Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma

 ,
,  ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Clinical Characteristics of GBM
3. Risk Factors
4. Etiology and Pathogenesis of GBM
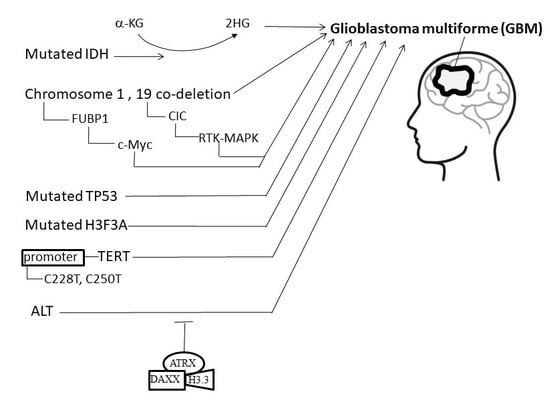
4.1. Isocitrate Dehydrogenase (IDH)
4.2. Co-Deletion at Chromosome Regions 1p/19q
4.3. Mutations of H3F3A
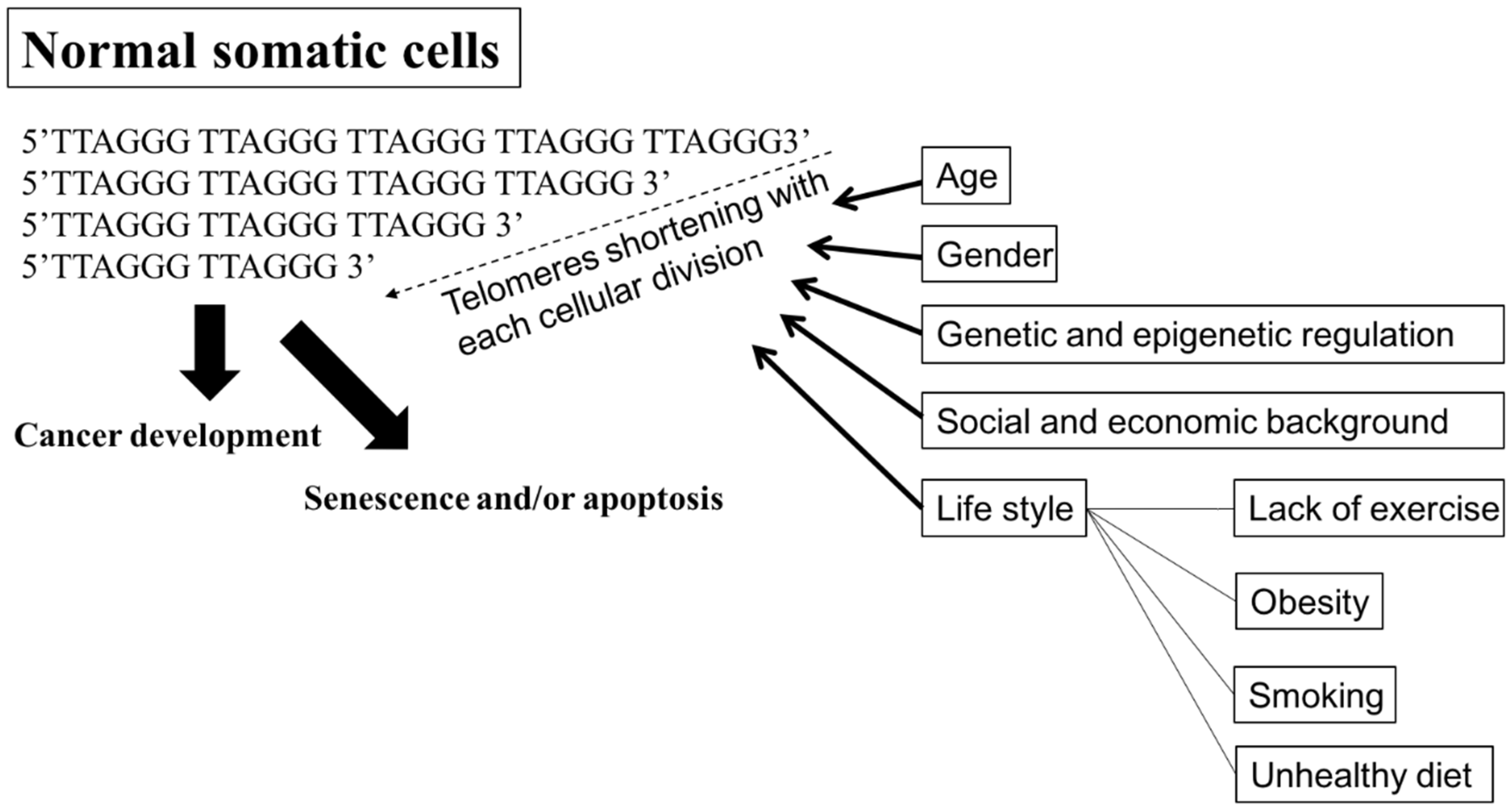
4.4. Telomeres and Telomerase
4.5. Alternative Lengthening of Telomeres, α-Thalassemia X-Linkedintellectual Disability, and Death-Domain-Associated Protein
5. Novel and Alternative Therapeutic Options for Treating GBM
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- de Robles, P.; Fiest, K.M.; Frolkis, A.D.; Pringsheim, T.; Atta, C.; St Germaine-Smith, C.; Day, L.; Lam, D.; Jette, N. The worldwide incidence and prevalence of primary brain tumors: A systematic review and meta-analysis. Neuro Oncol. 2015, 17, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: A clinician’s perspective. Acta Neuropathol. 2010, 120, 297–304. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- CBTRUS. Central Brain Tumor Registry of the United States CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2008; CBTRUS Central Brain Tumor Registry of the United States: Hinsdale, IL, USA, 2014. [Google Scholar]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Chien, L.N.; Gittleman, H.; Ostrom, Q.T.; Hung, K.S.; Sloan, A.E.; Hsieh, Y.C.; Kruchko, C.; Rogers, L.R.; Wang, Y.F.; Chiou, H.Y.; et al. Comparative Brain and Central Nervous System Tumor Incidence and Survival between the United States and Taiwan Based on Population-Based Registry. Front. Public Health 2016, 4, 151. [Google Scholar] [CrossRef]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol. 2012, 14 (Suppl. 5), v1–v49. [Google Scholar] [CrossRef]
- Rock, K.; McArdle, O.; Forde, P.; Dunne, M.; Fitzpatrick, D.; O’Neill, B.; Faul, C. A clinical review of treatment outcomes in glioblastoma multiforme—The validation in a non-trial population of the results of a randomised Phase III clinical trial: Has a more radical approach improved survival? Br. J. Radiol. 2012, 85, e729–e733. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Fan, H.C.; Chi, C.S.; Chang, Y.K.; Tung, M.C.; Lin, S.Z.; Harn, H.J. The Molecular Mechanisms of Plant-Derived Compounds Targeting Brain Cancer. Int. J. Mol. Sci. 2018, 19, 395. [Google Scholar] [CrossRef] [PubMed]
- Ohka, F.; Natsume, A.; Wakabayashi, T. Current trends in targeted therapies for glioblastoma multiforme. Neurol. Res. Int. 2012, 2012, 878425. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, A.; Gronberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef]
- Available online: https://brainfoundation.org.au/medical-information/c4-brain-tumour/diagnosis-of-brain-tumour (accessed on 12 December 2018).
- Nakada, M.; Kita, D.; Watanabe, T.; Hayashi, Y.; Teng, L.; Pyko, I.V.; Hamada, J. Aberrant signaling pathways in glioma. Cancers 2011, 3, 3242–3278. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Burrell, K.E.; Wolf, A.; Jalali, S.; Hawkins, C.; Rutka, J.T.; Zadeh, G. Glioblastoma, a brief review of history, molecular genetics, animal models and novel therapeutic strategies. Arch. Immunol. Ther. Exp. (Warsz) 2013, 61, 25–41. [Google Scholar] [CrossRef]
- Adamson, C.; Kanu, O.O.; Mehta, A.I.; Di, C.; Lin, N.; Mattox, A.K.; Bigner, D.D. Glioblastoma multiforme: A review of where we have been and where we are going. Expert Opin. Investig. Drugs 2009, 18, 1061–1083. [Google Scholar] [CrossRef]
- Fisher, J.L.; Schwartzbaum, J.A.; Wrensch, M.; Wiemels, J.L. Epidemiology of brain tumors. Neurol. Clin. 2007, 25, 867–890. [Google Scholar] [CrossRef]
- Inskip, P.D.; Tarone, R.E.; Hatch, E.E.; Wilcosky, T.C.; Shapiro, W.R.; Selker, R.G.; Fine, H.A.; Black, P.M.; Loeffler, J.S.; Linet, M.S. Cellular-telephone use and brain tumors. N. Engl. J. Med. 2001, 344, 79–86. [Google Scholar] [CrossRef]
- Ohgaki, H. Epidemiology of brain tumors. Methods Mol. Biol. 2009, 472, 323–342. [Google Scholar]
- Kabat, G.C.; Etgen, A.M.; Rohan, T.E. Do steroid hormones play a role in the etiology of glioma? Cancer Epidemiol. Biomarkers Prev. 2010, 19, 2421–2427. [Google Scholar] [CrossRef] [PubMed]
