Thyroxin Protects White Matter from Hypoxic-Ischemic Insult in the Immature Sprague–Dawley Rat Brain by Regulating Periventricular White Matter and Cortex BDNF and CREB Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. T4 Rescues HI-Mediated Hypomyelination in Injured WM
2.2. T4 Promoted MBP 23KDa Isoform Expression in Injured WM
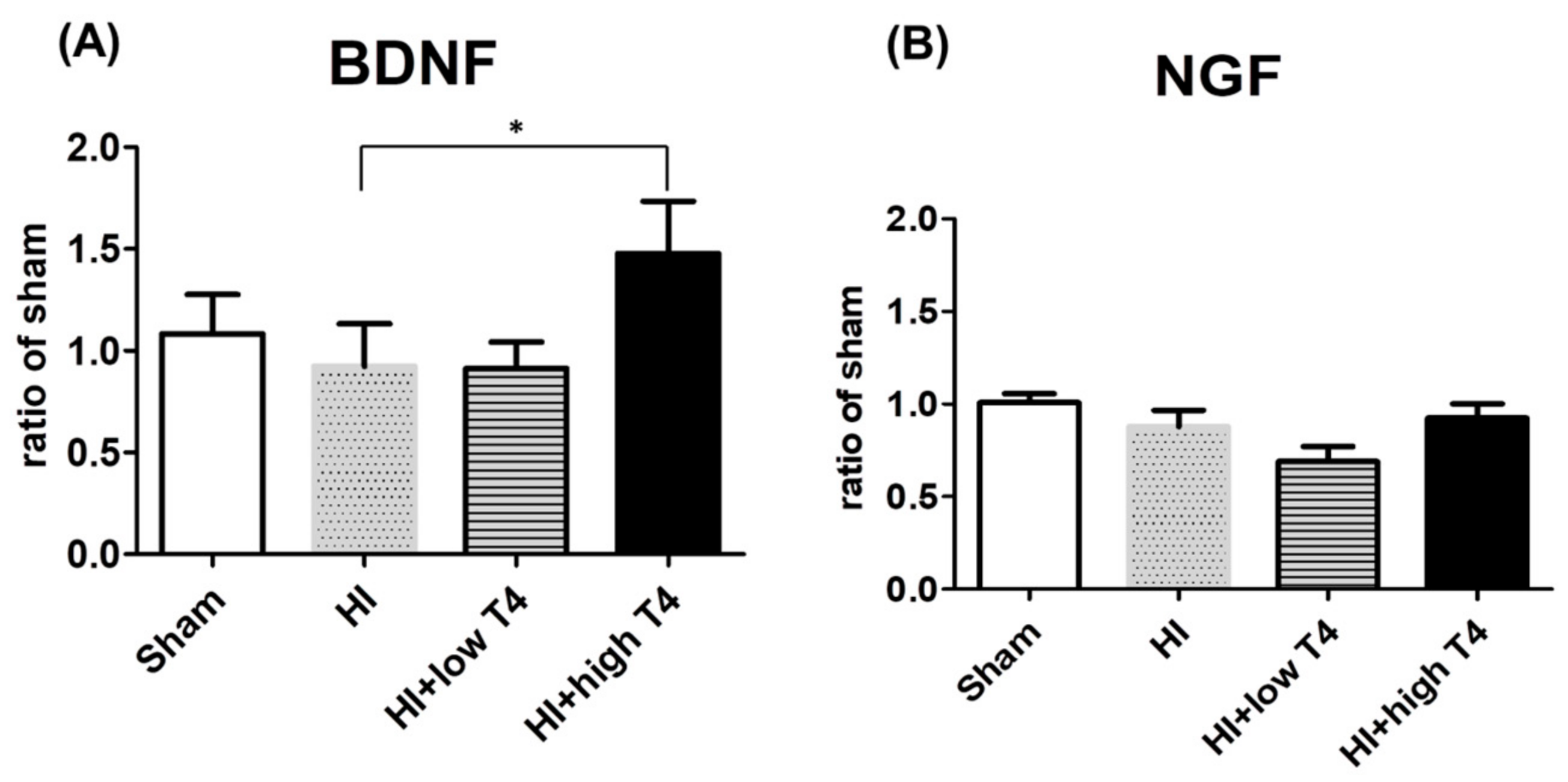
2.3. T4 Restored BDNF But Not NGF Expression of Injured WM Tissues
2.4. TrkB, Atk, p38, or JNK Did Not Actively Respond to T4 Treatment
2.5. T4 Reduced HI-Induced ERK Activation and CREB Phosphorylation
2.6. T4 Did Not Change Neointimal Formation in HI-Injured WM
2.7. T4 Increased BDNF Expression in Cortical Neurons
3. Discussion
4. Materials and Methods
4.1. Ischemia and Hypoxia-Induced in Premature Brains in Rat Pups
4.2. T4 Administration
4.3. Immunohistochemistry
4.4. Assessment of Immunohistochemical Staining
4.5. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.6. Immunofluorescence Staining
4.7. Morphometric Analysis
4.8. Western Blotting
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Volpe, J.J. Systemic inflammation, oligodendroglial maturation, and the encephalopathy of prematurity. Ann. Neurol. 2011, 70, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Gopagondanahalli, K.R.; Li, J.; Fahey, M.C.; Hunt, R.W.; Jenkin, G.; Miller, S.L.; Malhotra, A. Preterm Hypoxic-Ischemic Encephalopathy. Front. Pediatr. 2016, 4, 114. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.L.; Thompson, D.K.; Pascoe, L.; Leemans, A.; Inder, T.E.; Doyle, L.W.; Anderson, J.F.I.; Anderson, P.J. White matter abnormalities and impaired attention abilities in children born very preterm. Neuroimage 2016, 124, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.A.; Luo, N.L.; Mallinson, R.A.; O’Malley, J.P.; Wallen, L.D.; Frei, B.; Morrow, J.D.; Petito, C.K.; Roberts, C.T., Jr.; Murdoch, G.H.; et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann. Neurol. 2005, 58, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pena, A. Oligodendrocyte development and thyroid hormone. J. Neurobiol. 1999, 40, 497–512. [Google Scholar] [CrossRef]
- Calza, L.; Fernandez, M.; Giuliani, A.; Aloe, L.; Giardino, L. Thyroid hormone activates oligodendrocyte precursors and increases a myelin-forming protein and NGF content in the spinal cord during experimental allergic encephalomyelitis. Proc. Natl. Acad. Sci. USA 2002, 99, 3258–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leviton, A.; Paneth, N.; Reuss, M.L.; Susser, M.; Allred, E.N.; Dammann, O.; Kuban, K.; Van Marter, L.J.; Pagano, M. Hypothyroxinemia of prematurity and the risk of cerebral white matter damage. J. Pediatr. 1999, 134, 706–711. [Google Scholar] [CrossRef]
- Lucas, A.; Morley, R.; Fewtrell, M.S. Low triiodothyronine concentration in preterm infants and subsequent intelligence quotient (IQ) at 8 year follow up. BMJ 1996, 312, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Hung, P.L.; Huang, C.C.; Huang, H.M.; Tu, D.G.; Chang, Y.C. Thyroxin treatment protects against white matter injury in the immature brain via brain-derived neurotrophic factor. Stroke 2013, 44, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Barnabe-Heider, F.; Miller, F.D. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J. Neurosci. 2003, 23, 5149–5160. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Kanning, K.; Cavanaugh, J.E.; Xia, Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J. Biol. Chem. 1999, 274, 22569–22580. [Google Scholar] [CrossRef] [PubMed]
