Chromatographic Separation of Phenolic Compounds from Extra Virgin Olive Oil: Development and Validation of a New Method Based on a Biphenyl HPLC Column





Abstract
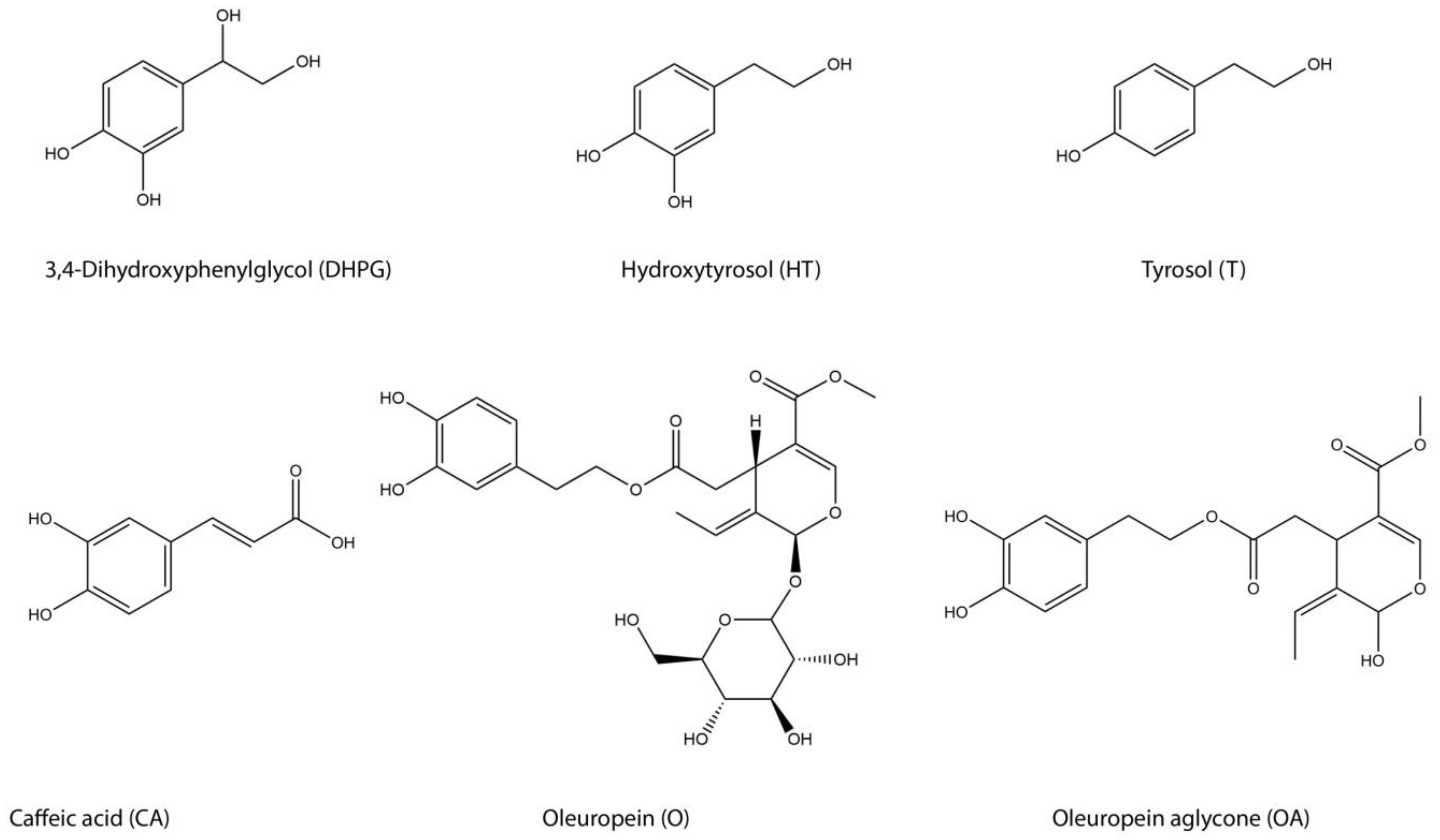
:1. Introduction
2. Results and Discussion
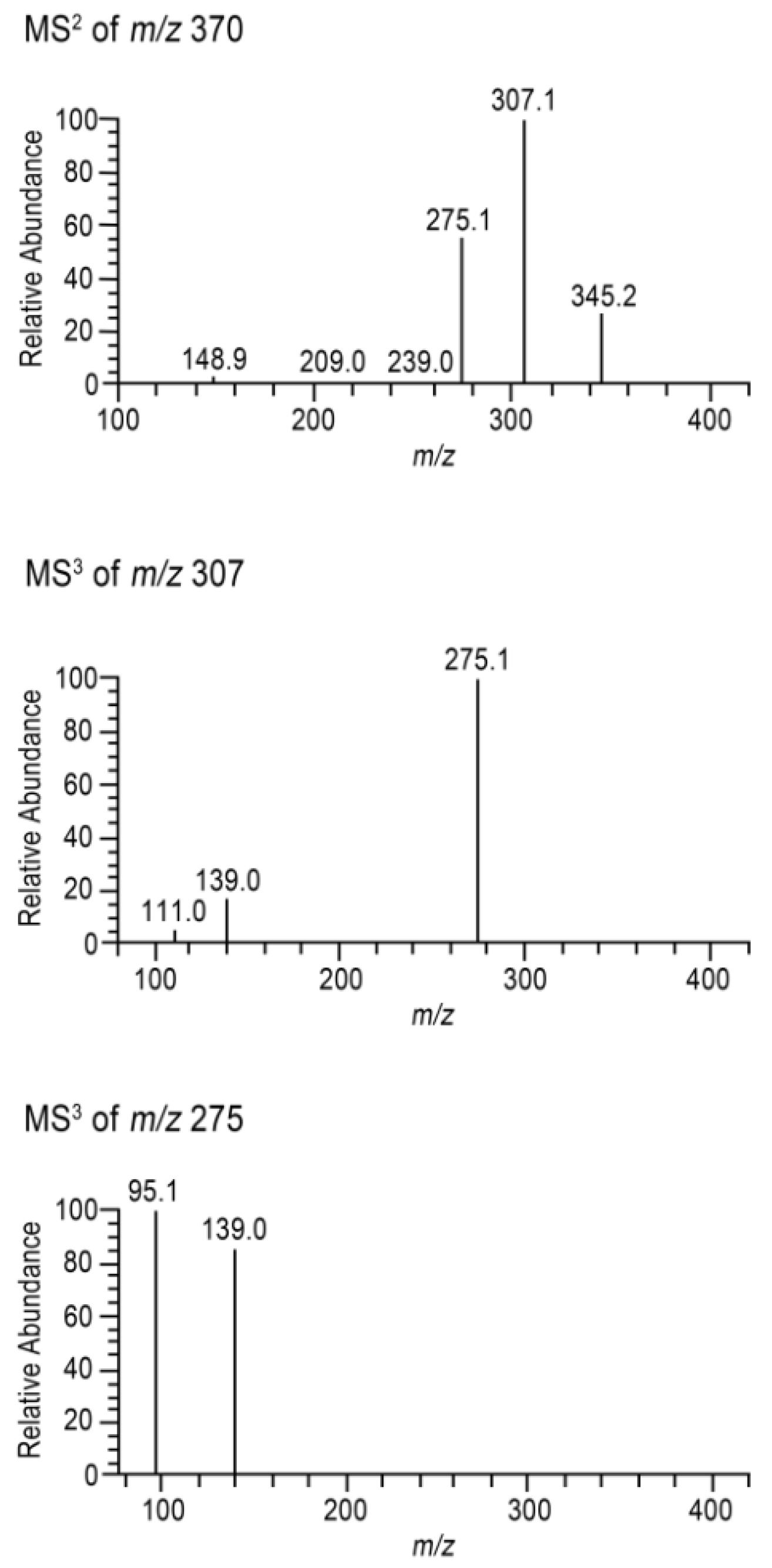
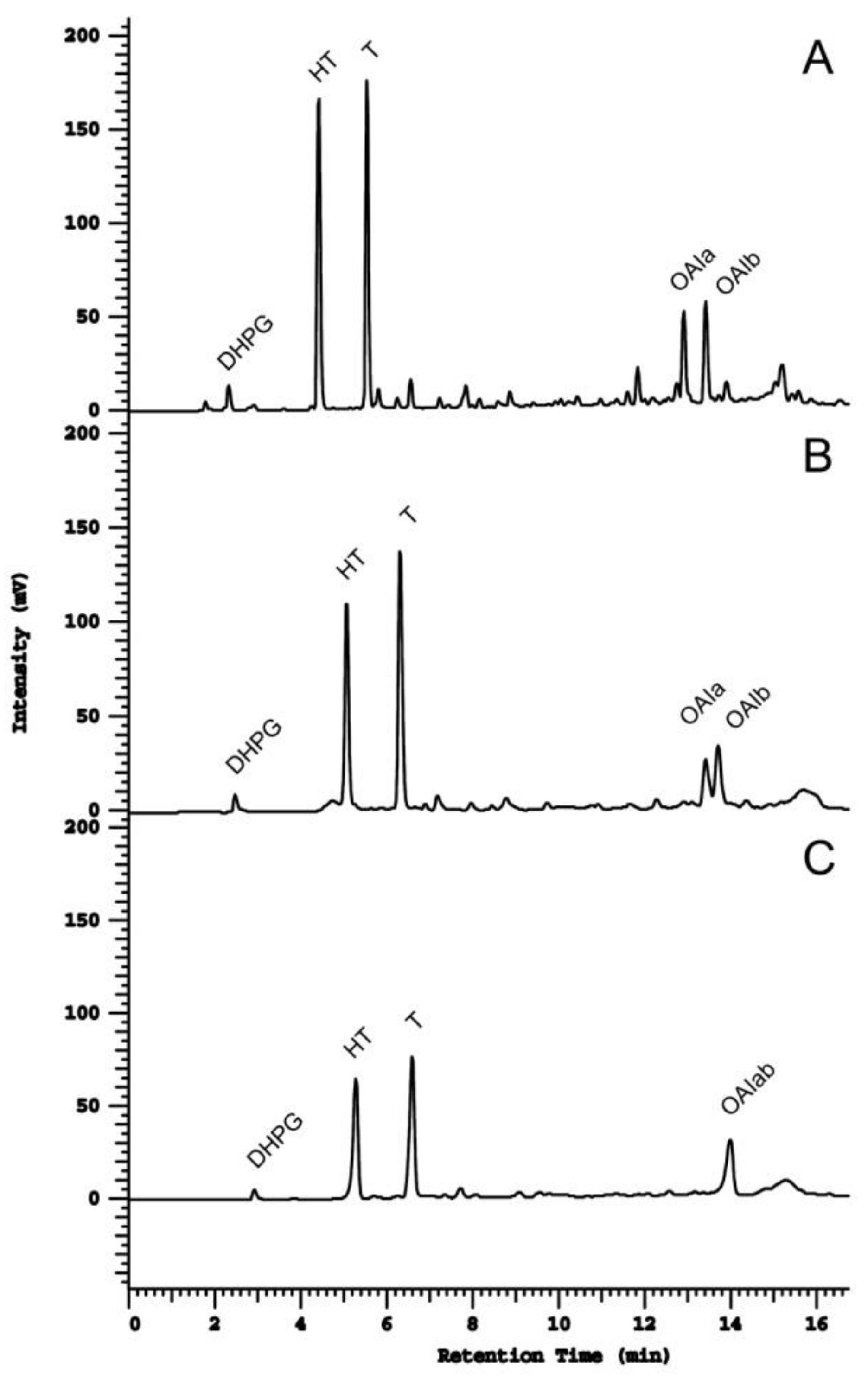
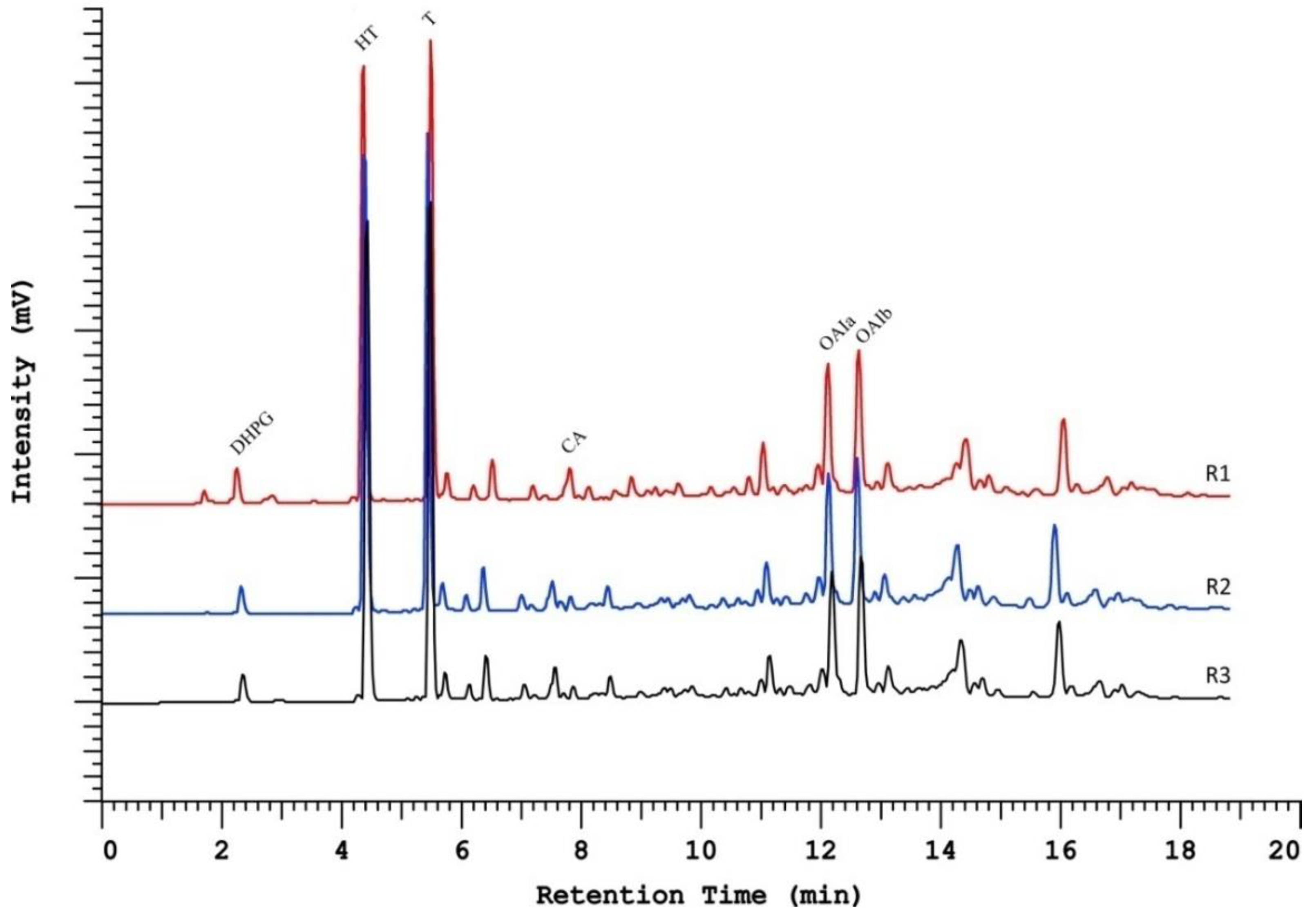
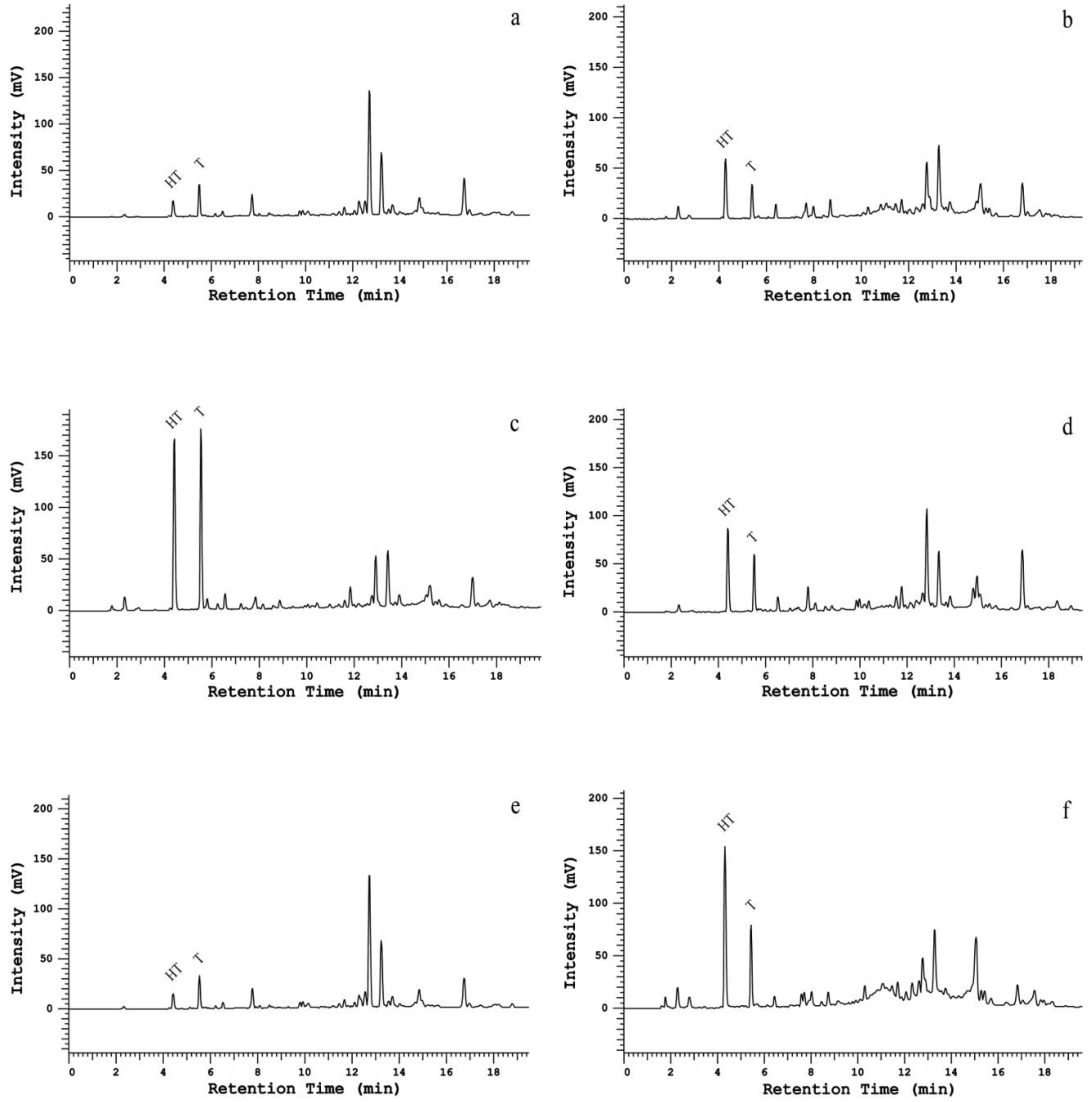
2.1. Column Comparison for Peak Identification
2.2. Resolution Parameters Comparison
2.3. Method Validation
2.3.1. Accuracy
2.3.2. Precision
2.3.3. Robustness
2.3.4. Linearity and Range
2.3.5. LOD and LOQ
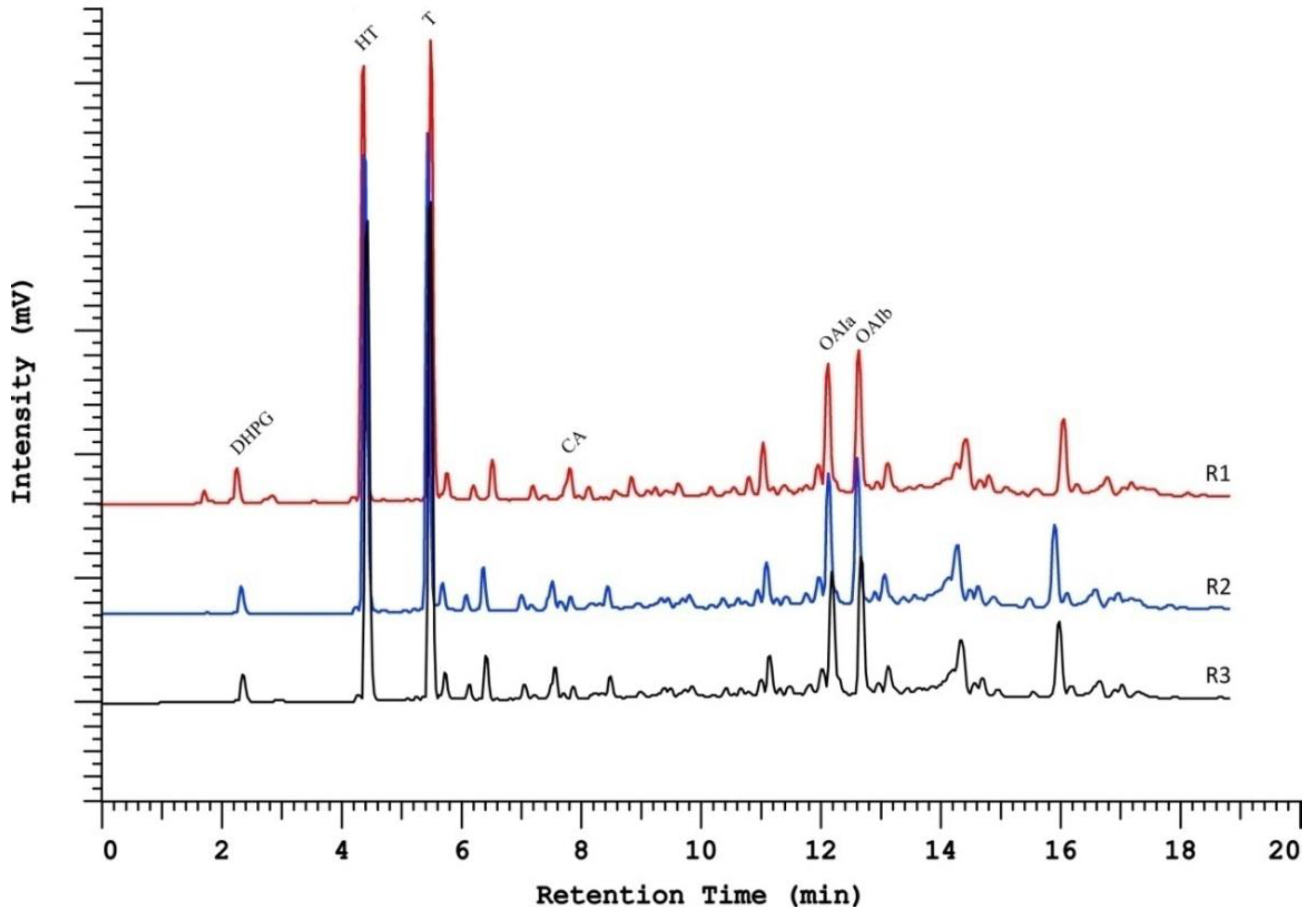
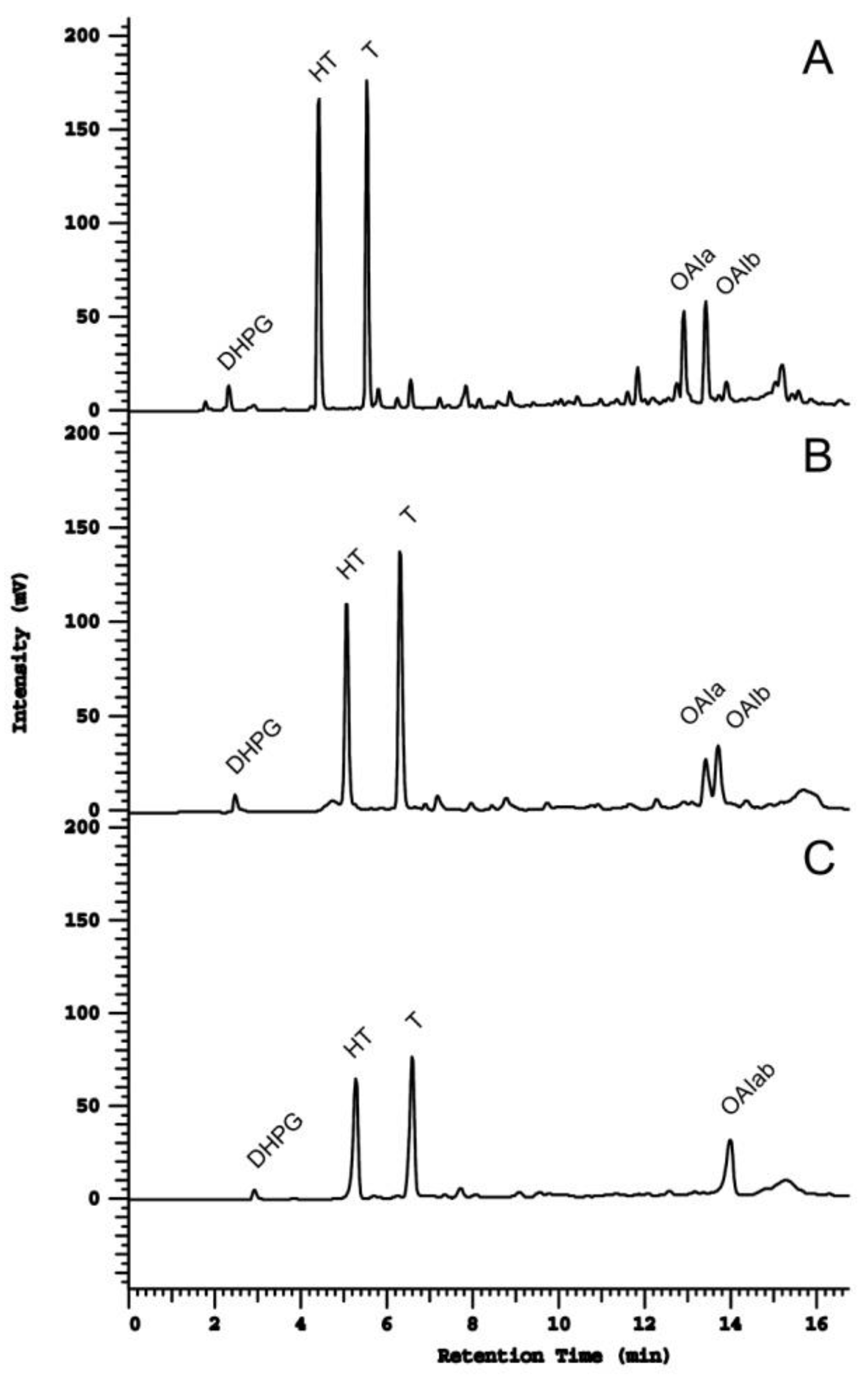
2.4. Method Application in a Set of Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. EVOO Sample
3.3. HPLC Columns
3.4. Sample Preparation
3.5. HPLC Analysis
3.6. UHPLC and Tandem Mass Spectrometry Analysis
3.7. Resolution Parameters and Asymmetry Determination
3.8. Method Validation
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CID | Charge injection device |
| DHPG | Dihydroxyphenyl glycol |
| DAD | Diode-array detection |
| EVOO | Extra virgin olive oil |
| HPLC | High performance liquid chromatography |
| HT | Hydroxytyrosol |
| ICH | International Conference on Harmonization |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| LLE | Liquid–liquid extraction |
| O | Oleuropein |
| OA | Oleuropeinaglycone |
| OAI | Oleuropeinaglycone isomers |
| OO | Olive oil |
| As | Peak asymmetry |
| nc | Peak capacity |
| PDA | photodiode array |
| PC | Phenolic compounds |
| PTFE | Polytetrafluoroethylene |
| RSD | Relative standard deviation |
| Rs | Resolution |
| k | Retention factor |
| RP | Reverse-phase |
| α | Selectivity |
| SPE | Solid phase extraction |