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il’yasova, D.; Kruchko, C.; McCarthy, B.J.; Rajaraman, P.; Schwartzbaum, J.A.; et al. Brain tumor epidemiology: Consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113 (Suppl. 7), 1953–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg-Beckhoff, G.; Schuz, J.; Blettner, M.; Munster, E.; Schlaefer, K.; Wahrendorf, J.; Schlehofer, B. History of allergic disease and epilepsy and risk of glioma and meningioma (INTERPHONE study group, Germany). Eur. J. Epidemiol. 2009, 24, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Il’yasova, D.; McCarthy, B.; Marcello, J.; Schildkraut, J.M.; Moorman, P.G.; Krishnamachari, B.; Ali-Osman, F.; Bigner, D.D.; Davis, F. Association between glioma and history of allergies, asthma, and eczema: A case-control study with three groups of controls. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 1232–1238. [Google Scholar] [CrossRef]
- McCarthy, B.J.; Rankin, K.; Il’yasova, D.; Erdal, S.; Vick, N.; Ali-Osman, F.; Bigner, D.D.; Davis, F. Assessment of type of allergy and antihistamine use in the development of glioma. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 370–378. [Google Scholar] [CrossRef]
- Turner, M.C.; Krewski, D.; Armstrong, B.K.; Chetrit, A.; Giles, G.G.; Hours, M.; McBride, M.L.; Parent, M.E.; Sadetzki, S.; Siemiatycki, J.; et al. Allergy and brain tumors in the INTERPHONE study: Pooled results from Australia, Canada, France, Israel, and New Zealand. Cancer Causes Control 2013, 24, 949–960. [Google Scholar] [CrossRef]
- Linos, E.; Raine, T.; Alonso, A.; Michaud, D. Atopy and risk of brain tumors: A meta-analysis. J. Natl. Cancer Inst. 2007, 99, 1544–1550. [Google Scholar] [CrossRef]
- Mulvihill, J.J.; Parry, D.M.; Sherman, J.L.; Pikus, A.; Kaiser-Kupfer, M.I.; Eldridge, R. NIH conference. Neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis). An update. Ann. Intern. Med. 1990, 113, 39–52. [Google Scholar] [CrossRef]
- Cristofori, E.; Colvecchio, A.; Rescaldani, R.; Micoli, G. Bilateral renal angiomyoliposarcoma and glioblastoma multiforme in tuberous sclerosis. Folia Hered. Pathol. (Milano) 1968, 17, 79–83. [Google Scholar]
- Therkildsen, C.; Ladelund, S.; Rambech, E.; Persson, A.; Petersen, A.; Nilbert, M. Glioblastomas, astrocytomas and oligodendrogliomas linked to Lynch syndrome. Eur. J. Neurol. 2015, 22, 717–724. [Google Scholar] [CrossRef]
- Bahuau, M.; Vidaud, D.; Jenkins, R.B.; Bieche, I.; Kimmel, D.W.; Assouline, B.; Smith, J.S.; Alderete, B.; Cayuela, J.M.; Harpey, J.P.; et al. Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res. 1998, 58, 2298–2303. [Google Scholar] [PubMed]
- Mellon, C.D.; Carter, J.E.; Owen, D.B. Ollier’s disease and Maffucci’s syndrome: Distinct entities or a continuum. Case report: Enchondromatosis complicated by an intracranial glioma. J. Neurol. 1988, 235, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; McComb, R.D.; Osborn, N.K.; Wolpert, P.A.; Lynch, J.F.; Wszolek, Z.K.; Sidransky, D.; Steg, R.E. Predominance of brain tumors in an extended Li-Fraumeni (SBLA) kindred, including a case of Sturge-Weber syndrome. Cancer 2000, 88, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Sanson, M.; Hosking, F.J.; Shete, S.; Zelenika, D.; Dobbins, S.E.; Ma, Y.; Enciso-Mora, V.; Idbaih, A.; Delattre, J.Y.; Hoang-Xuan, K.; et al. Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum. Mol. Genet. 2011, 20, 2897–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shete, S.; Hosking, F.J.; Robertson, L.B.; Dobbins, S.E.; Sanson, M.; Malmer, B.; Simon, M.; Marie, Y.; Boisselier, B.; Delattre, J.Y.; et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 2009, 41, 899–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacey, S.N.; Sulem, P.; Jonasdottir, A.; Masson, G.; Gudmundsson, J.; Gudbjartsson, D.F.; Magnusson, O.T.; Gudjonsson, S.A.; Sigurgeirsson, B.; Thorisdottir, K.; et al. A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet. 2011, 43, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Walsh, K.M.; Codd, V.; Smirnov, I.V.; Rice, T.; Decker, P.A.; Hansen, H.M.; Kollmeyer, T.; Kosel, M.L.; Molinaro, A.M.; McCoy, L.S.; et al. Variants near TERT and TERC influencing telomere length are associated with high-grade glioma risk. Nat. Genet. 2014, 46, 731–735. [Google Scholar] [CrossRef] [Green Version]
