Voltage-Dependent Calcium Channels, Calcium Binding Proteins, and Their Interaction in the Pathological Process of Epilepsy

1
Epilepsy Research Laboratory of Human Anatomy and Histoembryology Department, Key Laboratory of Environment and Genes Related to Diseases (Xi’an Jiaotong University), Xi’an Jiaotong University School of Basic Medical Sciences, Ministry of Education, Xi’an 710061, China
2
Radiation Physiology Laboratory, Singapore Nuclear Research and Safety Initiative, National University of Singapore, 1 CREATE Way #04-01, CREATE Tower, Singapore 138602, Singapore
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(9), 2735; https://doi.org/10.3390/ijms19092735
Submission received: 13 July 2018
/
Revised: 6 September 2018
/
Accepted: 7 September 2018
/
Published: 12 September 2018
(This article belongs to the Special Issue Calcium Binding Proteins)
Abstract
:As an important second messenger, the calcium ion (Ca2+) plays a vital role in normal brain function and in the pathophysiological process of different neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), and epilepsy. Ca2+ takes part in the regulation of neuronal excitability, and the imbalance of intracellular Ca2+ is a trigger factor for the occurrence of epilepsy. Several anti-epileptic drugs target voltage-dependent calcium channels (VDCCs). Intracellular Ca2+ levels are mainly controlled by VDCCs located in the plasma membrane, the calcium-binding proteins (CBPs) inside the cytoplasm, calcium channels located on the intracellular calcium store (particular the endoplasmic reticulum/sarcoplasmic reticulum), and the Ca2+-pumps located in the plasma membrane and intracellular calcium store. So far, while many studies have established the relationship between calcium control factors and epilepsy, the mechanism of various Ca2+ regulatory factors in epileptogenesis is still unknown. In this paper, we reviewed the function, distribution, and alteration of VDCCs and CBPs in the central nervous system in the pathological process of epilepsy. The interaction of VDCCs with CBPs in the pathological process of epilepsy was also summarized. We hope this review can provide some clues for better understanding the mechanism of epileptogenesis, and for the development of new anti-epileptic drugs targeting on VDCCs and CBPs.
1. Introduction
Neuronal intracellular calcium increase plays an important role in the triggering and propagation of seizure activity [1,2]. Ca2+ entry via voltage-dependent calcium channels (VDCCs) conveys the electric signals to intracellular transduction cascades in a wide variety of cells including neurons, muscle cells and endocrine cells [1,3,4]. It regulates contraction, secretion, synaptic transmission, enzyme activity, protein phosphorylation/dephosphorylation, gene transcription, and controls diverse functions including cell survival and death, as well as adaptive responses to synaptic activity [5,6,7,8,9,10,11,12,13]. Therefore, VDCCs are the key signal transducers of electrical excitability, and they convert the electrical signal of an action potential in the cell surface membrane to an intracellular Ca2+ transient. Alterations of VDCC functions can cause abnormality in cellular events, leading to pathological consequences. In neurons, VDCCs initiate synaptic transmission [3,14,15]. Enhanced VDCCs currents with altered properties occur in neurons of epileptic patients with Ammon’s horn sclerosis (AHS) and in the dentate gyrus granule cells of epileptic animal models [16,17,18,19]. The alteration of some VDCCs subunits in resected brain tissue of epileptic patients and samples of epileptic animal models [20] suggests the possible involvement of VDCCs in epilepsy. While some studies show severe side effects of VDCC antagonists or failure in the control of epilepsy [1,21], VDCCs are still considered as promising drug targets in the treatment of epileptic seizures, as some VDCCs antagonists are both anticonvulsive and neuroprotective [22,23,24].
Ca2+ dependent-signaling cascades are largely mediated by calcium-binding proteins (CBPs) [25,26], and they are essential for multiple cellular and sub-cellular processes in physiological conditions. CBPs may achieve their cellular effects through Ca2+-dependent or Ca2+-independent signaling mechanisms [27,28]. CBPs mediate Ca2+-dependent signaling transduction pathways and regulate Ca2+ influx via the VDCCs in Ca2+-dependent feedback mechanisms [29]. CBPs containing EF-hand Ca2+ binding motifs are verified to regulate high voltage-activated VDCCs [30,31,32,33,34]. Many CBPs alter Ca2+ kinetics directly through the regulation of VDCC properties [30,31,32,33,34]. VDCCs are co-localized with CBPs in some neurons (especially in the subpopulation of hippocampal principal cell and interneurons) [35,36], and Ca2+ in these neurons is controlled by the synergy of VDCCs and CBPs. With the availability of human genetic databases and advanced molecular technologies, accumulated evidence suggest that dysfunctions in CBP-mediated VDCC regulation may be one of the mechanisms leading to human diseases [29]. CBPs are also involved in buffering the intracellular calcium concentration, and they may counteract an intracellular “overload” with Ca2+, and protect neurons from over-excitation and neuronal damage [37].
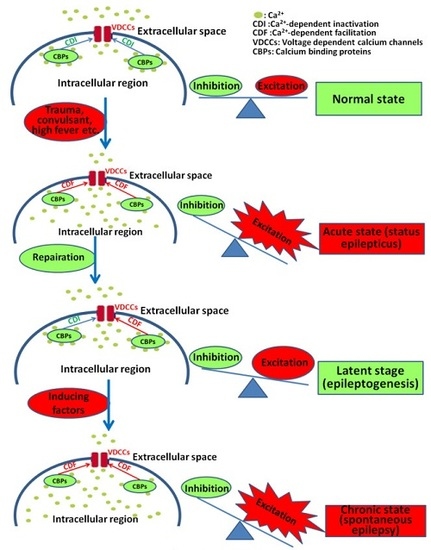
The pathological process of epilepsy includes three periods: an acute period with initiating factors (e.g., inborn brain malformations, acquired structural brain lesions etc.), including status epilepticus, a latent period (no seizure but often with the occurrence of interictal spikes), and a chronic stage with spontaneous recurrent seizures. VDCCs and CBPs have been reported to be involved in all stages of the pathogenesis of epilepsy. Epileptogenesis is mainly related to the latent period (the transformation of healthy brain tissue into hyperexcitable neuronal networks). Interictal spikes, as the omen of seizure activity, present during this period [38,39,40]. Interictal spikes include slow and fast interictal spikes [41]. The formation of the fast interictal spikes has been demonstrated repeatedly to crucially depend on VDCC-mediated Ca2+ influx and may be modulated by CBPs [40,42,43,44], indicating the vital roles of both VDCCs and CBPs in the pathological process of epilepsy.
2. The Function and Distribution of VDCCs in the Central Nervous System
Electrophysiological studies reveal different Ca2+ currents designated as L-, N-, P/Q-, R-, and T-type, based on the long-lasting current and intermediate voltage dependence, which were first recorded in Purkinje neurons and cerebellar granule neurons that were resistant to subtype-specific organic and peptide Ca2+ channel blockers, and transient current, respectively [45,46]. The L-type VDCC family has four members, i.e., voltage-gated calcium 1.1 (Cav1.1), Cav1.2, Cav1.3, and Cav1.4. P/Q-, N-, and R-type are corresponding to Cav2.1, Cav2.2, and Cav2.3, respectively. T-type VDCCs include Cav3.1, Cav3.2, and Cav3.3.
L-type VDCCs members, Cav1.2 and Cav1.3, shape neuronal firing and activate Ca2+-dependent pathways involved in the control of gene expression [47]. N-type, P/Q-type, and R-type VDCCs initiate rapid synaptic transmission, and are regulated primarily by direct interaction with G proteins and SNARE proteins, and secondarily by protein phosphorylation. T-type VDCCs, i.e., Cav3.1, Cav3.2, and Cav3.3 are important for the repetitive firing of action potentials in rhythmically firing cells, and could be activated and inactivated more rapidly and at more negative membrane potentials than other VDCCs mentioned above [45]. T-type VDCCs abnormalities in expression and function have been linked to a range of neurological diseases, including absence seizure, epilepsy, and neuropathic pain [48].
3. VDCCs in the Pathological Process of Epilepsy
The evidence to show VDCCs in epileptogenesis has been reported four decades ago, which indicated that decreases in extracellular free Ca2+ concentrations might trigger seizures in the brain tissues [72,73,74]. With the small volume of the extracellular space, the decrease in extracellular Ca2+ may indicate at least in part, the Ca2+ influx through neuronal VDCCs [75]. Pieces of evidence that VDCCs may take part in the pathological process of epilepsy have been reviewed in the following tables (Table 3, Table 4 and Table 5), and their possible roles in epilepsy are summarized in Table 1.
4. The Function and Distribution of CBPs in the Central Nervous System
Intracellular CBPs of the EF-hand superfamily are involved in the regulation of cell function and they contribute to the control of Ca2+ concentration in the cytosol, and participate in numerous cellular functions by acting as Ca2+ transporters across cell membranes or as Ca2+-modulated sensors, i.e., decoding Ca2+ signals. The members of intracellular CBPs with EF-hand [25,26,105], such as calmodulin (CaM), parvalbumin (PV), calretinin (CR), and calbindin-D28K (CB), contain 2 to 4 functioning EF-hand Ca2+ binding domains [29], have important function in brain, and have been indicated to be involved in many pathological process of the central nervous system. Among the intracellular CBPs superfamily, CaM, PV, CB, and CR are particularly important due to their abundance and the specificity of their distribution in the central nervous system. These CBPs are valuable markers of neuronal subpopulations for anatomical and developmental studies, although it remains unknown whether they all play a “triggering” role like CaM, or only act as buffers to modulate cytosolic calcium transients [106].
Among the EF-hand CBPs, the CaM family is very large and has been extensively characterized. CaM was firstly discovered in the bovine [107] and rat [108] brain and could be found in all eukaryotic organisms. A wide range of cellular processes, including cell division and differentiation, gene transcription, DNA synthesis, membrane fusion, and muscle contraction is mediated by CaM [109]. Due to its crucial role as Ca2+ sensor in all types of cells, it is assumed that CaM may be involved in many pathological processes, such as epilepsy [110,111], PD, AD or rheumatoid arthritis.
In the PV family of CBPs, PVs play an important role in cells by checking on calcium switching [112]. Because PV-expressing interneurons contribute to the maintenance and synchronization of cortical neurons activity through GABAergic synapses [113], PV has been associated with regulation of neuronal activity and PV-expressing interneurons may play a key role in numerous brain diseases, including epilepsy and complex psychiatric disorders, such as schizophrenia.
CR, as a member of the EF-hand CBPs family, was first identified in the retina. It is involved in intracellular calcium buffering, messenger targeting, cell cycle arrest, and apoptosis [114]. CR plays a crucial role in the modulation of intrinsic neuronal excitability [114] and the induction of LTP. Selective knockout of CR in mice resulted in an excess GABA release and impairment of LTP in the dentate gyrus (DG) [114,115], which was presumably due to an increased excitatory drive from CR-depleted mossy cells onto hilar interneurons [115,116].
As a high-affinity CBP, CB is expressed in the mammalian kidney, intestine, placenta, brain, peripheral nervous system, parathyroid gland, bone, and pancreas [117,118]. While the exact function of CB remains to be elucidated, current data suggest that it may play essential roles in neuronal Ca2+ buffer [119,120], ageing, and neurodegenerative diseases [121,122,123,124]. In the brain, CB may modulate intracellular Ca2+ transients and evoke Ca2+ signals in most neuronal groups [122,125], and therefore play a physiological role by sequestering free intracellular calcium, regulating intracellular responses to physiological stimuli and protecting neurons against Ca2+ mediated neurotoxicity by buffering intracellular Ca2+.
5. CBPs in the Pathological Process of Epilepsy
5.1. Background
Neuronal intracellular calcium increase plays an important role for the triggering and propagation of seizure activity [1]. CBPs contribute to the control of Ca2+ concentration in the cytosol and participate in numerous cellular functions by acting as Ca2+ transporters across cell membranes or as Ca2+-modulated sensors, i.e., decoding Ca2+ signals [126]. Therefore, CBPs are very likely involved in the pathological process of epilepsy. Indeed, some members of CBPs, such as CaM, CB, CR, and PV, are implicated in the generation of seizure or epileptogenesis.