| MSn | Tandem mass spectrometry |
| T | Tyrosol |
| UV | Ultraviolet |
References
- Panel on Dietetic Products, Nutrition and Allergies. EFSA Scientific Opinion on the substantiation of health claims related to polyphenols in olive and protection of LDL particles from oxidative damage (ID 1333, 1638, 1639, 1696, 2865), maintenance of normal blood HDL-cholesterol concentrations (ID 1639), mainte. EFSA J. 2011, 9, 1–25. [Google Scholar] [CrossRef]
- Elamin, M.; Daghestani, M.; Omer, S.; Elobeid, M.; Virk, P.; Al-Olayan, E.; Hassan, Z.; Mohammed, O.; Aboussekhra, A. Olive oil oleuropein has anti-breast cancer properties with higher efficiency on ER-negative cells. Food Chem. Toxicol. 2013, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Ruano, J.; Lopez-Miranda, J.; Fuentes, F.; Moreno, J.A.; Bellido, C.; Perez-Martinez, P.; Lozano, A.; Gómez, P.; Jiménez, Y.; Pérez Jiménez, F. Phenolic content of virgin olive oil improves ischemic reactive hyperemia in hypercholesterolemic patients. J. Am. Coll. Cardiol. 2005, 46, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Caruso, D.; Grande, S.; Bosisio, R.; Villa, M.; Galli, G.; Sirtori, C.; Galli, C. Virgin Olive Oil Study (VOLOS): Vasoprotective potential of extra virgin olive oil in mildly dyslipidemic patients. Eur. J. Nutr. 2005, 44, 1217. [Google Scholar] [CrossRef] [PubMed]
- Fitó, M.; Cladellas, M.; de la Torre, R.; Martí, J.; Alcántara, M.; Pujadas-Bastardes, M.; Marrugat, J.; Bruguera, J.; López-Sabater, M.C.; Vila, J.; et al. Antioxidant effect of virgin olive oil in patients with stable coronary heart disease: A randomized, crossover, controlled, clinical trial. Atherosclerosis 2005, 181, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Fitó, M.; de la Torre, R.; Covas, M.-I. Olive oil and oxidative stress. Mol. Nutr. Food Res. 2007, 51, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission Regulation (EU) No. 432/2012; European Commission: Brussels, Belgium, 2012. [Google Scholar]