- Wrensch, M.; Jenkins, R.B.; Chang, J.S.; Yeh, R.F.; Xiao, Y.; Decker, P.A.; Ballman, K.V.; Berger, M.; Buckner, J.C.; Chang, S.; et al. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat. Genet. 2009, 41, 905–908. [Google Scholar] [CrossRef] [Green Version]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Inagaki, A.; Silber, J.; Gorovets, D.; Zhang, J.; Kastenhuber, E.R.; Heguy, A.; Petrini, J.H.; Chan, T.A.; Huse, J.T. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 2012, 3, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Jr Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koelsche, C.; Sahm, F.; Capper, D.; Reuss, D.; Sturm, D.; Jones, D.T.; Kool, M.; Northcott, P.A.; Wiestler, B.; Bohmer, K.; et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013, 126, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.W.; Jensen, M.V.; Ilkayeva, O.; Palmieri, F.; Alarcon, C.; Rhodes, C.J.; Newgard, C.B. The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J. Biol. Chem. 2006, 281, 35624–35632. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jo, S.H.; Lee, S.M.; Koh, H.J.; Song, H.; Park, J.W.; Lee, W.H.; Huh, T.L. Role of NADP+-dependent isocitrate dehydrogenase (NADP+-ICDH) on cellular defence against oxidative injury by gamma-rays. Int. J. Radiat. Biol. 2004, 80, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Cairncross, J.G.; Ueki, K.; Zlatescu, M.C.; Lisle, D.K.; Finkelstein, D.M.; Hammond, R.R.; Silver, J.S.; Stark, P.C.; Macdonald, D.R.; Ino, Y.; et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J. Natl. Cancer Inst. 1998, 90, 1473–1479. [Google Scholar] [CrossRef]
- Kraus, J.A.; Koopmann, J.; Kaskel, P.; Maintz, D.; Brandner, S.; Schramm, J.; Louis, D.N.; Wiestler, O.D.; von Deimling, A. Shared allelic losses on chromosomes 1p and 19q suggest a common origin of oligodendroglioma and oligoastrocytoma. J. Neuropathol. Exp. Neurol. 1995, 54, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Polivka, J., Jr.; Polivka, J.; Repik, T.; Rohan, V.; Hes, O.; Topolcan, O. Co-deletion of 1p/19q as Prognostic and Predictive Biomarker for Patients in West Bohemia with Anaplastic Oligodendroglioma. Anticancer Res. 2016, 36, 471–476. [Google Scholar] [PubMed]
- Reifenberger, J.; Reifenberger, G.; Liu, L.; James, C.D.; Wechsler, W.; Collins, V.P. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am. J. Pathol. 1994, 145, 1175–1190. [Google Scholar] [PubMed]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, A.B.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Nagahashi Marie, S.K.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Labussiere, M.; Idbaih, A.; Wang, X.W.; Marie, Y.; Boisselier, B.; Falet, C.; Paris, S.; Laffaire, J.; Carpentier, C.; Criniere, E.; et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 2010, 74, 1886–1890. [Google Scholar] [CrossRef]
- Duan, J.; Bao, X.; Ma, X.; Zhang, Y.; Ni, D.; Wang, H.; Zhang, F.; Du, Q.; Fan, Y.; Chen, J.; et al. Upregulation of Far Upstream Element-Binding Protein 1 (FUBP1) Promotes Tumor Proliferation and Tumorigenesis of Clear Cell Renal Cell Carcinoma. PLoS ONE 2017, 12, e0169852. [Google Scholar] [CrossRef]
- Han, F.; Zhang, J.; Ma, S.; Chen, X.; Liu, W.; He, X.; Fei, Z.; Wang, Y. Altered capicua transcriptional repressor gene expression exhibits distinct prognostic value for isocitrate dehydrogenase-mutant oligodendroglial tumors. Oncol. Lett. 2018, 15, 1459–1468. [Google Scholar] [CrossRef]
- Cairncross, G.; Wang, M.; Shaw, E.; Jenkins, R.; Brachman, D.; Buckner, J.; Fink, K.; Souhami, L.; Laperriere, N.; Curran, W.; et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: Long-term results of RTOG 9402. J. Clin. Oncol. 2013, 31, 337–343. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Carpentier, A.F.; Brandes, A.A.; Sanson, M.; Taphoorn, M.J.; Bernsen, H.J.; Frenay, M.; Tijssen, C.C.; Grisold, W.; Sipos, L.; et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: A randomized European Organisation for Research and Treatment of Cancer phase III trial. J. Clin. Oncol. 2006, 24, 2715–2722. [Google Scholar] [PubMed]