- Klocker, N.; Kermer, P.; Weishaupt, J.H.; Labes, M.; Ankerhold, R.; Bahr, M. Brain-derived neurotrophic factor-mediated neuroprotection of adult rat retinal ganglion cells in vivo does not exclusively depend on phosphatidyl-inositol-3′-kinase/protein kinase B. signaling. J. Neurosci. 2000, 20, 6962–6967. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Gozdz, A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur. J. Biochem. 2004, 271, 2050–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, E.A.; Bamford, M. Role of mitogen- and stress-activated kinases in ischemic injury. J. Cereb. Blood Flow Metab. 2002, 22, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, H.; Hur, J.; Kim, S.Y.; Kim, H.; Park, J.H.; Cha, S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; et al. Hypoxia induces nitric oxide production in mouse microglia via p38 mitogen-activated protein kinase pathway. Brain Res. Mol. Brain Res. 2002, 107, 9–16. [Google Scholar] [CrossRef]
- Brazil, D.P.; Park, J.; Hemmings, B.A. PKB binding proteins. Getting in on the Akt. Cell 2002, 111, 293–303. [Google Scholar] [CrossRef]
- Sun, X.; Zhou, H.; Luo, X.; Li, S.; Yu, D.; Hua, J.; Mu, D.; Mao, M. Neuroprotection of brain-derived neurotrophic factor against hypoxic injury in vitro requires activation of extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. Int. J. Dev. Neurosci. 2008, 26, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Holtzman, D.M. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J. Neurosci. 2000, 20, 5775–5781. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.W.; Xiao, J.; Kemper, D.; Kilpatrick, T.J.; Murray, S.S. Oligodendroglial expression of TrkB independently regulates myelination and progenitor cell proliferation. J. Neurosci. 2013, 33, 4947–4957. [Google Scholar] [CrossRef] [PubMed]
- Mograbi, B.; Bocciardi, R.; Bourget, I.; Busca, R.; Rochet, N.; Farahi-Far, D.; Juhel, T.; Rossi, B. Glial cell line-derived neurotrophic factor-stimulated phosphatidylinositol 3-kinase and Akt activities exert opposing effects on the ERK pathway: Importance for the rescue of neuroectodermic cells. J. Biol. Chem. 2001, 276, 45307–45319. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Tamai, M.; Mori, N. Brain-derived neurotrophic factor prevents axotomized retinal ganglion cell death through MAPK and PI3K signaling pathways. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3319–3326. [Google Scholar]
- Vassall, K.A.; Bessonov, K.; De Avila, M.; Polverini, E.; Harauz, G. The effects of threonine phosphorylation on the stability and dynamics of the central molecular switch region of 18.5-kDa myelin basic protein. PLoS ONE 2013, 8, e68175. [Google Scholar] [CrossRef] [PubMed]
- Uruse, M.; Yamamoto, M.; Sugawa, M.; Matsuura, K.; Sato, Y.; Seiwa, C.; Watanabe, K.; Aiso, S.; Asou, H. Phase separation of myelin sheath in Triton X-114 solution: Predominant localization of the 21.5-kDa isoform of myelin basic protein in the lipid raft-associated domain. J. Biochem. 2014, 155, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.R.; Cosgaya, J.M.; Wu, Y.J.; Shooter, E.M. Neurotrophins are key mediators of the myelination program in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 2001, 98, 14661–14668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Fischer, T.Z.; Clinton-Luke, P.; Lercher, L.D.; Dreyfus, C.F. Distinct effects of p75 in mediating actions of neurotrophins on basal forebrain oligodendrocytes. Mol. Cell. Neurosci. 