- Reboredo-Rodríguez, P.; Valli, E.; Bendini, A.; Di Lecce, G.; Simal-Gándara, J.; Gallina Toschi, T. A widely used spectrophotometric assay to quantify olive oil biophenols according to the health claim (EU Reg. 432/2012). Eur. J. Lipid Sci. Technol. 2016, 118, 1593–1599. [Google Scholar] [CrossRef]
- Bartella, L.; Mazzotti, F.; Napoli, A.; Sindona, G.; Di Donna, L. A comprehensive evaluation of tyrosol and hydroxytyrosol derivatives in extra virgin olive oil by microwave-assisted hydrolysis and HPLC-MS/MS. Anal. Bioanal. Chem. 2018, 410, 2193–2201. [Google Scholar] [CrossRef]
- Servili, M.; Montedoro, G. Contribution of phenolic compounds to virgin olive oil quality. Eur. J. Lipid Sci. Technol. 2002, 104, 602–613. [Google Scholar] [CrossRef]
- El Riachy, M.; Priego-Capote, F.; León, L.; Rallo, L.; Luque de Castro, M.D. Hydrophilic antioxidants of virgin olive oil. Part 1: Hydrophilic phenols: A key factor for virgin olive oil quality. Eur. J. Lipid Sci. Technol. 2011, 113, 678–691. [Google Scholar] [CrossRef]
- Caponio, F.; Gomes, T.; Pasqualone, A. Phenolic compounds in virgin olive oils: Influence of the degree of olive ripeness on organoleptic characteristics and shelf-life. Eur. Food Res. Technol. 2001, 212, 329–333. [Google Scholar] [CrossRef]
- Vitaglione, P.; Savarese, M.; Paduano, A.; Scalfi, L.; Fogliano, V.; Sacchi, R. Healthy virgin olive oil: A matter of bitterness. Crit. Rev. Food Sci. Nutr. 2015, 55, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, I.; Esposto, S.; Taticchi, A.; Selvaggini, R.; Veneziani, G.; Urbani, S.; Servili, M. HPLC-ESI-MS investigation of tyrosol and hydroxytyrosol oxidation products in virgin olive oil. Food Chem. 2011, 125, 21–28. [Google Scholar] [CrossRef]