5.2. CaM in the Pathological Process of Epilepsy
The effects of Ca2+ are frequently mediated via its interaction with CaM, and strong evidence indicate that the effects of Ca2+/CaM are often achieved through the regulation of conductance of ion channels and the protein phosphorylation process [127]. A recent study suggests CaM can bind to multiple sites of voltage-gated Na+ channel (Nav) intracellular C-terminal domains (CTDs), and can limit persistent Na+ current and accelerate inactivation across the Nav family. Mutations located in Nav1.2 channel CTDs can reduce CaM binding and result in increased persistent Na+ current and the occurrence of epilepsy [110], suggesting that CaM deficit may contribute to the pathological process of epilepsy. Epilepsy is linked to mutations in KCNQ channels, and CaM is one of the auxiliary proteins that are necessary for activation of the KCNQ2 channel [128]. CaM over-expression enhanced the outward K+ current and decreased neuronal activity. Meanwhile, CaM knockdown reduced the KCNQ2 current and increased neuronal activity. It suggests that CaM may regulate neuronal excitability by KCNQ2, and it could be a target of gene therapy in epilepsy [111]. These data suggest that epileptogenic factors may induce the downregulation of CaM, which in turn increases neuronal activity and results in the occurrence of epilepsy. A significant increase in CaM content in cytosol and membrane fractions of both control and kindled rats during the dark phase was related to a protective mechanism against enhanced sensitivity to seizures observed during the night, which may indicate the vital antiepileptic role of CaM [129]. CaM may also be involved in the pathological process of epilepsy indirectly through its modulation on related kinases, especially calcium/CaM-dependent protein kinases (CaMK). Transgenic mice carrying a CaMKII alpha-subunit mutation exhibit limbic epilepsy, suggesting an important CaMKII involvement in epilepsy [130]. In various seizure models, the inactivation of CaMK II occurs during seizure activity that precedes neuronal cell death [131,132,133]. Decreased mRNA level of the alpha subunit of CaMKII was found in the hippocampus of patients with intractable TLE [134]. These data may indicate that functional down-regulation of CaMKII could result in the occurrence of epilepsy. However, increased CaMKII in postsynaptic neurons was reported in KA-induced epileptic adult rat brain, [135]. In the hippocampus of patients with TLE, CaMKII labeling was significantly increased in the granule cell somata and their proximal dendrites [136]. These regional up-regulations of CaMKII may be a compensatory protective reaction to seizure activity. Collectively, the precise mechanism of CaM role in epileptogenesis through the CaMKII pathway is still unclear and needs further investigation. Both alterations of CaMKII in phosphorylation [137,138,139] and subcellular distribution [140,141] are speculated to be involved in the regulation of neuronal activity, and both alpha [142] and beta [143] subunits may be involved in the pathological process of epilepsy. The schematic presentation for roles of CaM in the pathological process of epilepsy is indicated in Figure 1.
5.3. CB in the Pathological Process of Epilepsy
By a gene knockout study, the vulnerability of hippocampal CA1 neurons to seizure-induced injury was shown to be modified by CB [144]. In GEPRs, a lower level of expression in CB mRNA in the caudate putamen-accumbens nuclei was found before the induction of seizures, indicating that it might confer genetic susceptibility to, and the generalization of, seizures, in this rat strain [145]. In patients with tuberous sclerosis complex associated with refractory epilepsy, significant CB decrease was found in the cortical tubers [146]. CB immunoreactivity in the hippocampus of seizure-sensitive gerbils was significantly lower as compared with those seen in seizure resistant gerbils [147]. Loss of CB immunoreactivity in the dentate gyrus was found in patients with TLE [148,149]. Although all these data suggest the association between the decrease of CB and seizure occurrence, the causal relationship remains unknown. However, increased CB-positive cells in the epileptic brain were also reported by different research groups. Increased CB-positive neurons have been observed in the cortex of patients with focal cortical dysplasia, which might be responsible for the ongoing seizure activity [37]. In patients of TLE with microdysgenesis, many neurogliaform CB-positive nerve cells were observed in the microdysgenesis cortex [150], indicating that these CB-positive neurons might be involved in epilepsy. Furthermore, in knockout mice (PV/CB-deficient), CB did not appear to modulate the excitability of the hippocampus in a mouse model of TLE [151]. Overall, as a calcium buffer, high-level CB may bind the intracellular Ca2+ to reduce the overload-induced excitotoxicity and prevent the pathological process of epilepsy. The loss of CB from granule cells markedly increased the Ca2+-dependent inactivation (CDI) of VDCCs currents, thereby diminishing Ca2+ influx during repetitive neuronal firing in epilepsy [125] which may inhibit the pathological process of epilepsy. Other mechanisms, such as the synaptic reorganization of CB-positive neurons may also be involved in the pathological process of epilepsy [152].
5.4. CR in the Pathological Process of Epilepsy
In the cerebellum, granule cells produce CR to excite Purkinje cells through parallel fibers. In vivo electrophysiological recording indicated that knocking out CR induced dramatic alterations in motor coordination and Purkinje cell firing [153]. In acute slice preparation, CR-deficient granule cells exhibited faster action potentials and generated repetitive spike discharge due to decreased calcium buffering capacity [154], suggesting that CR modulated intrinsic neuronal excitability. In the hippocampus, CR plays an important role in the modulation of neuronal excitability as CR-/- mice show impaired LTP induction following tetanic stimulation of hippocampal inputs, which could be restored by using bicuculline (a GABA receptor blocker) [115]. At a cellular level, except the expression of CR on mossy cells which are excitatory neurons of the dentate gyrus, CR is expressed mainly by GABAergic interneurons [155,156]. CR-positive interneurons, a distinct subpopulation of inhibitory cells innervating other interneurons in rodents, and to some extent principal cells in humans, are suggested to play a key role in the hippocampal inhibitory network, due to their function of synchronizing inhibitory interneurons [157,158,159]. The sensitivity of CR-positive interneurons to epileptic seizures has been discussed in numerous animal models, and in patients with TLE [160,161,162,163,164,165]. Increased vulnerability of CR-positive cells in patients [160,161], and in various animal models of epilepsy [166,167,168,169,170,171,172,173,174,175,176] were extensively documented, suggesting that seizure generation might be associated with a loss of a certain amount of CR-positive cells, and prevention of the loss of CR-positive cells may have antiepileptic effects. Abnormal neural circuits formed by CR-positive cells may alter the gating function of the dentate gyrus, and thereby increase hippocampal epileptogenicity [162,163,177]. The causal relationship between changes of CR-positive cells and epileptogenesis remains to be further investigated [178]. Based on current data, the role of CR in the pathological process of epilepsy has been summarized in Figure 1.
5.5. PV in the Pathological Process of Epilepsy
PV is one of the CBPs that spatially and temporally controls calcium transients across the cell membranes and inside the cytoplasm. PV-positive GABAergic interneurons, which regulate the fast-spiking capability of GABAergic neurons, are responsible for mediating feed-forward inhibition within cortical networks and play an important role in the responsiveness of inhibitory neurons to an adaptation to repetitive spikes [179,180]. Therefore, changes in PV-positive interneurons may be involved in epilepsy. PV knock-out mice are more susceptible to pentylenetetrazol-induced seizures [181], suggesting the critical role of PV in the pathological process of epilepsy. A positive correlation between the presence of CB and/or PV in hippocampal neurons and their relative resistance to seizure-induced neuronal damage suggests their capability for intracellular calcium buffering [182]. Cryptogenic epileptics, who had hippocampal sclerosis as the only lesion associated with epilepsy, exhibited a preferential survival of hippocampal cells that were CB or PV positive, while other epileptics with specific aetiology lack this morphological change, indicating the possible relevance of CB and PV in epileptogenesis of cryptogenic epilepsy [183]. The Mongolian gerbil is used as a model in epilepsy studies, due to its seizure sensitivity which may be related to its specific GABAergic system, i.e., the extra (versus rat, mouse, cat and man) PV projection from the entorhinal perforant path to the hippocampus [184], which is involved in the regulation of excitatory postsynaptic potential [185]. The relative increase in the PV positive cell population in the brain may suggest the resistance of PV positive cells to cell death in certain brain regions and highlight the selective nature of neuronal loss [183,186,187]. Other studies suggest that PV-positive interneurons are extremely vulnerable to status epilepticus (SE). PV-positive cells have a homogeneous distribution in the limbic regions, including the subiculum, the entorhinal and perirhinal cortex, the hippocampus, and the dentate gyrus [187]. PV-positive interneurons are rapidly degenerated in the hilus of the dentate gyrus one day after SE [188]. A significant decrease in PV-positive neurons was found in subiculum of epileptic animals with increased subiculum network excitability [189,190]. PV-positive interneurons are also found decreased in the entorhinal cortex deep layers [191], and the perirhinal cortex [187,192] in epileptic animals with limbic network hyperexcitability. In KA-induced epileptic rat, the loss of PV interneurons may contribute to the development of spontaneous seizures as reduced PV interneuron numbers in the subiculum and entorhinal cortex is correlated with the severity of seizure occurrence [176]. The loss of PV-positive interneurons in the subiculum results in marked impairment of feed-forward inhibition of the temporo-ammonic pathway, which may significantly contribute to the occurrence of epilepsy. A decrease of PV-positive interneurons was regarded as possibly resulting from the down-regulation of PV [183,193] which might depend on the frequency by which a certain neuronal area is recruited by seizures [189]. Combining the finding that convulsive status epilepticus duration determines epileptogenesis and interictal discharge generation in the rat limbic system [194], loss functioning of the PV positive interneuron in the limbic system may play a vital role in the pathological process of epilepsy. The down-regulation of PV may lead to a profound alteration in the functional characteristics of these brain structures [189,191,192,195], such as shifting the balance of excitation and inhibition towards excitation [196], and causing an input-specific disturbance of the subicular inhibitory system [190]. Previous data also revealed a drastic loss of PV positive interneurons in the tissue from epileptic patients [187,197,198,199,200]. The loss of PV-positive interneurons in the polymorphic layer of both sclerotic and non-sclerotic epileptic patients may result in a reduced inhibition of granular cells, which in turn increases the excitability of these cells and leads to the development of uncontrolled discharges in the hippocampus. The preservation of PV positive neurons is generally attributed to the buffering capability of PV and the underlining mechanisms for a selective PV cell loss induced by SE remain to be clarified. Research indicates that PV positive cells are shown to be damaged by a mechanism independent from oxygen supply and PV positive cells in human specimens present with high mobility group box 1 (HMGB1) translocation in the cytoplasm, suggesting a mechanism involving inflammation [201]. A recent study indicated that mitochondrial fragmentation regulated by p47Phox/CDK5/DRP1 signaling pathways might be involved in the PV positive cell loss [202]. The functional impairment of the PV positive neuron was also documented [203]. In this case, the loss of AMPA receptors in surviving PV-positive neurons may impair feed-forward inhibitory outputs, and contribute to the generation of spike-wave discharges and absence seizures in stargazers. Furthermore, alterations of the specification [204], migration [205,206,207,208], maturation [172,209], excitability [210,211,212], and synaptic connectivity [213] of PV-positive interneurons in rodents with genetic disorders, and seizures and in patients with genetic epilepsy indicate the complexity of the involvement of PV in epilepsy. The generally accepted Ca2+ buffering and GABA release regulation are two pathways through which PV regulates neuronal excitability and epileptogenesis (Figure 1).
6. Regulation of CBPs on VDCCs and the Implication of the Interaction between CBPs and VDCCs in the Pathological Process of Epilepsy
CBPs such as CaM do not act as solely Ca2+ chelators, but they do exert an important modulatory role by regulating the delivery of Ca2+ signal to different substrates and contribute to Ca2+ homeostasis via Ca2+-dependent inactivation (CDI), facilitation (CDF), and Ca2+-independent regulation (CIR) of the channels. Pathogenesis of human diseases may occur once CBPs-mediated VDCCs are impaired [29]. The best studied CBP that regulates VDCCs is CaM. CaM contains four functional EF-hand motifs, and regulates VDCCs properties in a pattern that is similar to an enzyme inhibitor [214]. CaM binds to various high-voltage-activated VDCCs, and causes CDI [29,30,31,215,216] or CDF [29,32,34,217]. CaM-mediated Cav2.1 CDF was blocked in the familial hemiplegic migraine type 1(FHM-1). This disruption of Cav2.1 CDF may cause the cerebellar ataxia-associated FHM-1 due to an imbalance between the excitatory and inhibitory inputs to the cerebellar Purkinje cells [218]. Therefore, an abnormal interaction between VDCCs and CaM may be involved in the pathological process of epilepsy (Figure 1).
