Fibroblast Growth Factor 21 Stimulates Pancreatic Islet Autophagy via Inhibition of AMPK-mTOR Signaling


Abstract
1. Introduction
2. Results
2.1. FGF21 Analog CVX343 Improves Glucose Homeostasis and Body Weight in HFD-Induced T2DM Mice
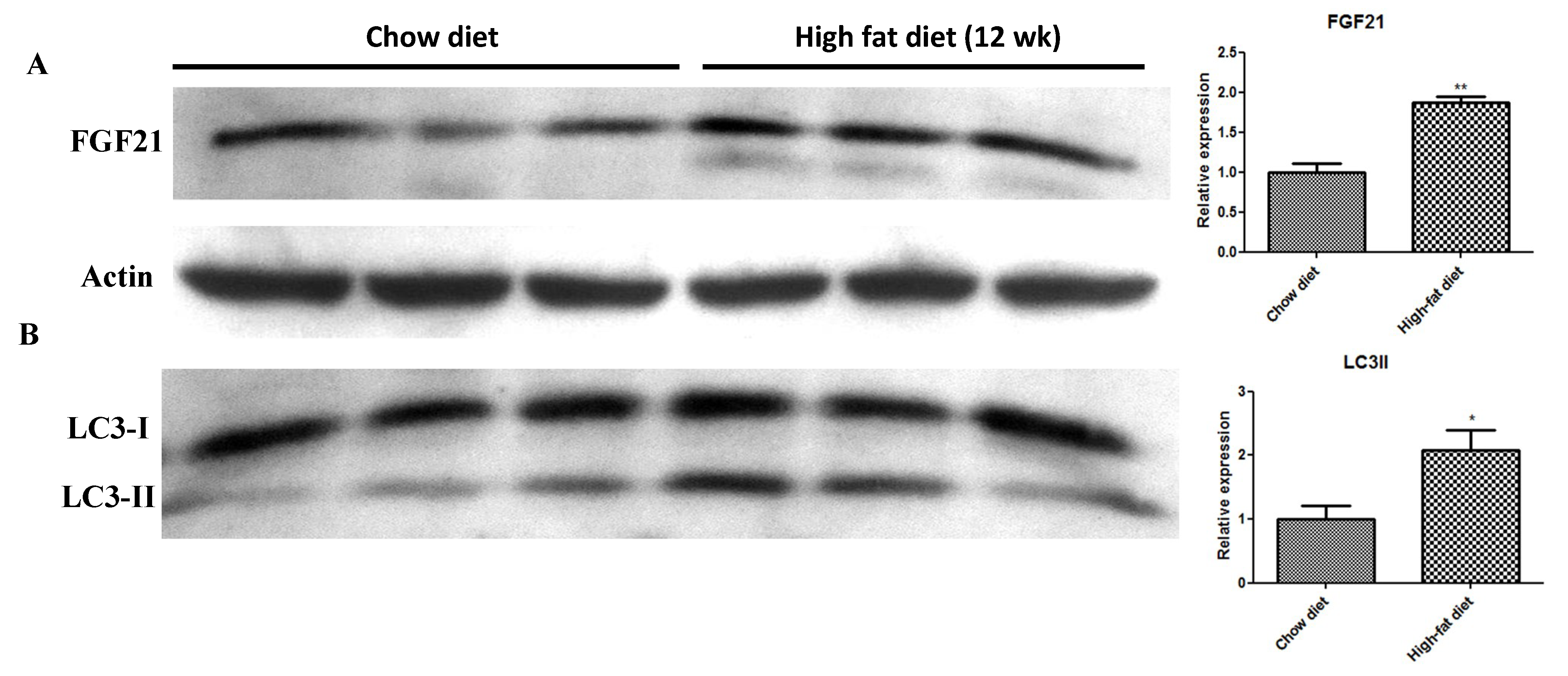
2.2. In Vivo HFD-Induced T2DM and Ex Vivo High-Glucose/High-PA Treatment Upregulate Protein Expression of FGF21 and LC3-II in Pancreatic Islets
2.3. Pancreatic Islets Isolated from FGF21 KO Mice and INS-1E β-Cells with FGF21 Knockdown Display Diminished Autophagy Induction
2.4. In Vivo HFD, Ex Vivo High-Glucose/High-PA, and Exogenous Recombinant FGF21 Treatments Each Reduced AMPK Phosphorylation in Pancreatic Islets
2.5. Compound C Suppressed mTOR Phosphorylation but Increased LC3-II Expression
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. FGF21 Analog Supplementation
4.3. Body Weight and In Vivo Glucose Homeostasis
4.4. Pancreatic Islet Isolation and Treatments
4.5. INS-1E Cell Culture and Treatments
4.6. Knockdown of FGF21 Transcription
4.7. Reverse Transcriptase (RT)-PCR and Real-Time PCR Analysis
4.8. Western Blot Analysis
4.9. Immunohistochemistry
4.10. Detection of Autophagosome Formation
4.11. Data Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FGF21 | Fibroblast Growth Factor 21 |
| HFD | High-fat Diet |
| KO | Knockout |
References
- International Diabetes Federation. Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017. [Google Scholar]
- Prentki, M.; Nolan, C.J. Islet β cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Del Prato, S. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet. Med. 2009, 26, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- So, W.Y.; Leung, P.S. Fibroblast Growth Factor 21 As an Emerging Therapeutic Target for Type 2 Diabetes Mellitus. Med. Res. Rev. 2016, 36, 672–704. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.O.; Molina-Carrion, M.; Abdul-Ghani, M.A.; Folli, F.; DeFronzo, R.A.; Tripathy, D. Circulating Fibroblast Growth Factor-21 Is Elevated in Impaired Glucose Tolerance and Type 2 Diabetes and Correlates with Muscle and Hepatic Insulin Resistance. Diabetes Care 2009, 32, 1542–1546. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Chui, P.C.; Antonellis, P.J.; Bina, H.A.; Kharitonenkov, A.; Flier, J.S.; Maratos-Flier, E. Obesity Is a Fibroblast Growth Factor 21 (FGF21)-Resistant State. Diabetes 2010, 59, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Wente, W.; Efanov, A.M.; Brenner, M.; Kharitonenkov, A.; Köster, A.; Sandusky, G.E.; Sewing, S.; Treinies, I.; Zitzer, H.; Gromada, J. Fibroblast Growth Factor-21 Improves Pancreatic β-Cell Function and Survival by Activation of Extracellular Signal–Regulated Kinase 1/2 and Akt Signaling Pathways. Diabetes 2006, 55, 2470–2478. [Google Scholar] [CrossRef]