- Brenes, M.; Garcia, A.; Garcia, P.; Garrido, A. Acid hydrolysis of secoiridoid aglycons during storage of virgin olive oil. J. Agric. Food Chem. 2001, 49, 5609–5614. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, G.; Lama, A.; Jaramillo, S.; Fuentes-Alventosa, J.M.; Guillen, R.; Ana, J.A.; Rocío, R.A.; Juan, F.B. 3,4-Dihydroxyphenylglycol (DHPG): An important phenolic compound present in natural table olives. J. Agric. Food Chem. 2009, 57, 6298–6304. [Google Scholar] [CrossRef]
- Lama-Muñoz, A.; Rodríguez-Gutiérrez, G.; Rubio-Senent, F.; Palacios-Díaz, R.; Fernández-Bolaños, J. A study of the precursors of the natural antioxidant phenol 3,4-dihydroxyphenylglycol in olive oil waste. Food Chem. 2013, 140, 154–160. [Google Scholar] [CrossRef]
- Sun-Waterhouse, D.; Zhou, J.; Miskelly, G.M.; Wibisono, R.; Wadhwa, S.S. Stability of encapsulated olive oil in the presence of caffeic acid. Food Chem. 2011, 126, 1049–1056. [Google Scholar] [CrossRef]
- Carrasco-Pancorbo, A.; Cerretani, L.; Bendini, A.; Segura-Carretero, A.; Gallina-Toschi, T.; Fernández-Gutiérrez, A. Analytical determination of polyphenols in olive oils. J. Sep. Sci. 2005, 28, 837–858. [Google Scholar] [CrossRef]
- Garcia, B.; Coelho, J.; Costa, M.; Pinto, J.; Paiva-Martins, F. A simple method for the determination of bioactive antioxidants in virgin olive oils. J. Sci. Food Agric. 2013, 93, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Sun, X.; Liu, P.; Li, X.; Jiang, R. Selectivity Differences Between ODS and Polar Modified Stationary Phases in RP-LC. Chromatographia 2010, 72, 23–32. [Google Scholar] [CrossRef]