- Kaneshiro, D.; Kobayashi, T.; Chao, S.T.; Suh, J.; Prayson, R.A. Chromosome 1p and 19q deletions in glioblastoma multiforme. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Yang, F.; Fujisawa, H.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. Loss of heterozygosity on chromosome 19 in secondary glioblastomas. J. Neuropathol. Exp. Neurol. 2000, 59, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Filipescu, D.; Muller, S.; Almouzni, G. Histone H3 variants and their chaperones during development and disease: Contributing to epigenetic control. Annu. Rev. Cell Dev. Biol. 2014, 30, 615–646. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Abbas, K.; Manan, M.; Ijaz, H.; Ahmed, B.; Ali, M.; Hanif, M.; Farooqi, A.A.; Qadir, M.I. Review-Epigenetic therapy for cancer. Pak. J. Pharm. Sci. 2015, 28, 1023–1032. [Google Scholar] [PubMed]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef]
- Martin, C.L.L. DH Telomeres. In Handbook of Human Molecular Evolution; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 721–724. [Google Scholar]
- Ruiz-Herrera, A.; Nergadze, S.G.; Santagostino, M.; Giulotto, E. Telomeric repeats far from the ends: Mechanisms of origin and role in evolution. Cytogenet. Genome Res. 2008, 122, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; O’Connor, M.S.; Qin, J.; Songyang, Z. Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J. Biol. Chem. 2004, 279, 51338–51342. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef]
- Gong, J.G.; Costanzo, A.; Yang, H.Q.; Melino, G.; Kaelin, W.G., Jr.; Levrero, M.; Wang, J.Y. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999, 399, 806–809. [Google Scholar] [CrossRef]
- Stiewe, T.; Putzer, B.M. p73 in apoptosis. Apoptosis 2001, 6, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dragowska, W.; Allsopp, R.C.; Thomas, T.E.; Harley, C.B.; Lansdorp, P.M. Evidence for a mitotic clock in human hematopoietic stem cells: Loss of telomeric DNA with age. Proc. Natl. Acad. Sci. USA 1994, 91, 9857–9860. [Google Scholar] [CrossRef] [PubMed]
- Brouilette, S.; Singh, R.K.; Thompson, J.R.; Goodall, A.H.; Samani, N.J. White cell telomere length and risk of premature myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Andrew, T.; Gardner, J.P.; Kimura, M.; Oelsner, E.; Cherkas, L.F.; Aviv, A.; Spector, T.D. Obesity, cigarette smoking, and telomere length in women. Lancet 2005, 366, 662–664. [Google Scholar] [CrossRef]
- Frenck, R.W., Jr.; Blackburn, E.H.; Shannon, K.M. The rate of telomere sequence loss in human leukocytes varies with age. Proc. Natl. Acad. Sci. USA 1998, 95, 5607–5610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgard, C.; Benetos, A.; Verhulst, S.; Labat, C.; Kark, J.D.; Christensen, K.; Kimura, M.; Kyvik, K.O.; Aviv, A. Leukocyte telomere length dynamics in women and men: Menopause vs age effects. Int. J. Epidemiol. 2015, 44, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Benetti, R.; Garcia-Cao, M.; Blasco, M.A. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat. Genet. 2007, 39, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Celli, G.B.; de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005, 7, 712–718. [Google Scholar] [CrossRef]
- Steinert, S.; Shay, J.W.; Wright, W.E. Modification of subtelomeric DNA. Mol. Cell. Biol. 2004, 24, 4571–4580. [Google Scholar] [CrossRef]
- Adams, J.; Martin-Ruiz, C.; Pearce, M.S.; White, M.; Parker, L.; von Zglinicki, T. No association between socio-economic status and white blood cell telomere length. Aging Cell 2007, 6, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Cherkas, L.F.; Hunkin, J.L.; Kato, B.S.; Richards, J.B.; Gardner, J.P.; Surdulescu, G.L.; Kimura, M.; Lu, X.; Spector, T.D.; Aviv, A. The association between physical activity in leisure time and leukocyte telomere length. Arch. Intern. Med. 2008, 168, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Nordfjall, K.; Eliasson, M.; Stegmayr, B.; Melander, O.; Nilsson, P.; Roos, G. Telomere length is associated with obesity parameters but with a gender difference. Obesity 2008, 16, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- van der Harst, P.; van der Steege, G.; de Boer, R.A.; Voors, A.A.; Hall, A.S.; Mulder, M.J.; van Gilst, W.H.; van Veldhuisen, D.J.; MERIT-HF Study Group. Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J. Am. Coll. Cardiol. 2007, 49, 1459–1464. [Google Scholar] [CrossRef]