- So, W.Y.; Cheng, Q.; Chen, L.; Evans-Molina, C.; Xu, A.; Lam, K.S.; Leung, P.S. High glucose represses β-klotho expression and impairs fibroblast growth factor 21 action in mouse pancreatic islets: Involvement of peroxisome proliferator-activated receptor gamma signaling. Diabetes 2013, 62, 3751–3759. [Google Scholar] [CrossRef]
- So, W.Y.; Cheng, Q.; Xu, A.; Lam, K.S.L.; Leung, P.S. Loss of fibroblast growth factor 21 action induces insulin resistance, pancreatic islet hyperplasia and dysfunction in mice. Cell Death Dis. 2015, 6, e1707. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Las, G.; Shirihai, O.S. The role of autophagy in β-cell lipotoxicity and type 2 diabetes. Diabetes Obes. Metab. 2010, 12, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.D.; Lee, M.-S.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.I.; Watada, H.; Wiley, J.W. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.Y.; O’Reilly, L.; Ramm, G.; Biden, T.J. High-fat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia 2015, 58, 2074–2078. [Google Scholar] [CrossRef] [PubMed]
- Martino, L.; Masini, M.; Novelli, M.; Beffy, P.; Bugliani, M.; Marselli, L.; Masiello, P.; Marchetti, P.; De Tata, V. Palmitate Activates Autophagy in INS-1E β-Cells and in Isolated Rat and Human Pancreatic Islets. PLoS ONE 2012, 7, e36188. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S. Role of islet β cell autophagy in the pathogenesis of diabetes. Trends Endocrinol. Metab. 2014, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of Beta Cell Mass in Response to High-Fat Diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Free Fatty Acids, Insulin Resistance, and Type 2 Diabetes Mellitus. Proc. Assoc. Am. Physicians 1999, 111, 241–248. [Google Scholar] [CrossRef]
- Boden, G.; She, P.; Mozzoli, M.; Cheung, P.; Gumireddy, K.; Reddy, P.; Xiang, X.; Luo, Z.; Ruderman, N. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-κB pathway in rat liver. Diabetes 2005, 54, 3458–3465. [Google Scholar] [CrossRef]
- Yin, J.J.; Li, Y.B.; Cao, M.M.; Wang, Y. Liraglutide Improves the Survival of INS-1 Cells by Promoting Macroautophagy. Int. J. Endocrinol. Metab. 2013, 11, 184–190. [Google Scholar]
- Jiang, Y.; Huang, W.; Wang, J.; Xu, Z.; He, J.; Lin, X.; Zhou, Z.; Zhang, J. Metformin Plays a Dual Role in MIN6 Pancreatic β Cell Function through AMPK-dependent Autophagy. Int. J. Boil. Sci. 2014, 10, 268–277. [Google Scholar] [CrossRef]