- Layne, J. Characterization and comparison of the chromatographic performance of conventional, polar-embedded, and polar-endcapped reversed-phase liquid chromatography stationary phases. J. Chromatogr. A 2002, 957, 149–164. [Google Scholar] [CrossRef]
- Baker, J.S.; Vinci, J.C.; Moore, A.D.; Colón, L.A. Physical characterization and evaluation of HPLC columns packed with superficially porous particles. J. Sep. Sci. 2010, 33, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Ricciutelli, M.; Marconi, S.; Boarelli, M.C.; Caprioli, G.; Sagratini, G.; Ballini, R.; Fiorini, D. Olive oil polyphenols: A quantitative method by high-performance liquid-chromatography-diode-array detection for their determination and the assessment of the related health claim. J. Chromatogr. A 2017, 1481, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, A.L.; Cavaliere, C.; Crescenzi, C.; Foglia, P.; Nescatelli, R.; Samperi, R.; Laganà, A. Comparison of extraction methods for the identification and quantification of polyphenols in virgin olive oil by ultra-HPLC-QToF mass spectrometry. Food Chem. 2014, 158, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Arráez-Román, D.; Menéndez, J.A.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Characterization of isomers of oleuropein aglycon in olive oils by rapid-resolution liquid chromatography coupled to electrospray time-of-flight and ion trap tandem. Rapid Commun. Mass Spectrom. 2009, 23, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Tsantili, E.; Kafkaletou, M.; Roussos, P.A.; Christopoulos, M.V. Phenolic compounds, maturation and quality in fresh green olives for table use during exposure at 20 degrees C after preharvest ReTain treatment. Sci. Hortic. (Amsterdam) 2012, 140, 26–32. [Google Scholar] [CrossRef]
