Theoretical Study on Zearalenol Compounds Binding with Wild Type Zearalenone Hydrolase and V153H Mutant

 and
and
Abstract
:
1. Introduction
2. Results and Discursion
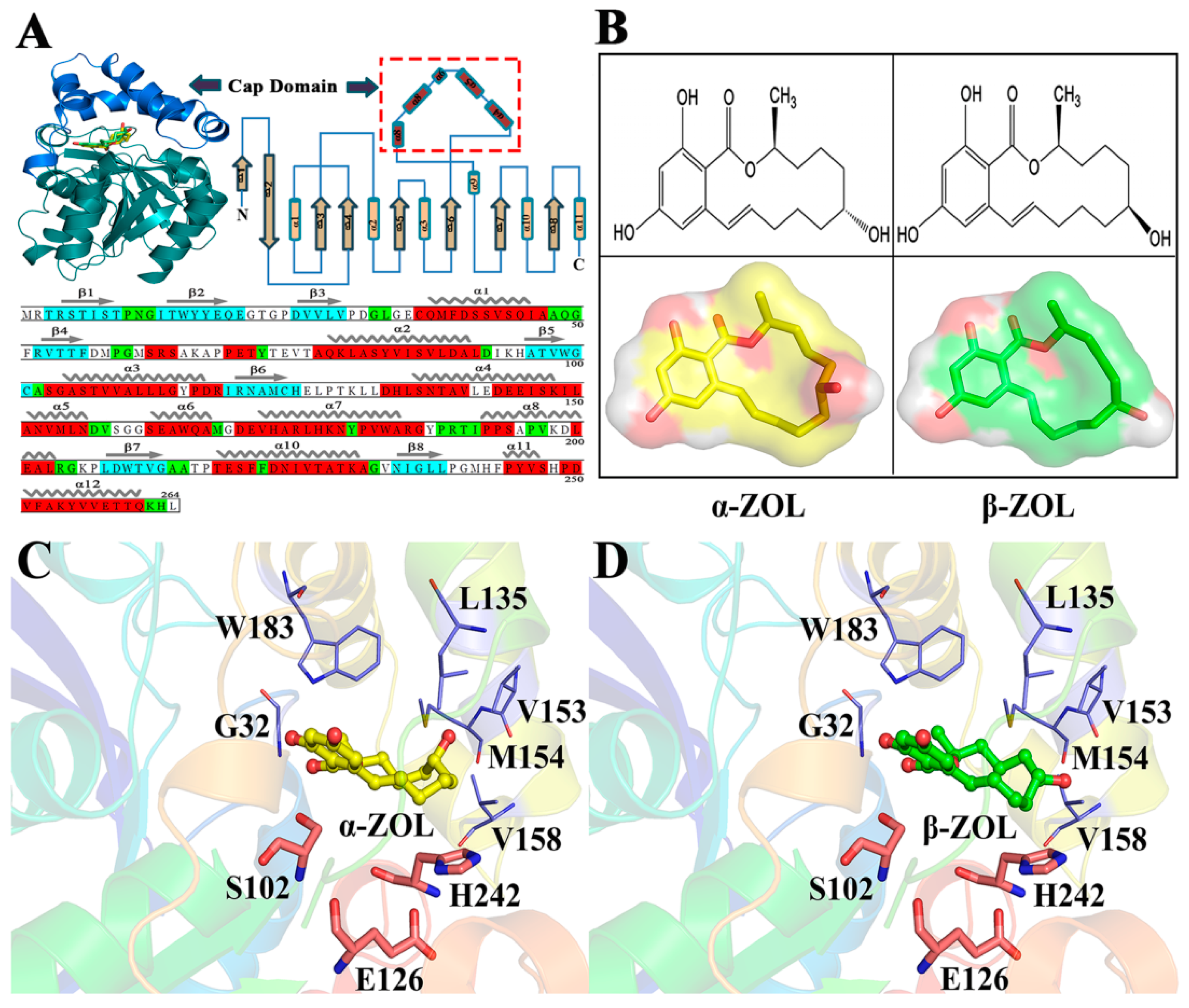
2.1. Structural Stability and Dynamics Properties of Four Complexes
2.2. Protein Network Analysis of the ZHD Structures: V153H Mutation Enhances Communication between Different Residues
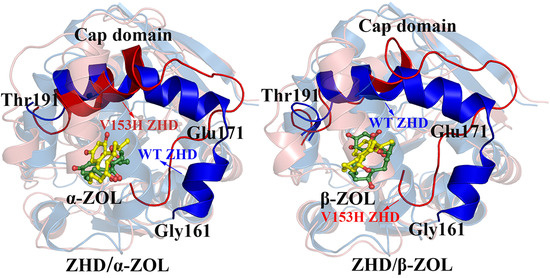
2.3. Comparison of the Conformational Changes of WT and V153H ZHD
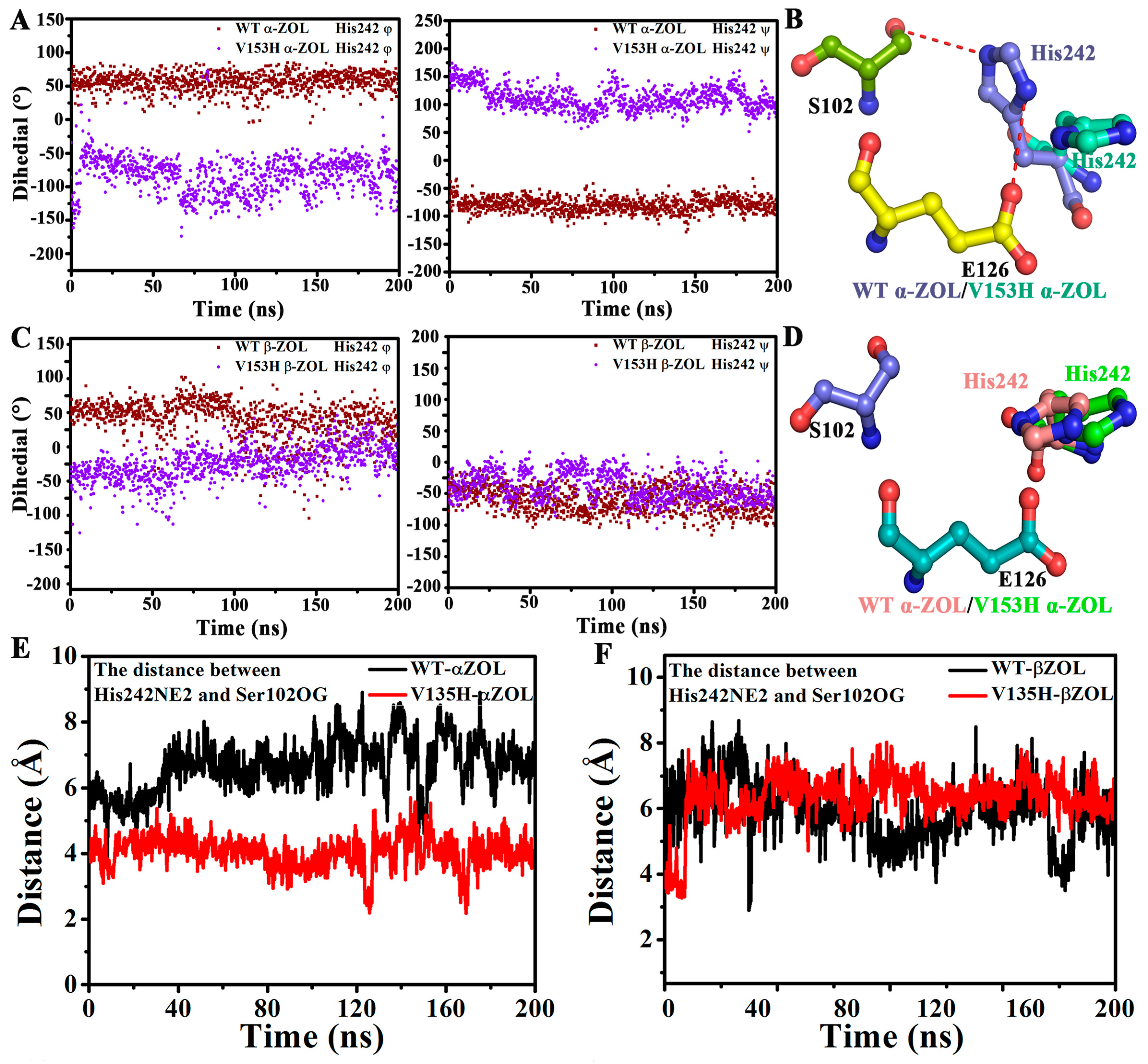
2.4. V153H Mutation in WT α-ZOL Could Lead to Catalytic Center of Be an Active Conformation
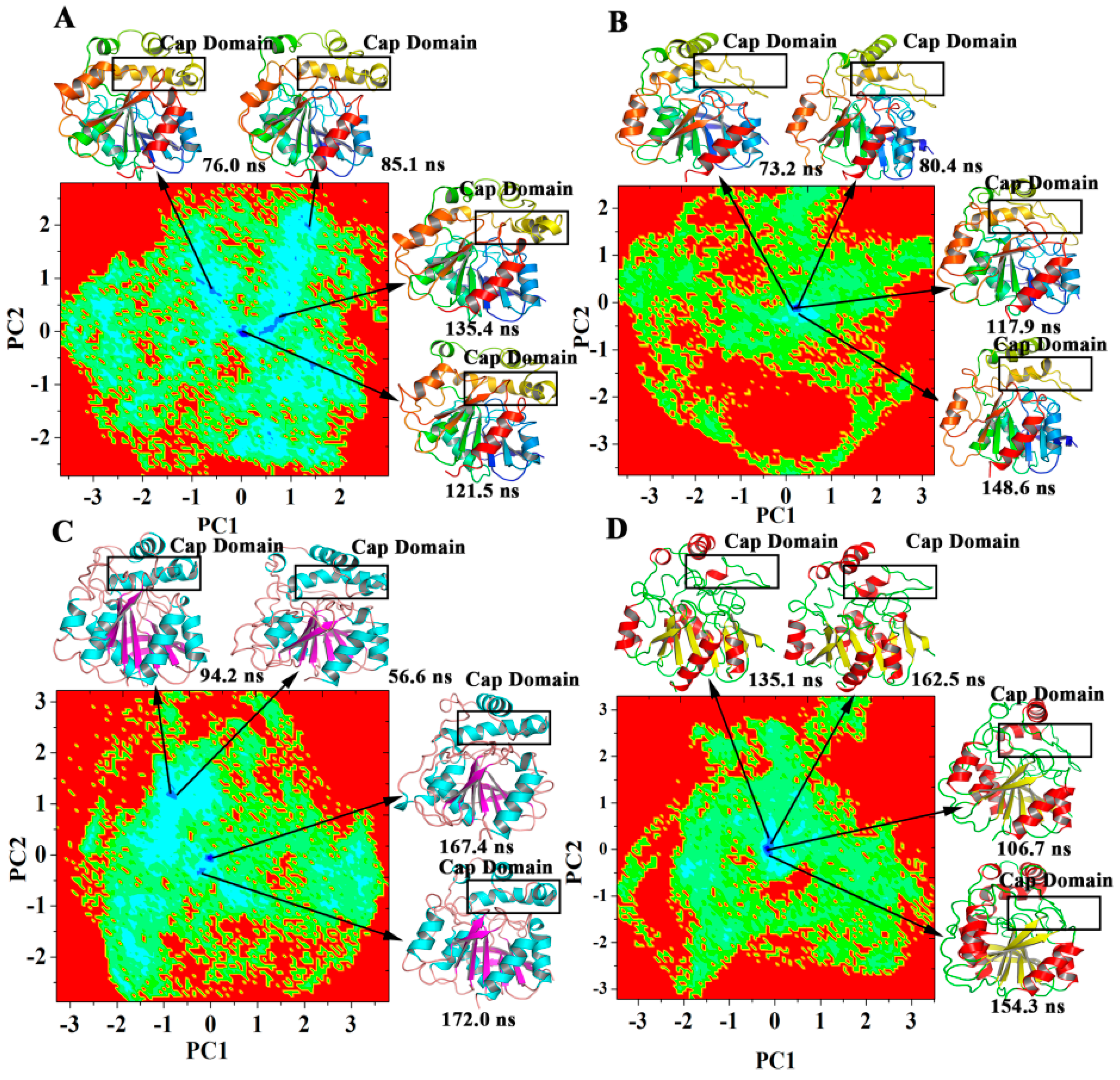
2.5. Dominant Domain Motions
2.6. Computational Mutagenesis of Active Site Residues
3. Materials and Methods
3.1. Structure Preparation
3.2. Molecular Dynamics Simulations
3.3. Computational Alanine Scanning
3.4. Protein Structure Network Analysis
3.5. Network Centrality Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Marin, S.; Ramos, A.J.; Canosancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Escrivá, L.; Font, G.; Manyes, L. In vivo toxicity studies of fusarium mycotoxins in the last decade: A review. Food Chem. Toxicol. 2015, 78, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, K.; Habrowskagórczyńska, D.E.; Piastowskaciesielska, A.W. Zearalenone as an endocrine disruptor in humans. Environ. Toxicol. Pharmacol. 2016, 48, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Liu, W.; Chen, C.C.; Li, Q.; Huang, J.W.; Ko, T.P.; Liu, G.; Liu, W.; Peng, W.; Cheng, Y.S.; et al. Enhanced α-zearalenol hydrolyzing activity of a mycoestrogen-detoxifying lactonase by structure-based engineering. ACS Catal. 2016, 6, 7657–7663. [Google Scholar] [CrossRef]