The interactions of CR or CB with VDCCs may also regulate neuronal excitability and be implicated in the pathological process of epilepsy. In cells expressing Cav2.1 in vitro, co-expression of CR inhibits CDI and enhances CDF via a direct interaction with Cav2.1. The direct modulation of Cav2.1 by CR affects intracellular Ca2+ signaling, and probably also the neuronal excitability, via a mechanism that is different from Ca2+ buffering function [116]. By controlling the CDI of the L-type VDCCs, CB can regulate the amount of Ca2+ entry into neurons (Figure 1). Loss of CB from granule cells could markedly increase the CDI of L-type VDCC currents, thereby reducing the Ca2+ influx during repetitive neuronal firing in epilepsy [125]. It suggests that a low level of CB may contribute to inhibiting the pathological process of epilepsy through the regulation of VDCCs. P/Q-type VDCCs regulate neurotransmitter release in most central synapses, but they have been demonstrated to be particularly critical to sustaining GABA release from PV-positive interneurons [219]. The synaptic connectivity and properties of PV positive interneurons are partially determined by the expression of Cav2.1 [220,221,222]. Many molecular determinants of PV-positive interneuron synaptic function have been associated with epilepsy [213]. Further study on the interaction between PV and P/Q type VDCC, as well as other types of VDCCs and their implication in the pathological process of epilepsy, is still needed.
7. Conclusions
As important pathways for Ca2+ entering into the cytosol and activating neuronal activity, the role of VDCCs in the pathological process of epilepsy has been well accepted. However, based on the diverse functions of VDCCs in various tissues (e.g., contraction coupling in muscles, secretion regulation in endocrine tissues, vasoactive function in the cardiovascular system, etc.) and the lack of isoform-specific inhibitors of VDCCs, clinical application of antagonists of VDCCs in patients with epilepsy has been compromised due to universal severe side effects. Recent studies have indicated that the redistribution of VDCCs may play an important role in the pathological process of epilepsy, understanding the trafficking mechanism of VDCCs becomes very important and may provide novel therapeutic targets for seizure control or prevention of epilepsy. CBPs (such as CaM, CB, PV, and CR) as Ca2+ buffers, can reduce Ca2+ overload and protect neurons from excitotoxicity, and regulate neuronal excitability through CDF and CDI of VDCCs, or other pathways. Further study to understand the molecular mechanisms of the interaction between CBPs and VDCCs in the pathological process of epilepsy will greatly facilitate the discovery of promising targets for the development of new antiepileptic drugs. By overexpression of CBPs to investigate their gain-function in reducing epileptogenesis, novel approaches in controlling or prevention of the pathological process of epilepsy may be developed.
Funding
This work was supported by the funds of National Natural Science Foundation of China (NSFC) and the Social Development Grant of Shaanxi Province (Nos. 81071053 and 2010K15-08).
Acknowledgments
This work was supported by the funds of National Natural Science Foundation of China (NSFC) and the Social Development Grant of Shaanxi Province (Nos. 81071053 and 2010K15-08), China (XJH), and the National Research Foundation of Singapore to the Singapore Nuclear Research and Safety Initiative, National University of Singapore (TFR).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Larkin, J.G.; Besag, F.M.; Cox, A.; Williams, J.; Brodie, M.J. Nifedipine for epilepsy? A double-blind, placebo-controlled trial. Epilepsia 1992, 33, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Steinlein, O.K. Calcium signaling and epilepsy. Cell Tissue Res. 2014, 357, 385–393. [Google Scholar] [CrossRef] [PubMed]
- William, A.; CatterallCatterall, W.A. Voltage-gated calcium channels. Cold Spring Harbor Perspect. Biol. 2011, 3, a003947. [Google Scholar]
- Spitzer, N.C. Calcium: First messenger. Nat. Neurosci. 2008, 11, 243–244. [Google Scholar] [CrossRef] [PubMed]
- Bengtson, C.P.; Bading, H. Nuclear calcium signaling. Adv. Exp. Med. Biol. 2012, 970, 377–405. [Google Scholar] [PubMed]
- Bading, H.; Ginty, D.D.; Greenberg, M.E. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 1993, 260, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Chawla, S.; Johnson, C.M.; Bading, H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature 1997, 385, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Bading, H. CREB/CBP and SRE-interacting transcriptional regulators are fast on-off switches: Duration of calcium transients specifies the magnitude of transcriptional responses. J. Neurochem. 2001, 79, 849–858. [Google Scholar] [CrossRef] [PubMed]
- West, A.E.; Chen, W.G.; Dalva, M.B.; Dolmetsch, R.E.; Kornhauser, J.M.; Shaywitz, A.J.; Takasu, M.A.; Tao, X.; Greenberg, M.E. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 11024–11031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellstrom, B.; Savignac, M.; Gomez-Villafuertes, R.; Naranjo, J.R. Ca2+-operated transcriptional networks: Molecular mechanisms and in vivo models. Physiol. Rev. 2008, 88, 421–449. [Google Scholar] [CrossRef] [PubMed]
- Redmond, L. Translating neuronal activity into dendrite elaboration: Signaling to the nucleus. Neuro-Signals 2008, 16, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Fearnley, C.; Smyrnias, I.; MacDonald, F.; Roderick, H.L. An update on nuclear calcium signalling. J. Cell Sci. 2009, 122, 2337–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.J.; Zou, M.; Lu, L.; Lau, D.; Ditzel, D.A.; Delucinge-Vivier, C.; Aso, Y.; Descombes, P.; Bading, H. Nuclear calcium signaling controls expression of a large gene pool: Identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009, 5, e1000604. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.W.; Lipscombe, D.; Madison, D.V.; Bley, K.R.; Fox, A.P. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988, 11, 431–438. [Google Scholar] [CrossRef]
- Catterall, W.A.; Few, A.P. Calcium channel regulation and presynaptic plasticity. Neuron 2008, 59, 882–901. [Google Scholar] [CrossRef] [PubMed]
- Djamshidian, A.; Grassl, R.; Seltenhammer, M.; Czech, T.; Baumgartner, C.; Schmidbauer, M.; Ulrich, W.; Zimprich, F. Altered expression of voltage-dependent calcium channel α1 subunits in temporal lobe epilepsy with Ammon’s horn sclerosis. Neuroscience 2002, 111, 57–69. [Google Scholar] [CrossRef]
- Faas, G.C.; Vreugdenhil, M.; Wadman, W.J. Calcium currents in pyramidal CA1 neurons in vitro after kindling epileptogenesis in the hippocampus of the rat. Neuroscience 1996, 75, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Jeub, M.; Lie, A.; Blumcke, I.; Elger, C.E.; Beck, H. Loss of dynorphin-mediated inhibition of voltage-dependent Ca2+ currents in hippocampal granule cells isolated from epilepsy patients is associated with mossy fiber sprouting. Neuroscience 1999, 94, 465–471. [Google Scholar] [CrossRef]
- Hendriksen, H.; Kamphuis, W.; Lopes da Silva, F.H. Changes in voltage-dependent calcium channel alpha1-subunit mRNA levels in the kindling model of epileptogenesis. Brain Res. Mol. Brain Res. 1997, 50, 257–266. [Google Scholar] [CrossRef]
- Lie, A.A.; Blumcke, I.; Volsen, S.G.; Wiestler, O.D.; Elger, C.E.; Beck, H. Distribution of voltage-dependent calcium channel beta subunits in the hippocampus of patients with temporal lobe epilepsy. Neuroscience 1999, 93, 449–456. [Google Scholar] [CrossRef]
- Sander, J.W.; Trevisol-Bittencourt, P.C. Nifedipine as an add-on drug in the management of refractory epilepsy. Epilepsy Res. 1990, 6, 82–84. [Google Scholar] [CrossRef]
- Narayanan, J.; Frech, R.; Walters, S.; Patel, V.; Frigerio, R.; Maraganore, D.M. Low dose verapamil as an adjunct therapy for medically refractory epilepsy—An open label pilot study. Epilepsy Res. 2016, 126, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Nicita, F.; Spalice, A.; Papetti, L.; Nikanorova, M.; Iannetti, P.; Parisi, P. Efficacy of verapamil as an adjunctive treatment in children with drug-resistant epilepsy: A pilot study. Seizure 2014, 23, 36–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otoom, S.; Hasan, Z. Nifedipine inhibits picrotoxin-induced seizure activity: Further evidence on the involvement of L-type calcium channel blockers in epilepsy. Fundam. Clin. Pharmacol. 2006, 20, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Schaub, M.C.; Heizmann, C.W. Calcium, troponin, calmodulin, S100 proteins: From myocardial basics to new therapeutic strategies. Biochem. Biophys. Res. Commun. 2008, 369, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Ames, J.B.; Swindells, M.B.; Ikura, M. Diversity of conformational states and changes within the EF-hand protein superfamily. Proteins 1999, 37, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, R.D.; O’Callaghan, D.W.; Hasdemir, B.; Haynes, L.P.; Tepikin, A.V. Neuronal Ca2+-sensor proteins: Multitalented regulators of neuronal function. Trends Neurosci. 2004, 27, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D. Neuronal calcium sensor proteins: Generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosc. 2007, 8, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Nejatbakhsh, N.; Feng, Z.P. Calcium binding protein-mediated regulation of voltage-gated calcium channels linked to human diseases. Acta Pharmacol. Sin. 2011, 32, 741–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuhlke, R.D.; Pitt, G.S.; Tsien, R.W.; Reuter, H. Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the α1C subunit. J. Biol. Chem. 2000, 275, 21121–21129. [Google Scholar] [CrossRef] [PubMed]
- Pitt, G.S.; Zuhlke, R.D.; Hudmon, A.; Schulman, H.; Reuter, H.; Tsien, R.W. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J. Biol. Chem. 2001, 276, 30794–30802. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; DeMaria, C.D.; Erickson, M.G.; Mori, M.X.; Alseikhan, B.A.; Yue, D.T. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron 2003, 39, 951–960. [Google Scholar] [CrossRef]
- Christel, C.; Lee, A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim. Biophys. Acta 2012, 1820, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Dick, I.E.; Tadross, M.R.; Liang, H.; Tay, L.H.; Yang, W.; Yue, D.T. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 2008, 451, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Long, L.; Tang, Y.C.; Hu, H.T.; Tang, F.R. Ca(v)1.2, Ca(v)1.3, and Ca(v)2.1 in the mouse hippocampus during and after pilocarpine-induced status epilepticus. Hippocampus 2007, 17, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Yang, Z.B.; Wang, H.; Tang, F.R. Co-localization of L-type voltage dependent calcium channel α1D subunit (Ca(v)1.3) and calbindin (CB) in the mouse central nervous system. Neurosci. Lett. 2014, 561, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Kuchukhidze, G.; Wieselthaler-Holzl, A.; Drexel, M.; Unterberger, I.; Luef, G.; Ortler, M.; Becker, A.J.; Trinka, E.; Sperk, G. Calcium-binding proteins in focal cortical dysplasia. Epilepsia 2015, 56, 1207–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, K.; Hellier, J.L.; Dudek, F.E. Do interictal spikes drive epileptogenesis? Neuroscientist 2005, 11, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Staley, K.J.; White, A.; Dudek, F.E. Interictal spikes: Harbingers or causes of epilepsy? Neurosci. Lett. 2011, 497, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, F.E.; Rogawski, M.A. Calcium currents burst back: A possible role for dendrites in epileptogenesis. Epilepsy Curr. 2007, 7, 140–141. [Google Scholar] [CrossRef] [PubMed]