- Gleason, C.E.; Lu, D.; Witters, L.A.; Newgard, C.B.; Birnbaum, M.J. The role of AMPK and mTOR in nutrient sensing in pancreatic B-cells. J. Biol. Chem. 2007, 282, 10341–10351. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Boil. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Leclerc, J.; Hébrard, S.; Lantier, L.; Mounier, R.; Andreelli, F.; Foretz, M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: From physiology to therapeutic perspectives. Acta Physiol. 2009, 196, 81–98. [Google Scholar] [CrossRef]
- Thomson, D.M.; Winder, W.W. AMPK Control of Fat Metabolism in Skeletal Muscle. Acta Physiol. 2009, 196, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ma, L.; Wu, Y.; Ye, X.; Zhang, T.; Rasoul, L.M.; Liu, Y.; Guo, M.; Zhou, B.; Ren, G.; et al. FGF21 treatment ameliorates alcoholic fatty liver through activation of AMPK-SIRT1 pathway. Acta Biochim. Biophys. Sin. 2014, 46, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.D.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK and autophagy get connected. EMBO J. 2011, 30, 634–635. [Google Scholar] [CrossRef]
- Sun, Y.; Ren, M.; Gao, G.Q.; Gong, B.; Xin, W.; Guo, H.; Zhang, X.J.; Gao, L.; Zhao, J.J. Chronic palmitate exposure inhibits AMPKα and decreases glucose-stimulated insulin secretion from β-cells: Modulation by fenofibrate. Acta Pharmacol. Sin. 2008, 29, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, L.; Lu, Y.; Zhang, J.; Jian, F.; Liu, Y.; Li, F.; Li, W.; Wang, X.; Li, G. Activation of SIRT1 protects pancreatic β-cells against palmitate-induced dysfunction. Biochim. Biophys. Acta Mol. Basis 2012, 1822, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Boil. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Palanivel, R.; Rai, E.; Park, M.; Gabor, T.V.; Scheid, M.P.; Xu, A.; Sweeney, G. Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high-fat diet feeding in mice. Diabetes 2015, 64, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, Y.; Gu, J.; Wang, S.; Zhou, S.; Wang, Y.; Tan, Y.; Feng, W.; Fu, Y.; Mellen, N.; et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin. Sci. 2016, 130, 625–641. [Google Scholar] [CrossRef]
- Zhu, S.; Wu, Y.; Ye, X.; Ma, L.; Qi, J.; Yu, D.; Wei, Y.; Lin, G.; Ren, G.; Li, D. FGF21 ameliorates nonalcoholic fatty liver disease by inducing autophagy. Mol. Cell. Biochem. 2016, 420, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ishino, T.; Chen, G.; Rolzin, P.; Osothprarop, T.F.; Retting, K.; Li, L.; Jin, P.; Matin, M.J.; Huyghe, B.; et al. Development of a Novel Long-Acting Antidiabetic FGF21 Mimetic by Targeted Conjugation to a Scaffold Antibody. J. Pharmacol. Exp. Ther. 2013, 346, 270–280. [Google Scholar] [CrossRef]