- Qi, Q.; Yang, W.J.; Zhou, H.J.; Ming, D.M.; Sun, K.L.; Xu, T.Y.; Hu, X.J.; Lv, H. The structure of a complex of the lactonohydrolase zearalenone hydrolase with the hydrolysis product of zearalenone at 1.60 Å resolution. Acta Crystallogr. 2017, 73, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.C.; Hsu, T.C.; Cheng, K.C.; Liu, J.R. Expression of the Clonostachys rosea lactonohydrolase gene by Lactobacillus reuteri to increase its zearalenone-removing ability. Microb. Cell Fact. 2017, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Popiel, D.; Koczyk, G.; Dawidziuk, A.; Gromadzka, K.; Blaszczyk, L.; Chelkowski, J. Zearalenone lactonohydrolase activity in hypocreales and its evolutionary relationships within the epoxide hydrolase subset of a/b-hydrolases. BMC Microbiol. 2014, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kosawang, C.; Karlsson, M.; Vélëz, H.; Rasmussen, P.H.; Collinge, D.B.; Jensen, B.; Jensen, D.F. Zearalenone detoxification by zearalenone hydrolase is important for the antagonistic ability of clonostachys rosea against mycotoxigenic fusarium graminearum. Fungal Biol. 2014, 118, 364. [Google Scholar] [CrossRef] [PubMed]
- Hui, R.; Hu, X.; Liu, W.; Liu, W.; Zheng, Y.; Chen, Y.; Guo, R.T.; Jin, J.; Chen, C.C. Characterization and crystal structure of a novel zearalenone hydrolase from cladophialophora bantiana. Acta Crystallogr. 2017, 73, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Ko, T.P.; Yang, Y.; Zheng, Y.; Chen, C.C.; Zhu, Z.; Huang, C.H.; Zeng, Y.F.; Huang, J.W.; Wang, H.J. Crystal structure and substrate-binding mode of the mycoestrogen-detoxifying lactonase ZHD from Clonostachys rosea. RSC Adv. 2014, 4, 62321–62325. [Google Scholar] [CrossRef]