2006, 31, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, J.C.; Wu, S.H. Neurotrophin signaling: Many exciting surprises! Cell. Mol. Life Sci. 2006, 63, 1523–1537. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Friedman, W.J.; Greene, L.A. Neurotrophin signaling via Trks and p75. Exp. Cell Res. 1999, 253, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Cosgaya, J.M.; Chan, J.R.; Shooter, E.M. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 2002, 298, 1245–1248. [Google Scholar] [CrossRef] [PubMed]
- Guthridge, M.A.; Stomski, F.C.; Thomas, D.; Woodcock, J.M.; Bagley, C.J.; Berndt, M.C.; Lopez, A.F. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells 1998, 16, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Itoh, T.; Arai, K.; Watanabe, S. Activation of c-Jun N-terminal kinase by human granulocyte macrophage-colony stimulating factor in BA/F3 cells. Biochem. Biophys. Res. Commun. 1997, 234, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Chang, Y.C.; Tu, Y.F.; Huang, C.C. VEGF-A/VEGFR-2 signaling leading to cAMP response element-binding protein phosphorylation is a shared pathway underlying the protective effect of preconditioning on neurons and endothelial cells. J. Neurosci. 2009, 29, 4356–4368. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.F.; Lu, P.J.; Huang, C.C.; Ho, C.J.; Chou, Y.P. Moderate dietary restriction reduces p53-mediated neurovascular damage and microglia activation after hypoxic ischemia in neonatal brain. Stroke 2012, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.F.; Wang, L.W.; Lin, S.H.; Jhuo, T.J.; Chiu, N.T.; Huang, C.C.; Hsia, C.C.; Shen, L.H. Tc-99m-HL91 imaging in the early detection of neuronal injury in a neonatal rat model of hypoxic ischemia. Crit. Care Med. 2012, 40, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Zhiling, W.; Hui, Z.; Shengfu, L.; Dan, Y.; Jiping, H. Cellular levels of TrkB and MAPK in the neuroprotective role of BDNF for embryonic rat cortical neurons against hypoxia in vitro. Int. J. Dev. Neurosci. 2005, 23, 515–521. [Google Scholar] [PubMed]
- Sato-Bigbee, C.; Pal, S.; Chu, A.K. Different neuroligands and signal transduction pathways stimulate CREB phosphorylation at specific developmental stages along oligodendrocyte differentiation. J. Neurochem. 1999, 72, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Shiga, H.; Asou, H.; Ito, E. Advancement of differentiation of oligodendrocyte progenitor cells by a cascade including protein kinase A and cyclic AMP-response element binding protein. Neurosci. Res. 2005, 53, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Pertuz, M.; Sanchez-Pacheco, A.; Aranda, A. The thyroid hormone receptor antagonizes CREB-mediated transcription. EMBO J. 2003, 22, 3102–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Gallardo, M.Y.; Cote-Velez, A.; Carreon-Rodriguez, A.; Charli, J.L.; Joseph-Bravo, P. Phosphorylated cyclic-AMP-response element-binding protein and thyroid hormone receptor have independent response elements in the rat thyrotropin-releasing hormone promoter: An analysis in hypothalamic cells. Neuroendocrinology 2010, 91, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Dugas, J.C.; Mandemakers, W.; Rogers, M.; Ibrahim, A.; Daneman, R.; Barres, B.A. A novel purification method for CNS projection neurons leads to the identification of brain vascular cells as a source of trophic support for corticospinal motor neurons. J. Neurosci. 