- Brouilette, S.W.; Moore, J.S.; McMahon, A.D.; Thompson, J.R.; Ford, I.; Shepherd, J.; Packard, C.J.; Samani, N.J.; West of Scotland Coronary Prevention Study Group. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: A nested case-control study. Lancet 2007, 369, 107–114. [Google Scholar] [CrossRef]
- Fitzpatrick, A.L.; Kronmal, R.A.; Gardner, J.P.; Psaty, B.M.; Jenny, N.S.; Tracy, R.P.; Walston, J.; Kimura, M.; Aviv, A. Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am. J. Epidemiol. 2007, 165, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Zee, R.Y.; Michaud, S.E.; Germer, S.; Ridker, P.M. Association of shorter mean telomere length with risk of incident myocardial infarction: A prospective, nested case-control approach. Clin. Chim. Acta 2009, 403, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.J.; Winterbone, M.S.; Hughes, J.C.; Dozio, N.; Hughes, D.A. Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care 2006, 29, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Valdes, A.M.; Richards, J.B.; Gardner, J.P.; Swaminathan, R.; Kimura, M.; Xiaobin, L.; Aviv, A.; Spector, T.D. Telomere length in leukocytes correlates with bone mineral density and is shorter in women with osteoporosis. Osteoporos. Int. 2007, 18, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395. [Google Scholar] [CrossRef]
- Farzaneh-Far, R.; Cawthon, R.M.; Na, B.; Browner, W.S.; Schiller, N.B.; Whooley, M.A. Prognostic value of leukocyte telomere length in patients with stable coronary artery disease: Data from the Heart and Soul Study. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1379–1384. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, X.; Jiang, H.; Zhang, Y.; Liu, H.; Qin, C.; Eisner, G.M.; Jose, P.A.; Rudolph, L.; Ju, Z. Short telomeres and prognosis of hypertension in a chinese population. Hypertension 2009, 53, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; DePinho, R.A. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97, 527–538. [Google Scholar] [CrossRef]
- De Lange, T. Telomere-related genome instability in cancer. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Meeker, A.K. Telomeres and telomerase in prostatic intraepithelial neoplasia and prostate cancer biology. Urol. Oncol. 2006, 24, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Role of telomeres and telomerase in cancer. Semin. Cancer Biol. 2011, 21, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Higashi, N.; Hansen, E.R.; Lund, M.; Bang, K.; Thestrup-Pedersen, K. Telomerase activity is increased and telomere length shortened in T cells from blood of patients with atopic dermatitis and psoriasis. J. Immunol. 2000, 165, 4742–4747. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef]
- Shippen-Lentz, D.; Blackburn, E.H. Functional evidence for an RNA template in telomerase. Science 1990, 247, 546–552. [Google Scholar] [CrossRef]
- Hiyama, K.; Hirai, Y.; Kyoizumi, S.; Akiyama, M.; Hiyama, E.; Piatyszek, M.A.; Shay, J.W.; Ishioka, S.; Yamakido, M. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J. Immunol. 1995, 155, 3711–3715. [Google Scholar]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Meyne, J.; Ratliff, R.L.; Moyzis, R.K. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc. Natl. Acad. Sci. USA 1989, 86, 7049–7053. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA component of human telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S.; Pooley, K.A.; Nitti, D. Telomerase and the search for the end of cancer. Trends Mol. Med. 2013, 19, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Smekalova, E.M.; Shubernetskaya, O.S.; Zvereva, M.I.; Gromenko, E.V.; Rubtsova, M.P.; Dontsova, O.A. Telomerase RNA biosynthesis and processing. Biochemistry 2012, 77, 1120–1128. [Google Scholar] [CrossRef]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Liu, X.; Wu, G.; Shan, Y.; Hartmann, C.; von Deimling, A.; Xing, M. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle 2013, 12, 1637–1638. [Google Scholar] [CrossRef]