- Zhu, J.; Lv, Y.; Han, X.; Xu, D.; Han, W. Understanding the differences of the ligand binding/unbinding pathways between phosphorylated and non-phosphorylated arh1 using molecular dynamics simulations. Sci. Rep. 2017, 7, 12439. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wang, Y.; Li, X.; Han, W.; Zhao, L. Understanding the interactions of different substrates with wild-type and mutant acylaminoacyl peptidase using molecular dynamics simulations. J. Biomol. Struct. Dyn. 2017. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Zhu, J.; Wang, S.; Xu, D. Understanding the phosphorylation mechanism by using quantum chemical calculations and molecular dynamics simulations. J. Phys. Chem. B 2016, 121, 3565–3573. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, Y.; Zhao, R.N.; Fan, S.; Han, J.G. Investigation on the mechanism for the binding and drug resistance of wild type and mutations of G86 residue in HIV-1 protease complexed with darunavir by molecular dynamic simulation and free energy calculation. J. Mol. Model. 2014, 20, 2122. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Zhu, W.; Li, G. A computational analysis of binding modes and conformation changes of MDM2 induced by p53 and inhibitor bindings. J. Comput. Aided Mol. Des. 2013, 27, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Rao, Z.H.; Liu, S.Q. Insight derived from molecular dynamics simulation into substrate-induced changes in protein motions of proteinase K. J. Biomol. Struct. Dyn. 2010, 28, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. Lincs: A linear constraint solver for molecular simulations. J. Chem. Theory Comput. 