- Granados-Principal, S.; Quiles, J.L.; Ramirez-Tortosa, C.L.; Sanchez-Rovira, P.; Ramirez-Tortosa, M.C. Hydroxytyrosol: From laboratory investigations to future clinical trials. Nutr. Rev. 2010, 68, 191–206. [Google Scholar] [CrossRef]
- Tsimidou, M.; Papadopoulos, G.; Boskou, D. Determination of phenolic compounds in virgin olive oil by reversed-phase HPLC with emphasis on UV detection. Food Chem. 1992, 44, 53–60. [Google Scholar] [CrossRef]
- Donato, P.; Dugo, P.; Cacciola, F.; Dugo, G.; Mondello, L. High peak capacity separation of peptides through the serial connection of LC shell-packed columns. J. Sep. Sci. 2009, 32, 1129–1136. [Google Scholar] [CrossRef]
- Reubsaet, E.; Vieskar, R.; Leon, J. Characterisation of p–p interactions which determine retention of aromatic compounds in reversed-phase liquid chromatography. J. Chromatogr. A 2000, 841, 147–154. [Google Scholar] [CrossRef]
- Bell, D.S.; Brandes, H.K.; Wallworth, D. Recent Trends and Advances in Superficially Porous Particle Technology: Application to Large Molecule Separations. Chromatogr. Today 2015, 8, 4–8. [Google Scholar]
- Rathore, A.S.; Kennedy, R.M.; O’Donnell, J.K.; Bemberis, I.; Kaltenbrunner, O. Qualification of a chromatographic column: Why and how to do it. BioPharm Int. 2003, 16, 30–40. [Google Scholar]
- ICH Expert Working Group. Validation of Analytical Procedures: Text and Methodology; ICH Secretariat: Geneva, Switzerland, 2005. [Google Scholar]
- Al-Rimawi, F. Development and validation of a simple reversed-phase HPLC-UV method for determination of oleuropein in olive leaves. J. Food Drug Anal. 2014, 22, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Taamalli, A.; Arráez Román, D.; Zarrouk, M.