- Panuccio, G.; D’Antuono, M.; de Guzman, P.; De Lannoy, L.; Biagini, G.; Avoli, M. In vitro ictogenesis and parahippocampal networks in a rodent model of temporal lobe epilepsy. Neurobiol. Dis. 2010, 39, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, K.J.; Dudek, F.E. Interictal spikes and epileptogenesis. Epilepsy Curr. 2006, 6, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Spacey, S.D.; Materek, L.A.; Szczygielski, B.I.; Bird, T.D. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch. Neurol. 2005, 62, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.P.; Carman, J.S.; Hoffer, B.J.; Wyatt, R.J. Effect of altered calcium ion concentration on interictal spike generation in the hippocampal slice. Exp. Neurol. 1980, 68, 489–499. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Ann. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Nowycky, M.C.; Fox, A.P.; Tsien, R.W. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 1985, 316, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Cohen, S.; Li, B.; Tsien, R.W. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci. Rep. 2012, 33, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Sallan, M.C.; Visa, A.; Shaikh, S.; Nager, M.; Herreros, J.; Canti, C. T-type Ca2+ Channels: T for Targetable. Cancer Res. 2018, 78, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Leal, K.; Nanou, E. Calcium channels and short-term synaptic plasticity. J. Biol. Chem. 2013, 288, 10742–10749. [Google Scholar] [CrossRef] [PubMed]
- Neher, E.; Sakaba, T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 2008, 59, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Luebke, J.I.; Dunlap, K.; Turner, T.J. Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron 1993, 11, 895–902. [Google Scholar] [CrossRef]
- Turner, T.J.; Adams, M.E.; Dunlap, K. Multiple Ca2+ channel types coexist to regulate synaptosomal neurotransmitter release. Proc. Natl. Acad. Sci. USA 1993, 90, 9518–9522. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.; Kirschstein, T.; Kukley, M.; Pereverzev, A.; von der Brelie, C.; Schneider, T.; Beck, H. Functional specialization of presynaptic Cav2.3 Ca2+ channels. Neuron 2003, 39, 483–496. [Google Scholar] [CrossRef]
- Kamp, M.A.; Krieger, A.; Henry, M.; Hescheler, J.; Weiergraber, M.; Schneider, T. Presynaptic ’Ca2.3-containing’ E-type Ca channels share dual roles during neurotransmitter release. Eur. J. Neurosci. 2005, 21, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Sinnegger-Brauns, M.J.; Huber, I.G.; Koschak, A.; Wild, C.; Obermair, G.J.; Einzinger, U.; Hoda, J.C.; Sartori, S.B.; Striessnig, J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol. Pharmacol. 2009, 75, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Hell, J.W.; Westenbroek, R.E.; Warner, C.; Ahlijanian, M.K.; Prystay, W.; Gilbert, M.M.; Snutch, T.P.; Catterall, W.A. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 1993, 123, 949–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Biase, V.; Obermair, G.J.; Szabo, Z.; Altier, C.; Sanguesa, J.; Bourinet, E.; Flucher, B.E. Stable membrane expression of postsynaptic CaV1.2 calcium channel clusters is independent of interactions with AKAP79/150 and PDZ proteins. J. Neurosci. 2008, 28, 13845–13855. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.A.; Christel, C.J.; Jiao, Y.; Abiria, S.; Kim, K.Y.; Usachev, Y.M.; Obermair, G.J.; Colbran, R.J.; Lee, A. Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 2010, 30, 5125–5135. [Google Scholar] [CrossRef] [PubMed]
- Westenbroek, R.E.; Ahlijanian, M.K.; Catterall, W.A. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature 1990, 347, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Vreugdenhil, M.; Wadman, W.J. Enhancement of calcium currents in rat hippocampal CA1 neurons induced by kindling epileptogenesis. Neuroscience 1992, 49, 373–381. [Google Scholar] [CrossRef]
- Vigues, S.; Gastaldi, M.; Chabret, C.; Massacrier, A.; Cau, P.; Valmier, J. Regulation of calcium channel α1A subunit splice variant mRNAs in kainate-induced temporal lobe epilepsy. Neurobiol. Dis. 1999, 6, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.M.; Ikonomovic, M.D.; Abrahamson, E.E.; Kharlamov, E.A.; Hentosz, T.M.; Armstrong, D.M. Alterations in hippocampal voltage-gated calcium channel alpha 1 subunit expression patterns after kainate-induced status epilepticus in aging rats. Epilepsy Res. 2003, 57, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Westenbroek, R.E.; Sakurai, T.; Elliott, E.M.; Hell, J.W.; Starr, T.V.; Snutch, T.P.; Catterall, W.A. Immunochemical identification and subcellular distribution of the alpha 1A subunits of brain calcium channels. J. Neurosci. 1995, 15, 6403–6418. [Google Scholar] [CrossRef] [PubMed]
- Westenbroek, R.E.; Hell, J.W.; Warner, C.; Dubel, S.J.; Snutch, T.P.; Catterall, W.A. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron 1992, 9, 1099–1115. [Google Scholar] [CrossRef]
- Chung, Y.H.; Shin, C.; Park, K.H.; Cha, C.I. Immunohistochemical study on the distribution of the voltage-gated calcium channel alpha(1B) subunit in the mature rat brain. Brain Res. 2000, 866, 274–280. [Google Scholar] [CrossRef]
- Xu, J.H.; Long, L.; Wang, J.; Tang, Y.C.; Hu, H.T.; Soong, T.W.; Tang, F.R. Nuclear localization of Ca(v)2.2 and its distribution in the mouse central nervous system, and changes in the hippocampus during and after pilocarpine-induced status epilepticus. Neuropathol. Appl. Neurobiol. 2010, 36, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Soong, T.W.; Stea, A.; Hodson, C.D.; Dubel, S.J.; Vincent, S.R.; Snutch, T.P. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science 1993, 260, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Marubio, L.M.; Deal, C.R.; Hans, M.; Brust, P.F.; Philipson, L.H.; Miller, R.J.; Johnson, E.C.; Harpold, M.M.; Ellis, S.B. Structure and functional characterization of neuronal alpha 1E calcium channel subtypes. J. Biol. Chem. 1994, 269, 22347–22357. [Google Scholar] [PubMed]
- Yokoyama, C.T.; Westenbroek, R.E.; Hell, J.W.; Soong, T.W.; Snutch, T.P.; Catterall, W.A. Biochemical properties and subcellular distribution of the neuronal class E calcium channel alpha 1 subunit. J. Neurosci. 1995, 15, 6419–6432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siwek, M.; Henseler, C.; Broich, K.; Papazoglou, A.; Weiergraber, M. Voltage-gated Ca(2+) channel mediated Ca(2+) influx in epileptogenesis. Adv. Exp. Med. Biol. 2012, 740, 1219–1247. [Google Scholar] [PubMed]
- Craig, P.J.; Beattie, R.E.; Folly, E.A.; Banerjee, M.D.; Reeves, M.B.; Priestley, J.V.; Carney, S.L.; Sher, E.; Perez-Reyes, E.; Volsen, S.G. Distribution of the voltage-dependent calcium channel alpha1G subunit mRNA and protein throughout the mature rat brain. Eur. J. Neurosci. 1999, 11, 2949–2964. [Google Scholar] [CrossRef] [PubMed]
- Traub, R.D.; Llinas, R. Hippocampal pyramidal cells: Significance of dendritic ionic conductances for neuronal function and epileptogenesis. J. Neurophysiol. 1979, 42, 476–496. [Google Scholar] [CrossRef] [PubMed]
- Yaari, Y.; Konnerth, A.; Heinemann, U. Spontaneous epileptiform activity of CA1 hippocampal neurons in low extracellular calcium solutions. Exp. Brain Res. 1983, 51, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Jones, O.T. Ca2+ channels and epilepsy. Eur. J. Pharmacol. 2002, 447, 211–225. [Google Scholar] [CrossRef]
- Heinemann, U.; Hamon, B. Calcium and epileptogenesis. Exp. Brain Res. 1986, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Blalock, E.M.; Chen, K.C.; Vanaman, T.C.; Landfield, P.W.; Slevin, J.T. Epilepsy-induced decrease of L-type Ca2+ channel activity and coordinate regulation of subunit mRNA in single neurons of rat hippocampal ‘zipper’ slices. Epilepsy Res. 2001, 43, 211–226. [Google Scholar] [CrossRef]
- Bernstein, G.M.; Mendonca, A.; Wadia, J.; Burnham, W.M.; Jones, O.T. Kindling induces an asymmetric enhancement of N-type Ca2+ channel density in the dendritic fields of the rat hippocampus. Neurosci. Lett. 1999, 268, 155–158. [Google Scholar] [CrossRef]
- Bernstein, G.M.; Mendonca, A.; Wadia, J.; Burnham, W.M.; Jones, O.T. Kindling induces a long-term enhancement in the density of N-type calcium channels in the rat hippocampus. Neuroscience 1999, 94, 1083–1095. [Google Scholar] [CrossRef]
- Beck, H.; Steffens, R.; Elger, C.E.; Heinemann, U. Voltage-dependent Ca2+ currents in epilepsy. Epilepsy Res. 1998, 32, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Sochivko, D.; Becker, A.; Chen, J.; Jiang, Y.; Yaari, Y.; Beck, H. Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J. Neurosci. 2002, 22, 3645–3655. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Steffens, R.; Heinemann, U.; Elger, C.E. Properties of voltage-activated Ca2+ currents in acutely isolated human hippocampal granule cells. J. Neurophysiol. 1997, 77, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Beuckmann, C.T.; Sinton, C.M.; Miyamoto, N.; Ino, M.; Yanagisawa, M. N-type calcium channel alpha1B subunit (Cav2.2) knock-out mice display hyperactivity and vigilance state differences. J. Neurosci. 2003, 23, 6793–6797. [Google Scholar] [CrossRef] [PubMed]
- N’Gouemo, P.; Yasuda, R.; Faingold, C.L. Seizure susceptibility is associated with altered protein expression of voltage-gated calcium channel subunits in inferior colliculus neurons of the genetically epilepsy-prone rat. Brain Res. 2010, 1308, 153–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Borman, B.; Lakaye, B.; Minet, A.; Zorzi, W.; Vergnes, M.; Marescaux, C.; Grisar, T. Expression of mRNA encoding alpha1E and alpha1G subunit in the brain of a rat model of absence epilepsy. Neuroreport 1999, 10, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Lakaye, B.; Thomas, E.; Minet, A.; Grisar, T. The genetic absence epilepsy rat from Strasbourg (GAERS), a rat model of absence epilepsy: Computer modeling and differential gene expression. Epilepsia 2002, 43 (Suppl. 5), 123–129. [Google Scholar] [CrossRef] [PubMed]
- Weiergraber, M.; Kamp, M.A.; Radhakrishnan, K.; Hescheler, J.; Schneider, T. The Ca(v)2.3 voltage-gated calcium channel in epileptogenesis--shedding new light on an enigmatic channel. Neurosci. Biobehav. Rev. 2006, 30, 1122–1144. [Google Scholar] [CrossRef] [PubMed]
- Weiergraber, M.; Henry, M.; Radhakrishnan, K.; Hescheler, J.; Schneider, T. Hippocampal seizure resistance and reduced neuronal excitotoxicity in mice lacking the Cav2.3 E/R-type voltage-gated calcium channel. J. Neurophysiol. 2007, 97, 3660–3669. [Google Scholar] [CrossRef] [PubMed]
- Weiergraber, M.; Henry, M.; Krieger, A.; Kamp, M.; Radhakrishnan, K.; Hescheler, J.; Schneider, T. Altered seizure susceptibility in mice lacking the Ca(v)2.3 E-type Ca2+ channel. Epilepsia 2006, 47, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Jun, K.; Piedras-Renteria, E.S.; Smith, S.M.; Wheeler, D.B.; Lee, S.B.; Lee, T.G.; Chin, H.; Adams, M.E.; Scheller, R.H.; Tsien, R.W.; et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc. Natl. Acad. Sci. USA 1999, 96, 15245–15250. [Google Scholar] [CrossRef] [PubMed]
- Noebels, J.L.; Sidman, R.L. Inherited epilepsy: Spike-wave and focal motor seizures in the mutant mouse tottering. Science 1979, 204, 1334–1336. [Google Scholar] [CrossRef] [PubMed]
- Bomben, V.C.; Aiba, I.; Qian, J.; Mark, M.D. Isolated P/Q Calcium Channel Deletion in Layer VI Corticothalamic Neurons Generates Absence Epilepsy. J. Neurosci. 2016, 36, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouvenceau, A.; Eunson, L.H.; Spauschus, A.; Ramesh, V.; Zuberi, S.M.; Kullmann, D.M.; Hanna, M.G. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet 2001, 358, 801–807. [Google Scholar] [CrossRef]
