinical responses to bone marrow transplantation in children with severe osteogenesis imperfecta. Blood 2001, 97, 1227–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, C.; He, Q.; Li, G. Allogenic peripheral blood derived mesenchymal stem cells (MSCs) enhance bone regeneration in rabbit ulna critical-sized bone defect model. J. Orthop. Res. 2006, 24, 610–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Zhang, Y.; Liu, B.; Sun, J.; Li, W.; Cui, L. Bone regeneration in a canine cranial model using allogeneic adipose derived stem cells and coral scaffold. Biomaterials 2013, 34, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Arinzeh, T.L.; Peter, S.J.; Archambault, M.P.; Van Den Bos, C.; Gordon, S.; Kraus, K.; Smith, A; Kadiyala, S. Allogeneic Mesenchymal Stem Cells Regenerate Bone in a Critical-Sized Canine Segmental Defect. JBJS 2003, 85, 1927–1935. [Google Scholar] [CrossRef]
- Berner, A.; Reichert, J.C.; Woodruff, M.A.; Saifzadeh, S.; Morris, A.J.; Epari, D.R.; Nerlich, M.; Schuetz, M.A.; Hutmacher, D.W. Autologous vs. allogenic mesenchymal progenitor cells for the reconstruction of critical sized segmental tibial bone defects in aged sheep. Acta Biomater. 2013, 9, 7874–7884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemeyer, P.; Schönberger, T.S.; Hahn, J.; Kasten, P.; Fellenberg, J.; Suedkamp, N.; Mehlhorn, A.T.; Milz, S.; Pearce, S. Xenogenic transplantation of human mesenchymal stem cells in a critical size defect of the sheep tibia for bone regeneration. Tissue Eng. Part A 2010, 16, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, P.; Szalay, K.; Luginbühl, R.; Südkamp, N.P.; Kasten, P. Transplantation of human mesenchymal stem cells in a non-autogenous setting for bone regeneration in a rabbit critical-size defect model. Acta Biomater. 2010, 6, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Dighe, A.S.; Yang, S.; Madhu, V.; Balian, G.; Cui, Q. Interferon gamma and T cells inhibit osteogenesis induced by allogeneic mesenchymal stromal cells. J. Orthop. Res. 2013, 31, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-scid IL2Rγnull Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed]
- Rapp, A.E.; Bindl, R.; Recknagel, S.; Erbacher, A.; Müller, I.; Schrezenmeier, H.; Ehrnthaller, C.; Gebhard, F.; Ignatius, A. Fracture Healing Is Delayed in Immunodeficient NOD/scidIL2Rγcnull Mice. PLoS ONE 2016, 11, e0147465. [Google Scholar] [CrossRef] [PubMed]
- Chatterjea, A.; LaPointe, V.L.; Alblas, J.; Chatterjea, S.; van Blitterswijk, C.A.; de Boer, J. Suppression of the immune system as a critical step for bone formation from allogeneic osteoprogenitors implanted in rats. J. Cell. Mol. Med. 2014, 18, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Kikuiri, T.; Akiyama, K.; Chen, C.; Xu, X.; Shi, S. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-γ and TNF-α. Nat. Med. 2011, 17, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.; Geissler, S.; Taylor, W.R.; Schmidt-Bleek, K.; Juelke, K.; Schwachmeyer, V.; Dahne, M.; Hartwig, T.; Akyüz, L.; Meisel, C.; et al. Terminally Differentiated CD8+ T Cells Negatively Affect Bone Regeneration in Humans. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- El Khassawna, T.; Serra, A.; Bucher, C.H.; Petersen, A.; Schlundt, C.; Könnecke, I.; Malhan, D.; Wendler, S.; Schell, H.; Volk, H.D.; et al. T Lymphocytes Influence the Mineralization Process of Bone. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wang, Q.; Fu, X.; Wu, X.; Gu, C.; Bi, J.; Xie, F.; Kang, N.; Liu, X.; Yan, L.; et al. Influence of Immunogenicity of Allogeneic Bone Marrow Mesenchymal Stem Cells on Bone Tissue Engineering. Cell Transpl. 2016, 25, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, J.; deGuzman, L.; Bao, M.; Bunting, S.; Peale, F.V.; Ferrara, N.; Steinmetz, H.; Hoeffel, J.; Cleland, J.L.; Daugherty, A.; et al. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc. Natl. Acad. Sci. USA 2002, 99, 9656–9661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasopoulos, A.N.; Schneider, D.; Keiper, T.; Alt, V.; Pendurthi, U.R.; Liegibel, U.M.; Sommer, U.; Nawroth, P.P.; Kasperk, C.; Chavakis, T. Vascular endothelial growth factor (VEGF)-induced up-regulation of CCN1 in osteoblasts mediates proangiogenic activities in endothelial cells and promotes fracture healing. J. Biol. Chem. 2007, 282, 26746–26753. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Olsen, B.R. Osteoblast-derived VEGF regulates osteoblast differentiation and bone formation during bone repair. J. Clin. Investig. 2016, 126, 509–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röntgen, V.; Blakytny, R.; Matthys, R.; Landauer, M.; Wehner, T.; Göckelmann, M.; Jermendy, P.; Amling, M.; Schinke, T.; Claes, L.; et al. Fracture healing in mice under controlled rigid and flexible conditions using an adjustable external fixator. J. Orthop. Res. 2010, 28, 1456–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pg/mL | Empty (n = 3) | Autologous (n = 2) | Allogeneic (n = 8) |
|---|---|---|---|
| CXCL1 | 19.2 ± 2.3 | 19.1 ± 4.5 | 20.0 ± 2.2 |
| MCP-1 | 66.7 ± 14.0 | 88.6 ± 38.5 | 84.3 ± 25.8 |
| IL-6 | 16.1 ± 12.4 | 23.4 ± 16.4 | 39.7 ± 20.0 |
| TNF-α | 1.5 ± 0.2 | 1.2 ± 0.4 | 1.6 ± 0.1 |
| IL-4 | 0.2 ± 0.03 | 0.2 ± 0.05 | 0.2 ± 0.06 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapp, A.E.; Bindl, R.; Erbacher, A.; Kruchen, A.; Rojewski, M.; Schrezenmeier, H.; Müller, I.; Ignatius, A. Autologous Mesenchymal Stroma Cells Are Superior to Allogeneic Ones in Bone Defect Regeneration. Int. J. Mol. Sci. 2018, 19, 2526. https://doi.org/10.3390/ijms19092526
Rapp AE, Bindl R, Erbacher A, Kruchen A, Rojewski M, Schrezenmeier H, Müller I, Ignatius A. Autologous Mesenchymal Stroma Cells Are Superior to Allogeneic Ones in Bone Defect Regeneration. International Journal of Molecular Sciences. 2018; 19(9):2526. https://doi.org/10.3390/ijms19092526
Chicago/Turabian StyleRapp, Anna E., Ronny Bindl, Annika Erbacher, Anne Kruchen, Markus Rojewski, Hubert Schrezenmeier, Ingo Müller, and Anita Ignatius. 2018. "Autologous Mesenchymal Stroma Cells Are Superior to Allogeneic Ones in Bone Defect Regeneration" International Journal of Molecular Sciences 19, no. 9: 2526. https://doi.org/10.3390/ijms19092526