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Classification of “Chemlali” accessions according to the geographical area using chemometric methods of phenolic profiles analysed by HPLC–ESI-TOF–MS. Food Chem. 2012, 132, 561–566. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Pacchiarotta, T.; Carrasco-Pancorbo, A.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Deelder, A.M.; Mayboroda, O.A. Gas chromatography-atmospheric pressure chemical ionization-time of flight mass spectrometry for profiling of phenolic compounds in extra virgin olive oil. J. Chromatogr. A 2011, 1218, 959–971. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization Geneva. Accuracy (Trueness and Precision) of Measurement Methods and Results; ISO 5725; International Organization for Standardization: Geneva, Switzerland, 1998. [Google Scholar]
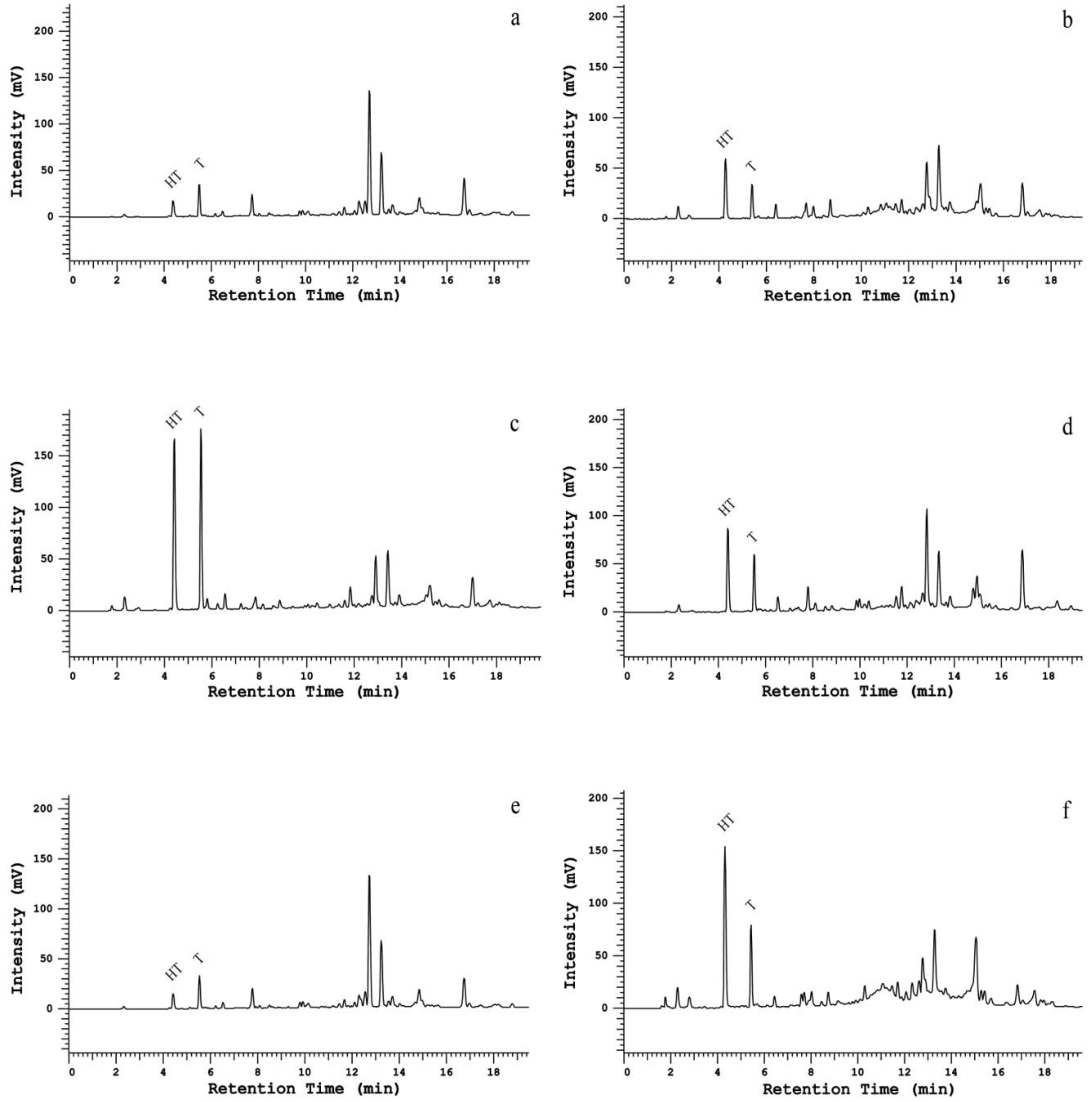

{kind=link}
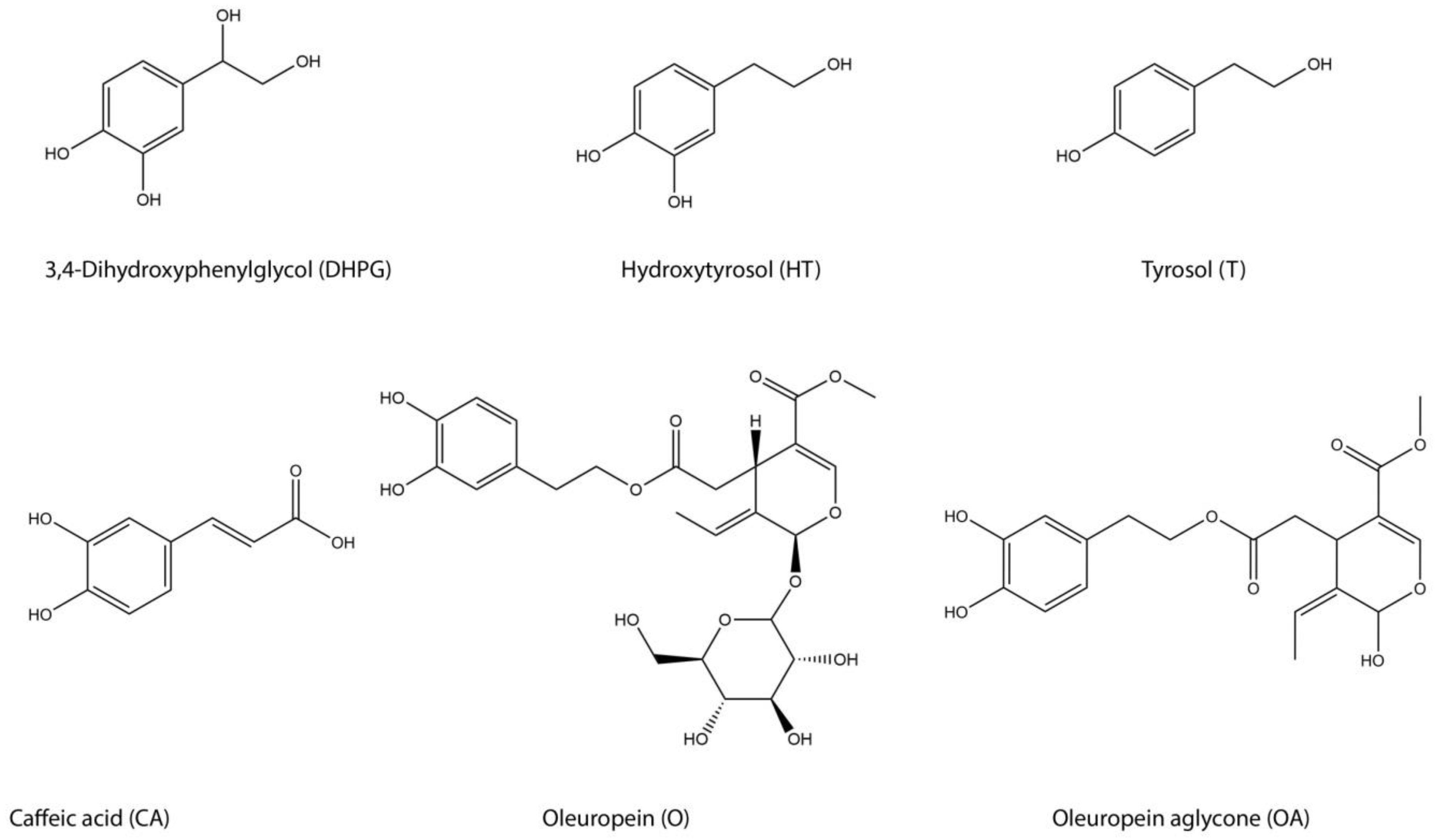
{kind=link}
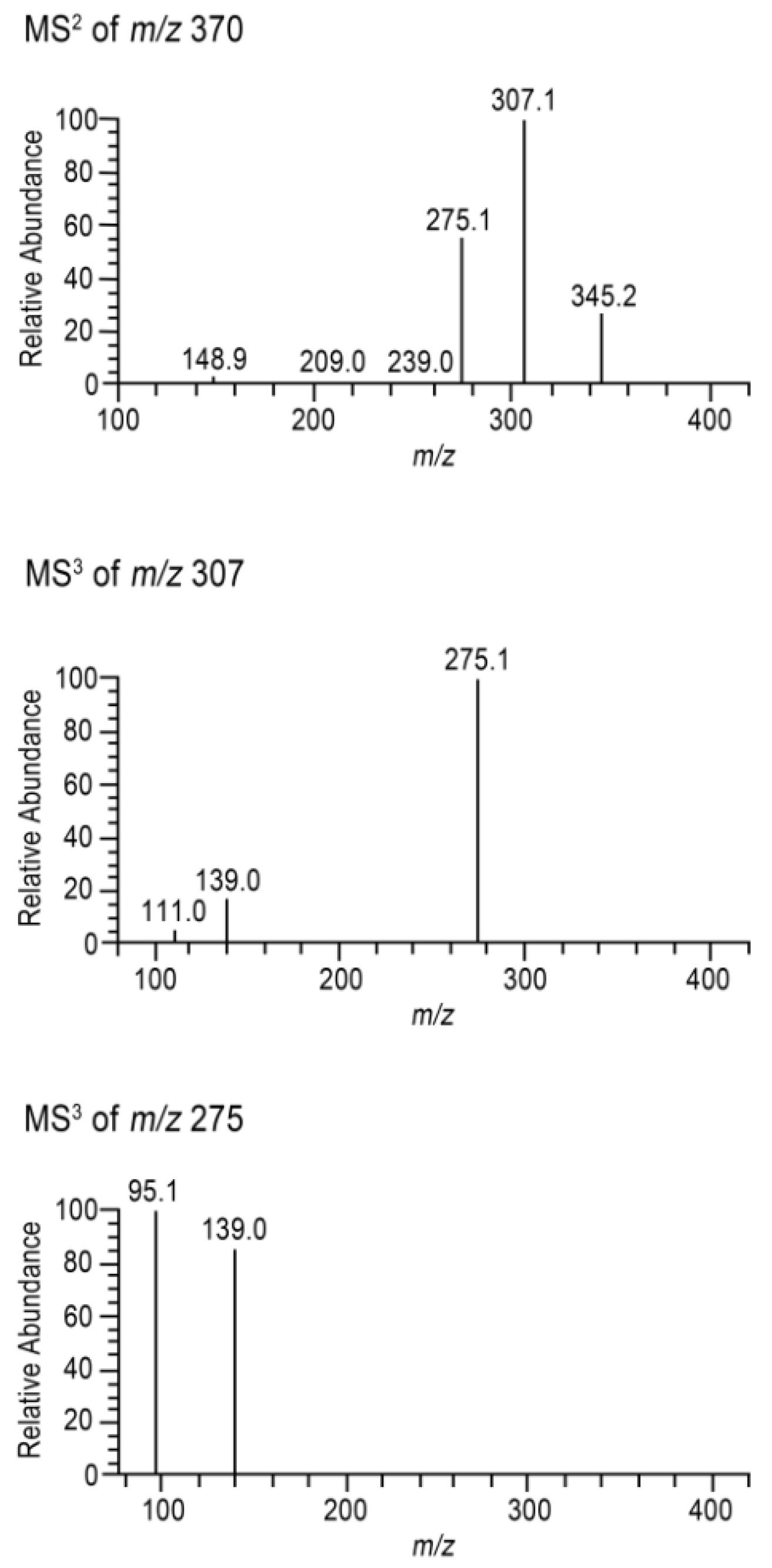
{kind=link}
{kind=link}
{kind=link}
| Column | nc | α | k | Rs | ||
|---|---|---|---|---|---|---|
| HT–T | OAIa–OAIb | HT | HT–T | OAa–OAb | ||
| Kinetex biphenyl | 277 | 1.43 | 1.045 | 1.46 | 5.60 | 2.68 |
| LiChrospher C18 | 192 | 1.38 | 1.024 | 1.81 | 5.39 | 1.12 |
| Spherisorb C18 | 179 | 1.37 | 1.0 | 1.94 | 3.91 | 0 |
| Identified EVOO PCs | Type of Column | |||
|---|---|---|---|---|
| Kinetex Biphenyl | LiChrospher C18 | Spherisorb C18 | ||
| DHPG | Dihydroxyphenyl glycol | 1.333 | 2.375 | 1.541 |
| HT | Hidroxytyrosol | 1.25 | 1.143 | 0.615 |
| T | Tyrosol | 1.364 | 1.500 | 1.437 |
| CA | Caffeic acid | 1.2 | 1.353 | n/d 1 |
| OAIa | Oleuropein aglycone isomer a | 1.333 | 1.555 | 0.468 2 |
| OAIb | Oleuropein aglycone isomer b | 1.25 | 1.368 | |
| Analyte Concentration (mg/L) | % Recovery ± SD | |
|---|---|---|