2008, 28, 8294–8305. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Kim, W.J.; Lok, J.; Lee, S.R.; Besancon, E.; Luo, B.H.; Stins, M.F.; Wang, X.; Dedhar, S.; Lo, E.H. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 7582–7587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Lo, E.H. Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke 2009, 40 (Suppl. 3), 4–7. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, S.; Lambrechts, D.; Carmeliet, P. Neurovascular signalling defects in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Chang, Y.C.; Tu, Y.F.; Huang, C.C. CREB activation mediates VEGF-A’s protection of neurons and cerebral vascular endothelial cells. J. Neurochem. 2010, 113, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cooper-Kuhn, C.M.; Nannmark, U.; Blomgren, K.; Kuhn, H.G. Stimulatory effects of thyroid hormone on brain angiogenesis in vivo and in vitro. J. Cereb. Blood Flow Metab. 2010, 30, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.J.; Watts, R.J. Connecting vascular and nervous system development: Angiogenesis and the blood-brain barrier. Annu. Rev. Neurosci. 2010, 33, 379–408. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, N.; Timiras, P.S. A Stereotaxic Atlas of the Developing Rat Brain; University of California Press: Berkeley/Los Angeles, CA, USA; London, UK, 1970; pp. 181–183. [Google Scholar]
- Wang, L.; Cai, R.; Lv, G.; Huang, Z.; Wang, Z. Hypoxia during pregnancy in rats leads to the changes of the cerebral white matter in adult offspring. Biochem. Biophys. Res. Commun. 2010, 396, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.J., Jr.; Goodman, G.P.; Zacchilli, M.; Posner, M.; Evans, C.; Owens, B.D. Incidence and epidemiology of combat injuries sustained during “the surge” portion of operation Iraqi Freedom by a U.S. Army brigade combat team. J. Trauma Acute Care Surg. 2010, 68, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Xu, H.; Hu, F.; Braun, A.; Smith, K.; Rivera, A.; Lou, N.; Ungvari, Z.; Goldman, S.A.; Csiszar, A.; et al. Angiogenic inhibition reduces germinal matrix hemorrhage. Nat. Med. 2007, 13, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Khazipov, R.; Zaynutdinova, D.; Ogievetsky, E.; Valeeva, G.; Mitrukhina, O.; Manent, J.B.; Represa, A. Atlas of the Postnatal Rat Brain in Stereotaxic Coordinates. Front. Neuroanat. 2015, 9, 161. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, P.-L.; Hsu, M.-H.; Yu, H.-R.; Wu, K.L.H.; Wang, F.-S. Thyroxin Protects White Matter from Hypoxic-Ischemic Insult in the Immature Sprague–Dawley Rat Brain by Regulating Periventricular White Matter and Cortex BDNF and CREB Pathways. Int. J. Mol. Sci. 2018, 19, 2573. https://doi.org/10.3390/ijms19092573
Hung P-L, Hsu M-H, Yu H-R, Wu KLH, Wang F-S. Thyroxin Protects White Matter from Hypoxic-Ischemic Insult in the Immature Sprague–Dawley Rat Brain by Regulating Periventricular White Matter and Cortex BDNF and CREB Pathways. International Journal of Molecular Sciences. 2018; 19(9):2573. https://doi.org/10.3390/ijms19092573
Chicago/Turabian StyleHung, Pi-Lien, Mei-Hsin Hsu, Hong-Ren Yu, Kay L. H. Wu, and Feng-Sheng Wang. 2018. "Thyroxin Protects White Matter from Hypoxic-Ischemic Insult in the Immature Sprague–Dawley Rat Brain by Regulating Periventricular White Matter and Cortex BDNF and CREB Pathways" International Journal of Molecular Sciences 19, no. 9: 2573. https://doi.org/10.3390/ijms19092573