- Arita, H.; Narita, Y.; Takami, H.; Fukushima, S.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Shibui, S.; Ichimura, K. TERT promoter mutations rather than methylation are the main mechanism for TERT upregulation in adult gliomas. Acta Neuropathol. 2013, 126, 939–941. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Nonoguchi, N.; Ohta, T.; Oh, J.E.; Kim, Y.H.; Kleihues, P.; Ohgaki, H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 2013, 126, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Molano, M.; Martin, D.C.; Moreno-Acosta, P.; Hernandez, G.; Cornall, A.; Buitrago, O.; Gamboa, O.; Garland, S.; Tabrizi, S.; Munoz, N. Telomerase activity in cervical scrapes of women with high-grade cervical disease: A nested case-control study. Oncol. Lett. 2018, 15, 354–360. [Google Scholar] [CrossRef]
- Wang, N.; Xu, D.; Sofiadis, A.; Hoog, A.; Vukojevic, V.; Backdahl, M.; Zedenius, J.; Larsson, C. Telomerase-dependent and independent telomere maintenance and its clinical implications in medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, E1571–E1579. [Google Scholar] [CrossRef] [PubMed]
- Bornstein-Quevedo, L.; Garcia-Hernandez, M.L.; Camacho-Arroyo, I.; Herrera, M.F.; Angeles, A.A.; Trevino, O.G.; Gamboa-Dominguez, A. Telomerase activity in well-differentiated papillary thyroid carcinoma correlates with advanced clinical stage of the disease. Endocr. Pathol. 2003, 14, 213–219. [Google Scholar] [CrossRef]
- Fernandez-Marcelo, T.; Gomez, A.; Pascua, I.; de Juan, C.; Head, J.; Hernando, F.; Jarabo, J.R.; Calatayud, J.; Torres-Garcia, A.J.; Iniesta, P. Telomere length and telomerase activity in non-small cell lung cancer prognosis: Clinical usefulness of a specific telomere status. J. Exp. Clin. Cancer Res. 2015, 34, 78. [Google Scholar] [CrossRef]
- Satyanarayana, A.; Manns, M.P.; Rudolph, K.L. Telomeres and telomerase: A dual role in hepatocarcinogenesis. Hepatology 2004, 40, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef]
- Slatter, T.L.; Tan, X.; Yuen, Y.C.; Gunningham, S.; Ma, S.S.; Daly, E.; Packer, S.; Devenish, C.; Royds, J.A.; Hung, N.A. The alternative lengthening of telomeres pathway may operate in non-neoplastic human cells. J. Pathol. 2012, 226, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Wiestler, B.; Capper, D.; Holland-Letz, T.; Korshunov, A.; von Deimling, A.; Pfister, S.M.; Platten, M.; Weller, M.; Wick, W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013, 126, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, J.W.; Reddel, R.R.; Wright, W.E. Cancer. Cancer and telomeres—An ALTernative to telomerase. Science 2012, 336, 1388–1390. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, R. Alpha thalassaemia-mental retardation, X linked. Orphanet J. Rare Dis. 2006, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Qadeer, Z.A.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026567. [Google Scholar] [CrossRef]
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016. [Google Scholar] [CrossRef]
- Clynes, D.; Higgs, D.R.; Gibbons, R.J. The chromatin remodeller ATRX: A repeat offender in human disease. Trends Biochem. Sci. 2013, 38, 461–466. [Google Scholar] [CrossRef]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Taylor, S.; Mitson, M.; Bachrati, C.Z.; Higgs, D.R.; Gibbons, R.J. ATRX dysfunction induces replication defects in primary mouse cells. PLoS ONE 2014, 9, e92915. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.; Demattei, M.V.; Episkopou, H.; Auge-Gouillou, C.; Decottignies, A.; Grandin, N.; Charbonneau, M. Genetic Inactivation of ATRX Leads to a Decrease in the Amount of Telomeric Cohesin and Level of Telomere Transcription in Human Glioma Cells. Mol. Cell. Biol. 2015, 35, 2818–2830. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Khosravi-Far, R.; Chang, H.Y.; Baltimore, D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 1997, 89, 1067–1076. [Google Scholar] [CrossRef]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [Green Version]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes 2016, 7, 66. [Google Scholar] [CrossRef]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm614128.htm (accessed on 12 December 2018).
- Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm569482.htm (accessed on 12 December 2018).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02977689 (accessed on 12 December 2018).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02746081 (accessed on 12 December 2018).
- Lee, W.Y.; Chen, K.C.; Chen, H.Y.; Chen, C.Y. Potential mitochondrial isocitrate dehydrogenase R140Q mutant inhibitor from traditional Chinese medicine against cancers. Biomed. Res. Int. 2014, 2014, 364625. [Google Scholar] [CrossRef] [PubMed]
- Parsch, D.; Brassat, U.; Brummendorf, T.H.; Fellenberg, J. Consequences of telomerase inhibition by BIBR1532 on proliferation and chemosensitivity of chondrosarcoma cell lines. Cancer Investig. 2008, 26, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Harley, C.B.; Baerlocher, G.M. Imetelstat (GRN163L)—Telomerase-based cancer therapy. Recent Results Cancer Res. 2010, 184, 221–234. [Google Scholar] [PubMed]
- Nava-Parada, P.; Emens, L.A. GV-1001, an injectable telomerase peptide vaccine for the treatment of solid cancers. Curr. Opin. Mol. Ther. 2007, 9, 490–497. [Google Scholar] [PubMed]
- Ouellette, M.M.; Wright, W.E.; Shay, J.W. Targeting telomerase-expressing cancer cells. J. Cell. Mol. Med. 2011, 15, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Kosmatopoulos KB, E.; Aggouraki, D.; Nikoloudi, E.; Kanellou, P.; Papadimitraki, E.; Kotsakis, A.; Mavroudis, D.; Georgoulias, V. Clinical efficacy and immunogenicity of the optimized cryptic peptide TERT572Y vaccine (Vx-001) in patients with advanced non-small cell lung cancer (NSCLC). In Proceedings of the ASCO Meeting, Atlanta, GA, USA, 2–6 June 2006. [Google Scholar]
- Taetz, S.; Baldes, C.; Murdter, T.E.; Kleideiter, E.; Piotrowska, K.; Bock, U.; Haltner-Ukomadu, E.; Mueller, J.; Huwer, H.; Schaefer, U.F.; et al. Biopharmaceutical characterization of the telomerase inhibitor BRACO19. Pharm. Res. 2006, 23, 1031–1037. [Google Scholar] [CrossRef]
- Grenert, J.P.; Sullivan, W.P.; Fadden, P.; Haystead, T.A.; Clark, J.; Mimnaugh, E.; Krutzsch, H.; Ochel, H.J.; Schulte, T.W.; Sausville, E.; et al. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J. Biol. Chem. 1997, 272, 23843–23850. [Google Scholar] [CrossRef]
- Lee, J.H.; Chung, I.K. Curcumin inhibits nuclear localization of telomerase by dissociating the Hsp90 co-chaperone p23 from hTERT. Cancer Lett. 2010, 290, 76–86. [Google Scholar] [CrossRef]
- Tauchi, T.; Shin-ya, K.; Sashida, G.; Sumi, M.; Okabe, S.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: In vitro and in vivo studies in acute leukemia. Oncogene 2006, 25, 5719–5725. [Google Scholar] [CrossRef]
- Li, G.D.; Kawashima, H.; Ogose, A.; Ariizumi, T.; Hotta, T.; Kuwano, R.; Urata, Y.; Fujiwara, T.; Endo, N. Telomelysin shows potent antitumor activity through apoptotic and non-apoptotic cell death in soft tissue sarcoma cells. Cancer Sci. 2013, 104, 1178–1188. [Google Scholar] [CrossRef] [Green Version]
- Burchett, K.M.; Yan, Y.; Ouellette, M.M. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS ONE 2014, 9, e85155. [Google Scholar] [CrossRef] [PubMed]
- Salloum, R.; Hummel, T.R.; Kumar, S.S.; Dorris, K.; Li, S.; Lin, T.; Daryani, V.M.; Stewart, C.F.; Miles, L.; Poussaint, T.Y.; et al. A molecular biology and phase II study of imetelstat (GRN163L) in children with recurrent or refractory central nervous system malignancies: A pediatric brain tumor consortium study. J. Neurooncol. 2016, 129, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.M.S.; Fukuoka, K.; Maida, Y.; Hayashi, M.; Hamada, A.; Nishikawa, R.; Nagane, M.; Maruyama, T.; Mukasa, A.; Arakawa, Y.; et al. EXTH-50. Development of investigator initiated clinical trial of TERT-targeting therapy using eribulin mesylate in patients with recurrent glioblastoma. Neuro Oncol. 2017, 19, vi83. [Google Scholar] [CrossRef]
- Gibbons, R.J.; McDowell, T.L.; Raman, S.; O’Rourke, D.M.; Garrick, D.; Ayyub, H.; Higgs, D.R. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat. Genet. 2000, 24, 368–371. [Google Scholar] [CrossRef]