| HT | T | |
| 60 | 101.4 ± 1.6 | 104.2 ± 1.8 |
| 150 | 105.1 ± 2.2 | 105.2 ± 0.5 |
| 260 | 99.2 ± 0.8 | 100.2 ± 1.3 |
| Method Changes | Rs (HT-T) ± SD |
|---|---|
| No change | 5.9 ± 0.2 |
| Flow rate = 1.4 mL/min | 5.91± 0.03 |
| Flow rate = 1.6 mL/min | 4.99 ± 0.06 |
| Wavelength = 278 nm | 5.6 ± 0.2 |
| Wavelength = 282 nm | 5.7 ± 0.2 |
| Gradient method = −2% for eluent B | 5.0 ± 0.1 |
| Gradient method = +2% for eluent B | 6.15 ± 0.05 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferro, M.D.; Santos, S.A.O.; Silvestre, A.J.D.; Duarte, M.F. Chromatographic Separation of Phenolic Compounds from Extra Virgin Olive Oil: Development and Validation of a New Method Based on a Biphenyl HPLC Column. Int. J. Mol. Sci. 2019, 20, 201. https://doi.org/10.3390/ijms20010201
Ferro MD, Santos SAO, Silvestre AJD, Duarte MF. Chromatographic Separation of Phenolic Compounds from Extra Virgin Olive Oil: Development and Validation of a New Method Based on a Biphenyl HPLC Column. International Journal of Molecular Sciences. 2019; 20(1):201. https://doi.org/10.3390/ijms20010201
Chicago/Turabian StyleFerro, Miguel D., Sónia A. O. Santos, Armando J. D. Silvestre, and Maria F. Duarte. 2019. "Chromatographic Separation of Phenolic Compounds from Extra Virgin Olive Oil: Development and Validation of a New Method Based on a Biphenyl HPLC Column" International Journal of Molecular Sciences 20, no. 1: 201. https://doi.org/10.3390/ijms20010201
APA StyleFerro, M. D., Santos, S. A. O., Silvestre, A. J. D., & Duarte, M. F. (2019). Chromatographic Separation of Phenolic Compounds from Extra Virgin Olive Oil: Development and Validation of a New Method Based on a Biphenyl HPLC Column. International Journal of Molecular Sciences, 20(1), 201. https://doi.org/10.3390/ijms20010201