- Tsakiridou, E.; Bertollini, L.; de Curtis, M.; Avanzini, G.; Pape, H.C. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J. Neurosci. 1995, 15, 3110–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talley, E.M.; Solorzano, G.; Depaulis, A.; Perez-Reyes, E.; Bayliss, D.A. Low-voltage-activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Brain Res. Mol. Brain Res. 2000, 75, 159–165. [Google Scholar] [CrossRef]
- Kim, D.; Song, I.; Keum, S.; Lee, T.; Jeong, M.J.; Kim, S.S.; McEnery, M.W.; Shin, H.S. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron 2001, 31, 35–45. [Google Scholar] [CrossRef]
- Ernst, W.L.; Zhang, Y.; Yoo, J.W.; Ernst, S.J.; Noebels, J.L. Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J. Neurosci. 2009, 29, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.J.; Pitsch, J.; Sochivko, D.; Opitz, T.; Staniek, M.; Chen, C.C.; Campbell, K.P.; Schoch, S.; Yaari, Y.; Beck, H. Transcriptional upregulation of Cav3.2 mediates epileptogenesis in the pilocarpine model of epilepsy. J. Neurosci. 2008, 28, 13341–13353. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravani, H.; Bladen, C.; Parker, D.B.; Snutch, T.P.; McRory, J.E.; Zamponi, G.W. Effects of Cav3.2 channel mutations linked to idiopathic generalized epilepsy. Ann. Neurol. 2005, 57, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Khosravani, H.; Varela, D.; Bladen, C.; Williams, T.C.; Newman, M.R.; Scheffer, I.E.; Berkovic, S.F.; Mulley, J.C.; Zamponi, G.W. Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann. Neurol. 2007, 62, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, J.; Pan, H.; Zhang, Y.; Wu, H.; Xu, K.; Liu, X.; Jiang, Y.; Bao, X.; Yao, Z.; et al. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann. Neurol. 2003, 54, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Eckle, V.S.; Shcheglovitov, A.; Vitko, I.; Dey, D.; Yap, C.C.; Winckler, B.; Perez-Reyes, E. Mechanisms by which a CACNA1H mutation in epilepsy patients increases seizure susceptibility. J. Physiol. 2014, 592, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, O. Genes and molecular mechanisms involved in the epileptogenesis of idiopathic absence epilepsies. Seizure 2012, 21, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, Y.; Wang, J.; Pan, H.; Wu, H.; Xu, K.; Liu, X.; Jiang, Y.; Shen, Y.; Wu, X. New variants in the CACNA1H gene identified in childhood absence epilepsy. Neurosci. Lett. 2006, 406, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D.; Weiss, J.L. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem. J. 2001, 353, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Baimbridge, K.G.; Celio, M.R.; Rogers, J.H. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992, 15, 303–308. [Google Scholar] [CrossRef]
- Cheung, W.Y. Cyclic 3′,5′-nucleotide phosphodiesterase. Demonstration of an activator. Biochem. Biophys. Res. Commun. 1970, 38, 533–538. [Google Scholar] [CrossRef]
- Kakiuchi, S.; Yamazaki, R. Calcium dependent phosphodiesterase activity and its activating factor (PAF) from brain studies on cyclic 3′,5′-nucleotide phosphodiesterase (3). Biochem. Biophys. Res. Commun. 1970, 41, 1104–1110. [Google Scholar] [CrossRef]
- Kawasaki, H.; Kretsinger, R.H. Calcium-binding proteins. 1: EF-hands. Protein Profile 1994, 1, 343–517. [Google Scholar] [PubMed]
- Yan, H.; Wang, C.; Marx, S.O.; Pitt, G.S. Calmodulin limits pathogenic Na+ channel persistent current. J. Gen. Physiol. 2017, 149, 277–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhuang, F.; Li, H.; Zheng, K.; Hong, Z.; Feng, W.; Zhou, W.; Chen, J. Calmodulin regulates KCNQ2 function in epilepsy. Am. J. Transl. Res. 2016, 8, 5610–5618. [Google Scholar] [PubMed]
- Arif, S.H. A Ca(2+)-binding protein with numerous roles and uses: Parvalbumin in molecular biology and physiology. BioEssays 2009, 31, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Baude, A.; Bleasdale, C.; Dalezios, Y.; Somogyi, P.; Klausberger, T. Immunoreactivity for the GABAA receptor alpha1 subunit, somatostatin and Connexin36 distinguishes axoaxonic, basket, and bistratified interneurons of the rat hippocampus. Cereb. Cortex 2007, 17, 2094–2107. [Google Scholar] [CrossRef] [PubMed]
- Camp, A.J.; Wijesinghe, R. Calretinin: Modulator of neuronal excitability. Int. J. Biochem. Cell Biol. 2009, 41, 2118–2121. [Google Scholar] [CrossRef] [PubMed]
- Schurmans, S.; Schiffmann, S.N.; Gurden, H.; Lemaire, M.; Lipp, H.P.; Schwam, V.; Pochet, R.; Imperato, A.; Bohme, G.A.; Parmentier, M. Impaired long-term potentiation induction in dentate gyrus of calretinin-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 10415–10420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaller, B. Calretinin: From a “simple” Ca(2+) buffer to a multifunctional protein implicated in many biological processes. Front. Neuroanat. 2014, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Morita, Y.; Hironaka, T.; Emson, P.C.; Tohyama, M. Ontological study of calbindin-D28k-like and parvalbumin-like immunoreactivities in rat spinal cord and dorsal root ganglia. J. Comparat. Neurol. 1990, 302, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Maskey, D.; Pradhan, J.; Kim, H.J.; Park, K.S.; Ahn, S.C.; Kim, M.J. Immunohistochemical localization of calbindin D28-k, parvalbumin, and calretinin in the cerebellar cortex of the circling mouse. Neurosci. Lett. 2010, 483, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Rychlik, B.; Chu, C.; Christakos, S. Evidence for calcium-reducing and excito-protective roles for the calcium-binding protein calbindin-D28k in cultured hippocampal neurons. Neuron 1991, 6, 41–51. [Google Scholar] [CrossRef]
- Chard, P.S.; Bleakman, D.; Christakos, S.; Fullmer, C.S.; Miller, R.J. Calcium buffering properties of calbindin D28k and parvalbumin in rat sensory neurones. J. Physiol. 1993, 472, 341–357. [Google Scholar] [CrossRef] [PubMed]
- Iacopino, A.; Christakos, S.; German, D.; Sonsalla, P.K.; Altar, C.A. Calbindin-D28K-containing neurons in animal models of neurodegeneration: Possible protection from excitotoxicity. Brain Res. Mol. Brain Res. 1992, 13, 251–261. [Google Scholar] [CrossRef]
- Bu, J.; Sathyendra, V.; Nagykery, N.; Geula, C. Age-related changes in calbindin-D28k, calretinin, and parvalbumin-immunoreactive neurons in the human cerebral cortex. Exp. Neurol. 2003, 182, 220–231. [Google Scholar] [CrossRef]
- Heizmann, C.W.; Braun, K. Changes in Ca(2+)-binding proteins in human neurodegenerative disorders. Trends Neurosci. 1992, 15, 259–264. [Google Scholar] [CrossRef]
- Ahmadian, S.S.; Rezvanian, A.; Peterson, M.; Weintraub, S.; Bigio, E.H.; Mesulam, M.M.; Geula, C. Loss of calbindin-D28K is associated with the full range of tangle pathology within basal forebrain cholinergic neurons in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3163–3170. [Google Scholar] [CrossRef] [PubMed]
- Nagerl, U.V.; Mody, I.; Jeub, M.; Lie, A.A.; Elger, C.E.; Beck, H. Surviving granule cells of the sclerotic human hippocampus have reduced Ca(2+) influx because of a loss of calbindin-D(28k) in temporal lobe epilepsy. J. Neurosci. 2000, 20, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Yanez, M.; Gil-Longo, J.; Campos-Toimil, M. Calcium binding proteins. Adv. Exp. Med. Biol. 2012, 740, 461–482. [Google Scholar] [PubMed]
- Nairn, A.C.; Picciotto, M.R. Calcium/calmodulin-dependent protein kinases. Semin. Cancer Biol. 1994, 5, 295–303. [Google Scholar] [PubMed]
- Hani, A.J.; Mikati, H.M.; Mikati, M.A. Genetics of pediatric epilepsy. Pediatr. Clin. N. Am. 2015, 62, 703–722. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Benitez-King, G. Variations of rat brain calmodulin content in dark and light phases: Effect of pentylenetetrazol-induced kindling. Neurochem. Res. 1998, 23, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.S.; Silva, A.J.; Abeliovich, A.; Watanabe, Y.; Tonegawa, S.; McNamara, J.O. Limbic epilepsy in transgenic mice carrying a Ca2+/calmodulin-dependent kinase II alpha-subunit mutation. Proc. Natl. Acad. Sci. USA 1995, 92, 6852–6855. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Jones, E.G. Differential and time-dependent changes in gene expression for type II calcium/calmodulin-dependent protein kinase, 67 kDa glutamic acid decarboxylase, and glutamate receptor subunits in tetanus toxin-induced focal epilepsy. J. Neurosci. 1997, 17, 2168–2180. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, Y.; Imoto, K.; Obata, K. A mechanism for the inactivation of Ca2+/calmodulin-dependent protein kinase II during prolonged seizure activity and its consequence after the recovery from seizure activity in rats in vivo. Neuroscience 2006, 140, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, J.M.; Micevych, P.; Popper, P.; Huez, G.; Farber, D.B.; Wasterlain, C.G. Long-lasting decreases of type II calmodulin kinase expression in kindled rat brains. Brain Res. 1992, 584, 257–260. [Google Scholar] [CrossRef]
- Murray, K.D.; Isackson, P.J.; Eskin, T.A.; King, M.A.; Montesinos, S.P.; Abraham, L.A.; Roper, S.N. Altered mRNA expression for brain-derived neurotrophic factor and type II calcium/calmodulin-dependent protein kinase in the hippocampus of patients with intractable temporal lobe epilepsy. J. Comparat. Neurol. 2000, 418, 411–422. [Google Scholar] [CrossRef]
- Lee, M.C.; Ban, S.S.; Woo, Y.J.; Kim, S.U. Calcium/calmodulin kinase II activity of hippocampus in kainate-induced epilepsy. J. Korean Med. Sci. 2001, 16, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Lie, A.A.; Blumcke, I.; Beck, H.; Schramm, J.; Wiestler, O.D.; Elger, C.E. Altered patterns of Ca2+/calmodulin-dependent protein kinase II and calcineurin immunoreactivity in the hippocampus of patients with temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 1998, 57, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.R.; Suzuki, T.; Shimizu, H.; Nishino, H. Amygdala kindling activates the phosphorylation of Ca2+/calmodulin-dependent protein kinase II in rat hippocampus. Neurosci. Lett. 1994, 171, 45–48. [Google Scholar] [CrossRef]
- Wu, K.; Wasterlain, C.; Sachs, L.; Siekevitz, P. Effect of septal kindling on glutamate binding and calcium/calmodulin-dependent phosphorylation in a postsynaptic density fraction isolated from rat cerebral cortex. Proc. Natl. Acad. Sci. USA 1990, 87, 5298–5302. [Google Scholar] [CrossRef] [PubMed]
- Wasterlain, C.G.; Farber, D.B. Kindling alters the calcium/calmodulin-dependent phosphorylation of synaptic plasma membrane proteins in rat hippocampus. Proc. Natl. Acad. Sci. USA 1984, 81, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Rosenberg, H.C. Brief seizure activity alters Ca2+/calmodulin dependent protein kinase II dephosphorylation and subcellular distribution in rat brain for several hours. Neurosci. Lett. 2004, 357, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, J.M.; Farber, D.B.; Micevych, P.E.; Lasher, R.; Wasterlain, C.G. Kindling induced changes in calmodulin kinase II immunoreactivity. Brain Res. 1990, 524, 49–53. [Google Scholar] [CrossRef]
- Savina, T.A.; Shchipakina, T.G.; Godukhin, O.V. Effect of seizure activity on subunit composition of Ca2+/calmodulin-dependent protein kinase II in hippocampus of Krushinskii-Molodkina rats. Rossiiskii Fiziologicheskii Zhurnal Imeni I.M. Sechenova 2011, 97, 590–600. [Google Scholar] [PubMed]
- Matsu-ura, T.; Nakadai, T.; Oda, Y.; Nagasu, T.; Mikoshiba, K.; Tamura, T.A. Seizure-mediated accumulation of the beta subunit of Ca2+/calmodulin-dependent protein kinase II in nuclei of mouse brain cells. Neurosci. Lett. 2002, 322, 149–152. [Google Scholar] [CrossRef]