2008, 4, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Zhao, L.; Jin, H.; Shan, N.; Han, W.; Wang, S.; Shan, Y. Binding modes of phosphotriesterase-like lactonase complexed with δ-nonanoic lactone and paraoxon using molecular dynamics simulations. J. Biomol. Struct. Dyn. 2016, 35, 1. [Google Scholar] [CrossRef] [PubMed]
- Schuttelkopf, A.W.; van Aalten, D.M.F. Prodrg: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Nf, V.D.V. Hydration thermodynamic properties of amino acid analogues: A systematic comparison of biomolecular force fields and water models. J. Phys. Chem. B 2006, 110, 17616–17626. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Liu, J.; Li, D.Z.; Liu, X.Z.J. A simple and accurate algorithm for path integral molecular dynamics with the Langevin thermostat. J. Chem. Phys. 2016, 145, 024103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza-Fonseca, L.M.; Ramírez-Salinas, G.L. Microsecond molecular simulations reveal a transient proton pathway in the calcium pump. J. Am. Chem. Soc. 2015, 137, 7055–7058. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Liu, X.; Peng, L.; Zhou, Y.; Zhang, Y.; Zhang, J. Computational alanine scanning with interaction entropy for protein–ligand binding free energies. J. Chem. Theory Comput. 2018, 14, 1772–1780. [Google Scholar] [CrossRef] [PubMed]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Tokuriki, N.; Stricher, F.; Schymkowitz, J.; Serrano, L.; Tawfik, D.S. The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol. 2007, 369, 1318–1332. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The foldx web server: An online force field. Nucleic Acids Res. 2005, 33, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A graphical interface for the foldx forcefield. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.J.; Kepp, K.P. Stability mechanisms of laccase isoforms using a modified foldx protocol applicable to widely different proteins. J. Chem. Theory Comput. 2013, 9, 3210. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.J.; Kepp, K.P. Accurate stabilities of laccase mutants predicted with a modified foldx protocol. J. Chem. Inf. Model. 2012, 52, 3028–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA: Protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [PubMed]
- Stetz, G.; Verkhivker, G.M. Probing allosteric inhibition mechanisms of the Hsp70 chaperone proteins using molecular dynamics simulations and analysis of the residue interaction networks. J. Chem. Inf. Model. 2016, 56, 1490. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.W. Algorithm 97, shortest path algorithms. Commun. ACM 1962, 5, 345. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Klein, K.; Domingues, F.S.; Albrecht, M. Analyzing and visualizing residue networks of protein structures. Trends Biochem. Sci. 2011, 36, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Blacklock, K.; Verkhivker, G.M. Computational modeling of allosteric regulation in the hsp90 chaperones: A statistical ensemble analysis of protein structure networks and allosteric communications. PLoS Comput. Biol. 2014, 10, e1003679. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.; Schult, D.; Swart, P. Exploring Network Structure, Dynamics, and Function; Scipy: Los Alamos, NM, USA, 2008; pp. 11–15. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Cap Domain | Turn Occupancy |
|---|---|---|
| WT α-ZOL | G161 to R175 | 3.40% |
| V153H α-ZOL | G161 to R175 | 62.8% |