- Morrice, N.; Mcilroy, G.D.; Tammireddy, S.R.; Reekie, J.; Shearer, K.D.; Doherty, M.K.; Delibegović, M.; Whitfield, P.D.; Mody, N. Elevated Fibroblast growth factor 21 in obese, insulin resistant states is normalised by the synthetic retinoid Fenretinide in mice. Sci. Rep. 2017, 7, 43782. [Google Scholar] [CrossRef] [PubMed]
- Mraz, M.; Bartlova, M.; Lacinová, Z.; Michalsky, D.; Kasalický, M.; Haluzíková, D.; Matoulek, M.; Dostalova, I.; Humenanska, V.; Haluzik, M. Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin. Endocrinol. 2009, 71, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast Growth Factor 21 Corrects Obesity in Mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef]
- Véniant, M.M.; Komorowski, R.; Chen, P.; Stanislaus, S.; Winters, K.; Hager, T.; Zhou, L.; Wada, R.; Hecht, R.; Xu, J. Long-Acting FGF21 Has Enhanced Efficacy in Diet-Induced Obese Mice and in Obese Rhesus Monkeys. Endocrinology 2012, 153, 4192–4203. [Google Scholar] [CrossRef]
- Xu, J.; Stanislaus, S.; Chinookoswong, N.; Lau, Y.Y.; Hager, T.; Patel, J.; Ge, H.; Weiszmann, J.; Lu, S.-C.; Graham, M.; et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models—Association with liver and adipose tissue effects. Am. J. Physiol. Metab. 2009, 297, E1105–E1114. [Google Scholar] [CrossRef] [PubMed]
- Singhal, G.; Fisher, F.M.; Chee, M.J.; Tan, T.G.; Ouaamari, A.E.; Adams, A.C.; Najarian, R.; Kulkarni, R.N.; Benoist, C.; Flier, J.S.; et al. Fibroblast growth factor 21protects against high fat diet induced inflammation and islet hyperplasia in pancreas. PLoS ONE 2016, 11, e0148252. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, X.H.; Ebersole, B.; Ribnicky, D.; Wang, Z.Q. Bitter melon extract attenuating hepatic steatosis may be mediated by FGF21 and AMPK/Sirt1 signaling in mice. Sci. Rep. 2013, 3, 3142. [Google Scholar] [CrossRef]
- Vucicevic, L.; Misirkic, M.; Kristina, J.; Vilimanovich, U.; Sudar-Milovanovic, E.; Isenović, E.; Prica, M.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Vladimir, B.; et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy 2011, 7, 40–50. [Google Scholar] [CrossRef]
- Bartolomé, A.; Kimura-Koyanagi, M.; Asahara, S.; Guillén, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; García-Aguilar, A.; Koike, M.; et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef]
- Chen, Z.-F.; Li, Y.-B.; Han, J.-Y.; Wang, J.; Yin, J.-J.; Li, J.-B.; Tian, H. The double-edged effect of autophagy in pancreatic beta cells and diabetes. Autophagy 2011, 7, 12–16. [Google Scholar] [CrossRef]