- Gary, D.S.; Sooy, K.; Chan, S.L.; Christakos, S.; Mattson, M.P. Concentration- and cell type-specific effects of calbindin D28k on vulnerability of hippocampal neurons to seizure-induced injury. Brain Res. Mol. Brain Res. 2000, 75, 89–95. [Google Scholar] [CrossRef]
- Montpied, P.; Winsky, L.; Dailey, J.W.; Jobe, P.C.; Jacobowitz, D.M. Alteration in levels of expression of brain calbindin D-28k and calretinin mRNA in genetically epilepsy-prone rats. Epilepsia 1995, 36, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Valencia, I.; Legido, A.; Yelin, K.; Khurana, D.; Kothare, S.V.; Katsetos, C.D. Anomalous inhibitory circuits in cortical tubers of human tuberous sclerosis complex associated with refractory epilepsy: Aberrant expression of parvalbumin and calbindin-D28k in dysplastic cortex. J. Child Neurol. 2006, 21, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.K.; Lee, H.Y.; Seong, N.S.; Chung, H.G.; Kim, J.H.; Lee, H.J.; Kim, J.D.; Kang, T.C.; Won, M.H. Changes of calbindin D-28k immunoreactivity in the hippocampus after adrenalectomy in the seizure sensitive gerbil. Anat. Histol. Embryol. 2004, 33, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Magloczky, Z.; Halasz, P.; Vajda, J.; Czirjak, S.; Freund, T.F. Loss of Calbindin-D28K immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience 1997, 76, 377–385. [Google Scholar] [CrossRef]
- Karadi, K.; Janszky, J.; Gyimesi, C.; Horvath, Z.; Lucza, T.; Doczi, T.; Kallai, J.; Abraham, H. Correlation between calbindin expression in granule cells of the resected hippocampal dentate gyrus and verbal memory in temporal lobe epilepsy. Epilepsy Behav. 2012, 25, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Thom, M.; Holton, J.L.; D’Arrigo, C.; Griffin, B.; Beckett, A.; Sisodiya, S.; Alexiou, D.; Sander, J.W. Microdysgenesis with abnormal cortical myelinated fibres in temporal lobe epilepsy: A histopathological study with calbindin D-28-K immunohistochemistry. Neuropathol. Appl. Neurobiol. 2000, 26, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Bouilleret, V.; Schwaller, B.; Schurmans, S.; Celio, M.R.; Fritschy, J.M. Neurodegenerative and morphogenic changes in a mouse model of temporal lobe epilepsy do not depend on the expression of the calcium-binding proteins parvalbumin, calbindin, or calretinin. Neuroscience 2000, 97, 47–58. [Google Scholar] [CrossRef]
- Wittner, L.; Eross, L.; Szabo, Z.; Toth, S.; Czirjak, S.; Halasz, P.; Freund, T.F.; Magloczky, Z.S. Synaptic reorganization of calbindin-positive neurons in the human hippocampal CA1 region in temporal lobe epilepsy. Neuroscience 2002, 115, 961–978. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Cheron, G.; Lohof, A.; d’Alcantara, P.; Meyer, M.; Parmentier, M.; Schurmans, S. Impaired motor coordination and Purkinje cell excitability in mice lacking calretinin. Proc. Natl. Acad. Sci. USA 1999, 96, 5257–5262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, D.; Roussel, C.; Nieus, T.; Cheron, G.; Servais, L.; D’Angelo, E.; Schiffmann, S.N. Role of calcium binding proteins in the control of cerebellar granule cell neuronal excitability: Experimental and modeling studies. Prog. Brain Res. 2005, 148, 321–328. [Google Scholar] [PubMed]
- Miettinen, R.; Gulyas, A.I.; Baimbridge, K.G.; Jacobowitz, D.M.; Freund, T.F. Calretinin is present in non-pyramidal cells of the rat hippocampus—II. Co-existence with other calcium binding proteins and GABA. Neuroscience 1992, 48, 29–43. [Google Scholar] [CrossRef]
- Liu, Y.; Fujise, N.; Kosaka, T. Distribution of calretinin immunoreactivity in the mouse dentate gyrus. I. General description. Exp. Brain Res. 1996, 108, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Gulyas, A.I.; Hajos, N.; Freund, T.F. Interneurons containing calretinin are specialized to control other interneurons in the rat hippocampus. J. Neurosci. 1996, 16, 3397–3411. [Google Scholar] [CrossRef] [PubMed]
- Hajos, N.; Acsady, L.; Freund, T.F. Target selectivity and neurochemical characteristics of VIP-immunoreactive interneurons in the rat dentate gyrus. Eur. J. Neurosci. 1996, 8, 1415–1431. [Google Scholar] [CrossRef] [PubMed]
- Urban, Z.; Magloczky, Z.; Freund, T.F. Calretinin-containing interneurons innervate both principal cells and interneurons in the CA1 region of the human hippocampus. Acta Biol. Hung. 2002, 53, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Magloczky, Z.; Wittner, L.; Borhegyi, Z.; Halasz, P.; Vajda, J.; Czirjak, S.; Freund, T.F. Changes in the distribution and connectivity of interneurons in the epileptic human dentate gyrus. Neuroscience 2000, 96, 7–25. [Google Scholar] [CrossRef]
- Toth, K.; Eross, L.; Vajda, J.; Halasz, P.; Freund, T.F.; Magloczky, Z. Loss and reorganization of calretinin-containing interneurons in the epileptic human hippocampus. Brain 2010, 133, 2763–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumcke, I.; Beck, H.; Suter, B.; Hoffmann, D.; Fodisch, H.J.; Wolf, H.K.; Schramm, J.; Elger, C.E.; Wiestler, O.D. An increase of hippocampal calretinin-immunoreactive neurons correlates with early febrile seizures in temporal lobe epilepsy. Acta Neuropathol. 1999, 97, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Blumcke, I.; Beck, H.; Nitsch, R.; Eickhoff, C.; Scheffler, B.; Celio, M.R.; Schramm, J.; Elger, C.E.; Wolf, H.K.; Wiestler, O.D. Preservation of calretinin-immunoreactive neurons in the hippocampus of epilepsy patients with Ammon’s horn sclerosis. J. Neuropathol. Exp. Neurol. 1996, 55, 329–341. [Google Scholar] [PubMed]
- Toth, K.; Magloczky, Z. The vulnerability of calretinin-containing hippocampal interneurons to temporal lobe epilepsy. Front. Neuroanat. 2014, 8, 100. [Google Scholar] [PubMed]
- Barinka, F.; Druga, R.; Marusic, P.; Krsek, P.; Zamecnik, J. Calretinin immunoreactivity in focal cortical dysplasias and in non-malformed epileptic cortex. Epilepsy Res. 2010, 88, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Magloczky, Z.; Freund, T.F. Selective neuronal death in the contralateral hippocampus following unilateral kainate injections into the CA3 subfield. Neuroscience 1993, 56, 317–335. [Google Scholar] [CrossRef]
- Magloczky, Z.; Freund, T.F. Delayed cell death in the contralateral hippocampus following kainate injection into the CA3 subfield. Neuroscience 1995, 66, 847–860. [Google Scholar] [CrossRef]
- Bouilleret, V.; Loup, F.; Kiener, T.; Marescaux, C.; Fritschy, J.M. Early loss of interneurons and delayed subunit-specific changes in GABA(A)-receptor expression in a mouse model of mesial temporal lobe epilepsy. Hippocampus 2000, 10, 305–324. [Google Scholar] [CrossRef]
- Andre, V.; Marescaux, C.; Nehlig, A.; Fritschy, J.M. Alterations of hippocampal GAbaergic system contribute to development of spontaneous recurrent seizures in the rat lithium-pilocarpine model of temporal lobe epilepsy. Hippocampus 2001, 11, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, M.I.; Blasco-Ibanez, J.M.; Crespo, C.; Marques-Mari, A.I.; Martinez-Guijarro, F.J. Calretinin/PSA-NCAM immunoreactive granule cells after hippocampal damage produced by kainic acid and DEDTC treatment in mouse. Brain Res. 2003, 966, 206–217. [Google Scholar] [CrossRef]
- Van Vliet, E.A.; Aronica, E.; Tolner, E.A.; Lopes da Silva, F.H.; Gorter, J.A. Progression of temporal lobe epilepsy in the rat is associated with immunocytochemical changes in inhibitory interneurons in specific regions of the hippocampal formation. Exp. Neurol. 2004, 187, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Cobos, I.; Calcagnotto, M.E.; Vilaythong, A.J.; Thwin, M.T.; Noebels, J.L.; Baraban, S.C.; Rubenstein, J.L. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 2005, 8, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Tang, F. Survival of calbindin, calretinin and parvalbumin positive neurons in mouse hippocampal CA area at chronic stage of pilocarpine-induced epilepsy. Zhong Nan Da Xue Xue Bao 2013, 38, 437–442. [Google Scholar] [PubMed]
- Tang, F.R.; Chia, S.C.; Jiang, F.L.; Ma, D.L.; Chen, P.M.; Tang, Y.C. Calcium binding protein containing neurons in the gliotic mouse hippocampus with special reference to their afferents from the medial septum and the entorhinal cortex. Neuroscience 2006, 140, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Huusko, N.; Romer, C.; Ndode-Ekane, X.E.; Lukasiuk, K.; Pitkanen, A. Loss of hippocampal interneurons and epileptogenesis: A comparison of two animal models of acquired epilepsy. Brain Struct. Funct. 2015, 220, 153–191. [Google Scholar] [CrossRef] [PubMed]
- Drexel, M.; Preidt, A.P.; Kirchmair, E.; Sperk, G. Parvalbumin interneurons and calretinin fibers arising from the thalamic nucleus reuniens degenerate in the subiculum after kainic acid-induced seizures. Neuroscience 2011, 189, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Thom, M.; Sisodiya, S.M.; Beckett, A.; Martinian, L.; Lin, W.R.; Harkness, W.; Mitchell, T.N.; Craig, J.; Duncan, J.; Scaravilli, F. Cytoarchitectural abnormalities in hippocampal sclerosis. J. Neuropathol. Exp. Neurol. 2002, 61, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Volz, F.; Bock, H.H.; Gierthmuehlen, M.; Zentner, J.; Haas, C.A.; Freiman, T.M. Stereologic estimation of hippocampal GluR2/3- and calretinin-immunoreactive hilar neurons (presumptive mossy cells) in two mouse models of temporal lobe epilepsy. Epilepsia 2011, 52, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, M.; Losi, G.; Chiavegato, A.; Zonta, M.; Carmignoto, G. Fast spiking interneuron control of seizure propagation in a cortical slice model of focal epilepsy. J. Physiol. 2013, 591, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, C.; Kohler, J.; Bartos, M. Persistent discharges in dentate gyrus perisoma-inhibiting interneurons require hyperpolarization-activated cyclic nucleotide-gated channel activation. J. Neurosci. 2015, 35, 4131–4139. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, B.; Tetko, I.V.; Tandon, P.; Silveira, D.C.; Vreugdenhil, M.; Henzi, T.; Potier, M.C.; Celio, M.R.; Villa, A.E. Parvalbumin deficiency affects network properties resulting in increased susceptibility to epileptic seizures. Mol. Cell. Neurosci. 2004, 25, 650–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloviter, R.S. Calcium-binding protein (calbindin-D28k) and parvalbumin immunocytochemistry: Localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J. Comparat. Neurol. 1989, 280, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Sloviter, R.S.; Sollas, A.L.; Barbaro, N.M.; Laxer, K.D. Calcium-binding protein (calbindin-D28K) and parvalbumin immunocytochemistry in the normal and epileptic human hippocampus. J. Comparat. Neurol. 1991, 308, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Scotti, A.L.; Nitsch, C. The perforant path in the seizure sensitive gerbil contains the Ca(2+)-binding protein parvalbumin. Exp. Brain Res. 1991, 85, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Scotti, A.L.; Frank, C.; Sagratella, S.; Scotti de Carolis, A.; Nitsch, C. Absence of calcium-induced LTP-like response in the dentate area of seizure-prone gerbils and its relation to parvalbumin in the entorhinal perforant path synapse of this species. Brain Res. Bull. 1993, 31, 501–507. [Google Scholar] [CrossRef]
- Houser, C.R.; Swartz, B.E.; Walsh, G.O.; Delgado-Escueta, A.V. Granule cell disorganization in the dentate gyrus: Possible alterations of neuronal migration in human temporal lobe epilepsy. Epilepsy Res. Suppl. 1992, 9, 41–48, discussion 48–49. [Google Scholar] [PubMed]
- Curia, G.; Lucchi, C.; Vinet, J.; Gualtieri, F.; Marinelli, C.; Torsello, A.; Costantino, L.; Biagini, G. Pathophysiogenesis of mesial temporal lobe epilepsy: Is prevention of damage antiepileptogenic? Curr. Med. Chem. 2014, 21, 663–688. [Google Scholar] [CrossRef] [PubMed]