| WT β-ZOL | G161 to R175 | 4.60% |
| V153H β-ZOL | G161 to R175 | 84.1% |
| Hydrogen Bonds | WT α-ZOL | V153H α-ZOL | WT β-ZOL | V153H β-ZOL | |
|---|---|---|---|---|---|
| Donor | Accepter | ||||
| Ser162:OG | Gly240:O | 11.39 | 41.96 | 24.48 | |
| Ser162:OG | Gly240:C | 12.74 | 30.59 | 21.36 | 35.61 |
| Ser162:OG | His242:N | 23.49 | 42.59 | 15.67 | 36.86 |
| Ser162:OG | His242:CB | 25.53 | 13.64 | 47.25 | |
| Ser162:N | Met241:O | 16.38 | 33.52 | 22.52 | 36.61 |
| Ser162:OG | Phe243:N | 24.26 | 48.35 | 10.25 | 34.03 |
| Ser162:OG | Met241:O | 38.01 | 21.03 | ||
| Protein | Principle Component (PC) | Probility (%) |
|---|---|---|
| WT α-ZOL | PC1 | 43.52 |
| PC2 | 13.58 | |
| V153H α-ZOL | PC1 | 42.34 |
| PC2 | 14.93 | |
| WT β-ZOL | PC1 | 49.75 |
| PC2 | 12.43 | |
| V153H β-ZOL | PC1 | 47.18 |
| PC2 | 20.37 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Wan, Y.; Zhu, J.; Yu, Z.; Tian, X.; Han, J.; Zhang, Z.; Han, W. Theoretical Study on Zearalenol Compounds Binding with Wild Type Zearalenone Hydrolase and V153H Mutant. Int. J. Mol. Sci. 2018, 19, 2808. https://doi.org/10.3390/ijms19092808
Liu Y, Wan Y, Zhu J, Yu Z, Tian X, Han J, Zhang Z, Han W. Theoretical Study on Zearalenol Compounds Binding with Wild Type Zearalenone Hydrolase and V153H Mutant. International Journal of Molecular Sciences. 2018; 19(9):2808. https://doi.org/10.3390/ijms19092808
Chicago/Turabian StyleLiu, Ye, Youzhong Wan, Jingxuan Zhu, Zhengfei Yu, Xiaopian Tian, Jiarui Han, Zuoming Zhang, and Weiwei Han. 2018. "Theoretical Study on Zearalenol Compounds Binding with Wild Type Zearalenone Hydrolase and V153H Mutant" International Journal of Molecular Sciences 19, no. 9: 2808. https://doi.org/10.3390/ijms19092808