- Yang, Y.L.; Huang, P.H.; Chiu, H.C.; Kulp, S.K.; Chen, C.S.; Kuo, C.J.; Chen, H.D.; Chen, C.S. Histone deacetylase inhibitor AR42 regulates telomerase activity in human glioma cells via an Akt-dependent mechanism. Biochem. Biophys. Res. Commun. 2013, 435, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 827. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Murphy, B.; Miller, R.; Menon, V.; Banik, N.L.; Giglio, P.; Lindhorst, S.M.; Varma, A.K.; Vandergrift, W.A.; Patel, S.J.; et al. Mechanisms and clinical significance of histone deacetylase inhibitors: Epigenetic glioblastoma therapy. Anticancer Res. 2015, 35, 615–625. [Google Scholar]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef]
- Ashour, M.L.; Wink, M. Genus Bupleurum: A review of its phytochemistry, pharmacology and modes of action. J. Pharm. Pharmacol. 2011, 63, 305–321. [Google Scholar] [CrossRef]
- Adorisio, S.; Fierabracci, A.; Gigliarelli, G.; Muscari, I.; Cannarile, L.; Liberati, A.M.; Marcotullio, M.C.; Riccardi, C.; Curini, M.; Robles Zepeda, R.E.; et al. The Hexane Fraction of Bursera microphylla A Gray Induces p21-Mediated Antiproliferative and Proapoptotic Effects in Human Cancer-Derived Cell Lines. Integr. Cancer Ther. 2017, 16, 426–435. [Google Scholar] [CrossRef]
- Chen, Y.L.; Lin, P.C.; Chen, S.P.; Lin, C.C.; Tsai, N.M.; Cheng, Y.L.; Chang, W.L.; Lin, S.Z.; Harn, H.J. Activation of nonsteroidal anti-inflammatory drug-activated gene-1 via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase revealed a isochaihulactone-triggered apoptotic pathway in human lung cancer A549 cells. J. Pharmacol. Exp. Ther. 2007, 323, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.B.; Nair, P.K.; Melnick, S.J.; Resek, A.P.; Ramachandran, C. Anticancer effects of Annona glabra plant extracts in human leukemia cell lines. Anticancer Res. 2008, 28, 965–971. [Google Scholar] [PubMed]
- Tsai, N.M.; Lin, S.Z.; Lee, C.C.; Chen, S.P.; Su, H.C.; Chang, W.L.; Harn, H.J. The antitumor effects of Angelica sinensis on malignant brain tumors in vitro and in vivo. Clin. Cancer Res. 2005, 11, 3475–3484. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.L.; Su, K.J.; Chen, C.J.; Wei, C.W.; Lin, C.J.; Yiang, G.T.; Lin, S.Z.; Harn, H.J.; Chen, Y.L. Synergistic anti-tumor activity of isochaihulactone and paclitaxel on human lung cancer cells. J. Cell. Physiol. 2012, 227, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Lin, S.Z.; Chen, Y.L.; Chang, J.S.; Ho, L.I.; Liu, P.Y.; Chang, L.F.; Harn, Y.C.; Chen, S.P.; Sun, L.Y.; et al. Butylidenephthalide suppresses human telomerase reverse transcriptase (TERT) in human glioblastomas. Ann. Surg. Oncol. 2011, 18, 3514–3527. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.M.; Chen, Y.L.; Lee, C.C.; Lin, P.C.; Cheng, Y.L.; Chang, W.L.; Lin, S.Z.; Harn, H.J. The natural compound n-butylidenephthalide derived from Angelica sinensis inhibits malignant brain tumor growth in vitro and in vivo. J. Neurochem. 2006, 99, 1251–1262. [Google Scholar] [CrossRef]
- Yen, S.Y.; Chen, S.R.; Hsieh, J.; Li, Y.S.; Chuang, S.E.; Chuang, H.M.; Huang, M.H.; Lin, S.Z.; Harn, H.J.; Chiou, T.W. Biodegradable interstitial release polymer loading a novel small molecule targeting Axl receptor tyrosine kinase and reducing brain tumour migration and invasion. Oncogene 2016, 35, 2156–2165. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, H.-C.; Chen, C.-M.; Chi, C.-S.; Tsai, J.-D.; Chiang, K.-L.; Chang, Y.-K.; Lin, S.-Z.; Harn, H.-J. Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma. Int. J. Mol. Sci. 2019, 20, 200. https://doi.org/10.3390/ijms20010200
Fan H-C, Chen C-M, Chi C-S, Tsai J-D, Chiang K-L, Chang Y-K, Lin S-Z, Harn H-J. Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma. International Journal of Molecular Sciences. 2019; 20(1):200. https://doi.org/10.3390/ijms20010200
Chicago/Turabian StyleFan, Hueng-Chuen, Chuan-Mu Chen, Ching-Shiang Chi, Jeng-Dau Tsai, Kuo-Liang Chiang, Yu-Kang Chang, Shinn-Zong Lin, and Horng-Jyh Harn. 2019. "Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma" International Journal of Molecular Sciences 20, no. 1: 200. https://doi.org/10.3390/ijms20010200