- Soukupova, M.; Binaschi, A.; Falcicchia, C.; Zucchini, S.; Roncon, P.; Palma, E.; Magri, E.; Grandi, E.; Simonato, M. Impairment of GABA release in the hippocampus at the time of the first spontaneous seizure in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 2014, 257, 39–49. [Google Scholar] [CrossRef] [PubMed]
- De Guzman, P.; Inaba, Y.; Biagini, G.; Baldelli, E.; Mollinari, C.; Merlo, D.; Avoli, M. Subiculum network excitability is increased in a rodent model of temporal lobe epilepsy. Hippocampus 2006, 16, 843–860. [Google Scholar] [CrossRef] [PubMed]
- Knopp, A.; Frahm, C.; Fidzinski, P.; Witte, O.W.; Behr, J. Loss of GABAergic neurons in the subiculum and its functional implications in temporal lobe epilepsy. Brain 2008, 131, 1516–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Guzman, P.; Inaba, Y.; Baldelli, E.; de Curtis, M.; Biagini, G.; Avoli, M. Network hyperexcitability within the deep layers of the pilocarpine-treated rat entorhinal cortex. J. Physiol. 2008, 586, 1867–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benini, R.; Longo, D.; Biagini, G.; Avoli, M. Perirhinal cortex hyperexcitability in pilocarpine-treated epileptic rats. Hippocampus 2011, 21, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Vizi, S.; Bagosi, A.; Krisztin-Peva, B.; Gulya, K.; Mihaly, A. Repeated 4-aminopyridine seizures reduce parvalbumin content in the medial mammillary nucleus of the rat brain. Brain Res. Mol. Brain Res. 2004, 131, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortel, A.; Levesque, M.; Biagini, G.; Gotman, J.; Avoli, M. Convulsive status epilepticus duration as determinant for epileptogenesis and interictal discharge generation in the rat limbic system. Neurobiol. Dis. 2010, 40, 478–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biagini, G.; D’Antuono, M.; Benini, R.; de Guzman, P.; Longo, D.; Avoli, M. Perirhinal cortex and temporal lobe epilepsy. Front. Cell. Neurosci. 2013, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Represa, A. Brief seizure episodes induce long-term potentiation and mossy fibre sprouting in the hippocampus. Trends Neurosci. 1990, 13, 312–318. [Google Scholar] [CrossRef]
- Andrioli, A.; Alonso-Nanclares, L.; Arellano, J.I.; DeFelipe, J. Quantitative analysis of parvalbumin-immunoreactive cells in the human epileptic hippocampus. Neuroscience 2007, 149, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Wittner, L.; Magloczky, Z.; Borhegyi, Z.; Halasz, P.; Toth, S.; Eross, L.; Szabo, Z.; Freund, T.F. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 2001, 108, 587–600. [Google Scholar] [CrossRef]
- Arellano, J.I.; Munoz, A.; Ballesteros-Yanez, I.; Sola, R.G.; DeFelipe, J. Histopathology and reorganization of chandelier cells in the human epileptic sclerotic hippocampus. Brain 2004, 127, 45–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.Q.; Armstrong, D.L.; Hamilton, W.J.; Grossman, R.G. Disproportionate loss of CA4 parvalbumin-immunoreactive interneurons in patients with Ammon’s horn sclerosis. J. Neuropathol. Exp. Neurol. 1997, 56, 988–998. [Google Scholar] [CrossRef]
- Gualtieri, F.; Marinelli, C.; Longo, D.; Pugnaghi, M.; Nichelli, P.F.; Meletti, S.; Biagini, G. Hypoxia markers are expressed in interneurons exposed to recurrent seizures. Neuromol. Med. 2013, 15, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kang, T.C. p47Phox/CDK5/DRP1-Mediated Mitochondrial Fission Evokes PV Cell Degeneration in the Rat Dentate Gyrus Following Status Epilepticus. Front. Cell. Neurosci. 2017, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Adotevi, N.K.; Leitch, B. Synaptic Changes in AMPA Receptor Subunit Expression in Cortical Parvalbumin Interneurons in the Stargazer Model of Absence Epilepsy. Front. Cell. Neurosci. 2017, 10, 434. [Google Scholar] [CrossRef] [PubMed]
- Neves, G.; Shah, M.M.; Liodis, P.; Achimastou, A.; Denaxa, M.; Roalfe, G.; Sesay, A.; Walker, M.C.; Pachnis, V. The LIM homeodomain protein Lhx6 regulates maturation of interneurons and network excitability in the mammalian cortex. Cerebral Cortex 2013, 23, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Batista-Brito, R.; Rossignol, E.; Hjerling-Leffler, J.; Denaxa, M.; Wegner, M.; Lefebvre, V.; Pachnis, V.; Fishell, G. The cell-intrinsic requirement of Sox6 for cortical interneuron development. Neuron 2009, 63, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dye, C.A.; Sohal, V.; Long, J.E.; Estrada, R.C.; Roztocil, T.; Lufkin, T.; Deisseroth, K.; Baraban, S.C.; Rubenstein, J.L. Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J. Neurosci. 2010, 30, 5334–5345. [Google Scholar] [CrossRef] [PubMed]
- Gant, J.C.; Thibault, O.; Blalock, E.M.; Yang, J.; Bachstetter, A.; Kotick, J.; Schauwecker, P.E.; Hauser, K.F.; Smith, G.M.; Mervis, R.; et al. Decreased number of interneurons and increased seizures in neuropilin 2 deficient mice: Implications for autism and epilepsy. Epilepsia 2009, 50, 629–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canty, A.J.; Dietze, J.; Harvey, M.; Enomoto, H.; Milbrandt, J.; Ibanez, C.F. Regionalized loss of parvalbumin interneurons in the cerebral cortex of mice with deficits in GFRalpha1 signaling. J. Neurosci. 2009, 29, 10695–10705. [Google Scholar] [CrossRef] [PubMed]
- Yutsudo, N.; Kitagawa, H. Involvement of chondroitin 6-sulfation in temporal lobe epilepsy. Exp. Neurol. 2015, 274, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; DeCan, E.; Stoll, K.; Marceau, E.; Deisseroth, K.; Lawrence, J.J. Muscarinic excitation of parvalbumin-positive interneurons contributes to the severity of pilocarpine-induced seizures. Epilepsia 2015, 56, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Ball, J.; Stoll, K.E.; Satpute, V.C.; Mitchell, S.M.; Pauli, J.L.; Holloway, B.B.; Johnston, A.D.; Nathanson, N.M.; Deisseroth, K.; et al. Direct excitation of parvalbumin-positive interneurons by M1 muscarinic acetylcholine receptors: Roles in cellular excitability, inhibitory transmission and cognition. J. Physiol. 2014, 592, 3463–3494. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, E.; Kruglikov, I.; van den Maagdenberg, A.M.; Rudy, B.; Fishell, G. CaV 2.1 ablation in cortical interneurons selectively impairs fast-spiking basket cells and causes generalized seizures. Ann. Neurol. 2013, 74, 209–222. [Google Scholar] [PubMed]
- Liu, X.; Yang, P.S.; Yang, W.; Yue, D.T. Enzyme-inhibitor-like tuning of Ca(2+) channel connectivity with calmodulin. Nature 2010, 463, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.X.; Erickson, M.G.; Yue, D.T. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science 2004, 304, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Westenbroek, R.E.; Yu, F.H.; Clark, J.P., 3rd; Marshall, M.R.; Scheuer, T.; Catterall, W.A. Deletion of the distal C terminus of CaV1.2 channels leads to loss of beta-adrenergic regulation and heart failure in vivo. J. Biol. Chem. 2011, 286, 12617–12626. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, C.D.; Soong, T.W.; Alseikhan, B.A.; Alvania, R.S.; Yue, D.T. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 2001, 411, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.J.; Rungta, R.L.; Garcia, E.; van den Maagdenberg, A.M.; MacVicar, B.A.; Snutch, T.P. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 18694–18699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaitsev, A.V.; Povysheva, N.V.; Lewis, D.A.; Krimer, L.S. P/Q-type, but not N-type, calcium channels mediate GABA release from fast-spiking interneurons to pyramidal cells in rat prefrontal cortex. J. Neurophysiol. 2007, 97, 3567–3573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gupta, A.; Toledo-Rodriguez, M.; Wu, C.Z.; Markram, H. Anatomical, physiological, molecular and circuit properties of nest basket cells in the developing somatosensory cortex. Cerebral Cortex 2002, 12, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, E. Genetics and function of neocortical GABAergic interneurons in neurodevelopmental disorders. Neural Plast. 2011, 2011, 649325. [Google Scholar] [CrossRef] [PubMed]
- Rudy, B.; Fishell, G.; Lee, S.; Hjerling-Leffler, J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 2011, 71, 45–61. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation to show the role of calcium-binding proteins (CBPs) in the pathological process of epilepsy. The green pathways show the inhibitory role of CBPs, which may down-regulate neuronal excitability and prevent the pathological process of epilepsy; the red pathways show the excitatory role of CBPs, which likely increase neuronal excitability and promote the pathological process of epilepsy. Calmodulin (CaM) can exert inhibitory functions through three pathways, including 1. binding to intracellular C-terminal domains (CTDs) of voltage-gated Na+ channel (Nav) and limit persistent Na+ current and accelerate inactivation across the Nav family (inhibit Na+ influx); 2. activation of the KCNQ2 channel (promote K+ efflux); 3. down-regulating intracellular Ca2+ through a buffering function, CaMK II media, and regulating VDCCs via Ca2+-dependent inactivation (CDI). calbindin (CB), parvulbumin (PV), and calretinin (CR) all can reduce intracellular Ca2+ through their buffering functions. In addition, PV and CR offer an inhibitory effect through regulating Gamma-amino butyric acid (GABA) release and synchronizing inhibitory interneurons respectively. CaM and CB can increase neuronal excitability by elevating intracellular Ca2+ via Ca2+-dependent facilitation (CDF) and the CDI of VDCCs respectively. CR can exert an excitatory effect through both CDF and CDI of VDCCs. In addition, CR may facilitate the pathological process of epilepsy through promoting neuronal axonal sprouting and impaired circuitry formation in the epileptic focus. (+) and (−) indicate promoting and inhibiting effects, respectively. (↑) and (↓) in the textbox indicate increase and decrease respectively. Arrows outside the textbox indicate the pathways.
Figure 1.
Schematic representation to show the role of calcium-binding proteins (CBPs) in the pathological process of epilepsy. The green pathways show the inhibitory role of CBPs, which may down-regulate neuronal excitability and prevent the pathological process of epilepsy; the red pathways show the excitatory role of CBPs, which likely increase neuronal excitability and promote the pathological process of epilepsy. Calmodulin (CaM) can exert inhibitory functions through three pathways, including 1. binding to intracellular C-terminal domains (CTDs) of voltage-gated Na+ channel (Nav) and limit persistent Na+ current and accelerate inactivation across the Nav family (inhibit Na+ influx); 2. activation of the KCNQ2 channel (promote K+ efflux); 3. down-regulating intracellular Ca2+ through a buffering function, CaMK II media, and regulating VDCCs via Ca2+-dependent inactivation (CDI). calbindin (CB), parvulbumin (PV), and calretinin (CR) all can reduce intracellular Ca2+ through their buffering functions. In addition, PV and CR offer an inhibitory effect through regulating Gamma-amino butyric acid (GABA) release and synchronizing inhibitory interneurons respectively. CaM and CB can increase neuronal excitability by elevating intracellular Ca2+ via Ca2+-dependent facilitation (CDF) and the CDI of VDCCs respectively. CR can exert an excitatory effect through both CDF and CDI of VDCCs. In addition, CR may facilitate the pathological process of epilepsy through promoting neuronal axonal sprouting and impaired circuitry formation in the epileptic focus. (+) and (−) indicate promoting and inhibiting effects, respectively. (↑) and (↓) in the textbox indicate increase and decrease respectively. Arrows outside the textbox indicate the pathways.


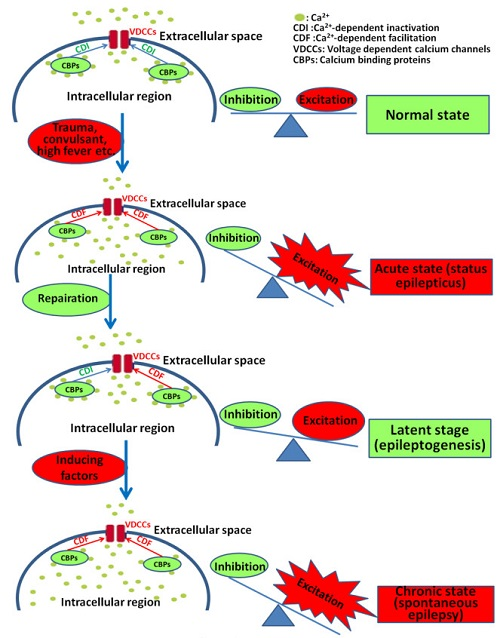{kind=link}
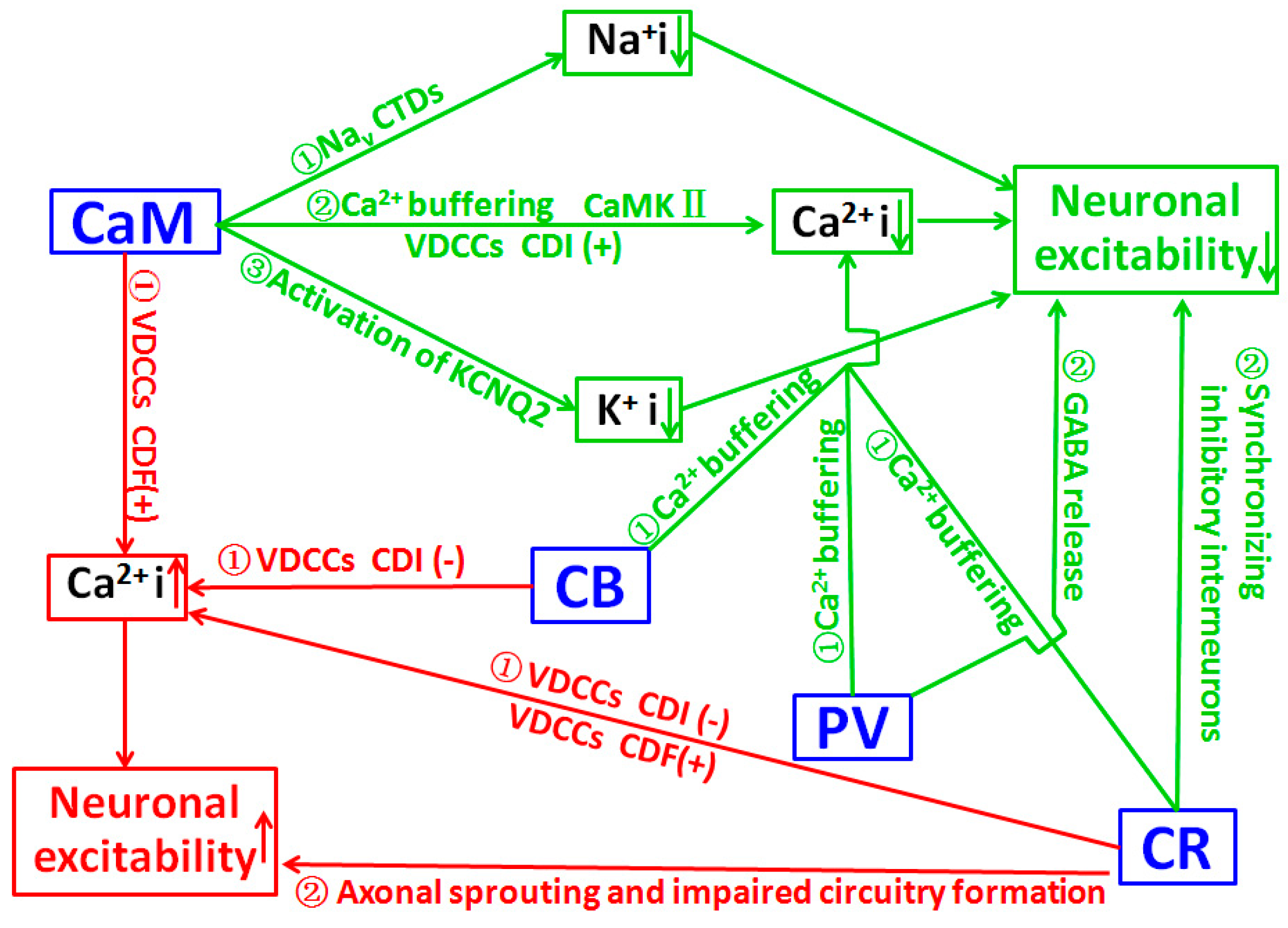{kind=link}
Table 1.
The distribution, physiological function, and possible roles of voltage-dependent calcium channels (VDCCs) in the pathological process of epilepsy.
Table 1.
The distribution, physiological function, and possible roles of voltage-dependent calcium channels (VDCCs) in the pathological process of epilepsy.
| VDCCs | Physiological Function | Possible Roles in Pathological Process of Epilepsy |
|---|---|---|
| L-Type | Shape neuronal firing and activate Ca2+-dependent pathways involved in the control of gene expression, and support neuronal plasticity [47] | Control neuronal excitability and likely provide the gene basis of epileptogenesis through regulation of gene expression |
| P/Q-Type | Regulate neurotransmitter release [49,50] | Inhibit epileptogenesis based on the fact that its null mutation can cause the occurrence of absence epilepsy |
| N-Type | Regulate neurotransmitter release [51,52] | Inhibit neuronal excitability through fast redistribution in the subcellullar organs of neurons |
| R-Type | Regulate long-term potentiation (LTP) and neurotransmitter release [53,54] | Trigger epileptiform activity in neuronal populations and promote epileptogenesis |
| T-Type | Regulate rhythmic firing of neurons [45] | Control burst firing of action potentials of neurons, and the plasticity of neurons induced by epiletogenic factors and promote the formation of epileptogenic focus |
Table 2.
The regional, cellular, and subcellular distribution of VDCCs in the nervous system.
| VDCCs | Regional and Cellular Distribution | Subcellular Distribution |
|---|---|---|
| L-Type | Cav1.1 and Cav1.4 are expressed in a limited subset of neurons in the brain [55]; 90% of the L-type VDCCs in the brain are Cav1.2, and only 10% are Cav1.3 [55,56] | Located postsynaptically, predominantly in the soma, dendritic spines, and shafts of dendrites [16,19,56,57,58,59,60,61,62] |
| P/Q-Type | Expressed in hippocampal principal cells of the human [16] and rat [19,56,60,61,62,63], and in both hippocampal principal cells and interneurons of mice [35] | Widely expressed at the presynaptic terminals [49,50] |
| N-Type | Expressed in the dorsal cortex and the hippocampal formation of rats [64,65] and in both the neuron and astrocyte of the mouse brain [66] | Localized in the dendrites, presynaptic membrane, and nucleus |
| R-Type | Expressed in the most basal ganglia regions, the thalamus, hypothalamus, amygdala, hippocampus, and cortex [67,68,69] | Localized in the presynaptic membrane [70] |
| T-Type | Present in neurons in both the central and peripheral nerve system | Localized in both soma and dendrites [71] |
Table 3.
The alterations of L-type VDCCs in the animal epileptic models and patients with temporal lobe epilepsy (TLE).
Table 3.
The alterations of L-type VDCCs in the animal epileptic models and patients with temporal lobe epilepsy (TLE).
| L-Type VDCCs | Epileptic Animal Model | Patients with TLE |
|---|---|---|
| Cav1.2 | Increased in the somata of the pyramidal cells and granule cells in the KA rat model [62]; in the granule cells of the mouse pilocarpine model [35] | Increased in the astrocytes in Ammon’s horn (or hippocampal) sclerosis (AHS) specimens [16] |
| Decreased in the neuropil of the CA3 stratum pyramidale and the part of CA1 regions in the KA rat model [62], in the hilar neurons of the mouse pilocarpine model [35]. | Decreased in the dentate gyrus granule cells and in the residual CA3 pyramidal neurons [16] | |
| No changes in the hippocampal subareas in the kindling model [19] | ||
| Cav1.3 | Increased in the hippocampal subareas in the kindling model [19], and in the granule cells of the dentate gyrus in the mouse pilocarpine model [35] | Increased in the neuropil of molecular layer of the dentate gyrus [16] |
| Decreased in CA3 and the hilus of the dentate gyrus of the KA rat model [62]; in the hippocampal neurons of the kindling model [76] |
Table 4.
The alterations of N- and R-type VDCCs in the pathological process of epilepsy and the effect of gene knockout.
Table 4.
The alterations of N- and R-type VDCCs in the pathological process of epilepsy and the effect of gene knockout.
| VDCCs | Epileptic Animal Model | Patients with TLE | Gene Knockout Outcomes |
|---|---|---|---|
| Cav2.2 | Increased in the dendritic fields of CA1 and CA3 areas of hippocampus in the rat kindling model [77,78], in the dentate granular cells of the animal KA model [79], and in the stratum lucidum of CA3 in the mouse pilocarpine model [66]. | Increased in the molecular layer [16] and granular cells of the dentate gyrus [81]; | Knockout mice displayed hyperactivity and vigilance state [82] |
| Decreased in the stratum lucidum of CA3 of the KA rat model [62], and in the stratum pyramidale of CA3 in the mouse pilocarpine model [66]. | |||
| No changes in CA1 neurons in the mouse pilocarpine model [80] | |||
| Cav2.3 | Increased in the inferior colliculus neurons of seizure-naïve rats [83]; | Increased in the molecular layer of the dentate gyrus [16]; | Knockout mice show hippocampal seizure resistance and reduced neuronal excitotoxicity [86,87,88] |
| Decreased in both cerebellum and medulla of genetic absence epilepsy rats from Strasbourg (GAERS) [84,85]; |
Table 5.
Alterations of P/Q- and T-type VDCCs in the pathological process of epilepsy, and gene interference, mutation, and knockout outcomes.
Table 5.
Alterations of P/Q- and T-type VDCCs in the pathological process of epilepsy, and gene interference, mutation, and knockout outcomes.
| VDCCs | Alterations in the Pathological Process of Epilepsy | Gene Interference, Mutation and Knockout Outcomes |
|---|---|---|
| Cav2.1 | Increased in the molecular layer of the dentate gyrus of patient with TLE [16]; different hippocampal subareas of kindling model [19] | Gene null mice exhibit ataxia and absence seizures [89]; point mutation (including tottering (tg), rocker (rkr), tottering leaner (tgla), and rolling Nagoya (tgrol)) mice exhibit reminiscent of tonic-clonic seizure, as well as electrographic and behavioral hallmarks of absence epilepsy [90]; isolated deletion in layer VI corticothalamic neurons generated absence epilepsy [91]; loss of function result in absence epilepsy [92] |
| Decreased in CA3 and the hilus of dentate gyrus of the rat KA model [62]; hippocampus and neocortex of KA rat model (6 h, 24 h and 7 days after KA treatment) [61] | ||
| No change in the cerebellum of the rat KA model (6 h, 24 h and 7 days after KA treatment) [61] | ||
| Cav3.1 | Increased in the reticular thalamic neurons of GAERS rats [93] and in neurons of the ventral posterior thalamic relay nuclei of adult GAERS [94] | Knockout mice did not show the burst firing of action potentials and were resistant to baclofen-induced seizures [95]; overexpression resulted in absence epilepsy [96] |
| Cav3.2 | Increased in both messenger RNA (mRNA) and protein level in the hippocampal CA1 area in the mouse pilocarpine model [97] | Mutation has been associated with seizure disorders, autism, and hyperaldosteronism [98]; single nucleotide mutation has been reported in patients with childhood absence epilepsy and other types of idiopathic generalized epilepsies [99,100,101,102,103,104] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Xu, J.-H.; Tang, F.-R. Voltage-Dependent Calcium Channels, Calcium Binding Proteins, and Their Interaction in the Pathological Process of Epilepsy. Int. J. Mol. Sci. 2018, 19, 2735. https://doi.org/10.3390/ijms19092735
AMA Style
Xu J-H, Tang F-R. Voltage-Dependent Calcium Channels, Calcium Binding Proteins, and Their Interaction in the Pathological Process of Epilepsy. International Journal of Molecular Sciences. 2018; 19(9):2735. https://doi.org/10.3390/ijms19092735
Chicago/Turabian StyleXu, Jie-Hua, and Feng-Ru Tang. 2018. "Voltage-Dependent Calcium Channels, Calcium Binding Proteins, and Their Interaction in the Pathological Process of Epilepsy" International Journal of Molecular Sciences 19, no. 9: 2735. https://doi.org/10.3390/ijms19092735
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.