- Fujimoto, K.; Hanson, P.T.; Tran, H.; Ford, E.L.; Han, Z.; Johnson, J.D.; Schmidt, R.E.; Green, K.G.; Wice, B.M.; Polonsky, K.S. Autophagy Regulates Pancreatic Beta Cell Death in Response to Pdx1 Deficiency and Nutrient Deprivation. J. Boil. Chem. 2009, 284, 27664–27673. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Bergmann, A. Autophagy and Cell Death: No Longer at Odds. Cell 2007, 131, 1032–1034. [Google Scholar] [CrossRef] [PubMed]
- Merglen, A.; Theander, S.; Rubi, B.; Chaffard, G.; Wollheim, C.B.; Maechler, P. Glucose Sensitivity and Metabolism-Secretion Coupling Studied during Two-Year Continuous Culture in INS-1E Insulinoma Cells. Endocrinology 2004, 145, 667–678. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| Mouse GAPDH | GCACAGTCAAGGCCGAGAAT | GCCTTCTCCATGGTGGTGAA |
| Mouse FGF21 | CQTCTGCCTCAGAAGGACTC | AAGGCTCTACCATGCTCAGG |
| Rat β-actin | TTTAATGTCACGCACGATTTC | CCCATCTATGAGGGTTACGC |
| Rat FGF21 | AGATCAGGGAGGACGGAACA | TCAGGATCAAAGTGAGGCGAT |
| Negative control siRNA | UUCUCCGAACGUGUCACGUTT; ACGUGACACGUUCGGAGAATT | |
| Rat siRNA-FGF21 (#1) | CAACCAGAUGGAACUCUCUAUGGAU; AUCCAUAGAGAGUUCCAUCUGGUUG | |
| Rat siRNA-FGF21 (#2) | GCAGUUUCAGAGAGCUGCUGCUUAA; UUAAGCAGCAGCUCUCUGAAACUGC | |
| Rat siRNA-FGF21 (#3) | CCCUGAGCAUGGUAGAGCCUUUGCA; UGCAAAGGCUCUACCAUGCUCAGGG |
| Antibody | Dilution | Host Species | Supplier |
|---|---|---|---|
| FGF21 | 1:1000 | Rabbit | Abcam |
| LC3 | 1:1000 | Rabbit | Novus |
| p-AMPK | 1:1000 | Rabbit | Cell Signaling |
| t-AMPK | 1:1000 | Rabbit | Cell Signaling |
| β-actin | 1:1000 | Mouse | Santa Cruz |
| p-mTOR | 1:1000 | Rabbit | Cell Signaling |
| t-mTOR | 1:1000 | Rabbit | Cell Signaling |
| HRP-anti-rabbit IgG | 1:1000 | Donkey | Amersham |
| HRP-anti-mouse IgG | 1:1000 | Sheep | GE Healthcare |
| Alexa Fluor® 568 anti-rabbit IgG | 1:2000 | Donkey | Life Technologies |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.T.W.; Li, S.Y.T.; Leung, P.S. Fibroblast Growth Factor 21 Stimulates Pancreatic Islet Autophagy via Inhibition of AMPK-mTOR Signaling. Int. J. Mol. Sci. 2019, 20, 2517. https://doi.org/10.3390/ijms20102517
Cheng STW, Li SYT, Leung PS. Fibroblast Growth Factor 21 Stimulates Pancreatic Islet Autophagy via Inhibition of AMPK-mTOR Signaling. International Journal of Molecular Sciences. 2019; 20(10):2517. https://doi.org/10.3390/ijms20102517
Chicago/Turabian StyleCheng, Sam Tsz Wai, Stephen Yu Ting Li, and Po Sing Leung. 2019. "Fibroblast Growth Factor 21 Stimulates Pancreatic Islet Autophagy via Inhibition of AMPK-mTOR Signaling" International Journal of Molecular Sciences 20, no. 10: 2517. https://doi.org/10.3390/ijms20102517
APA StyleCheng, S. T. W., Li, S. Y. T., & Leung, P. S. (2019). Fibroblast Growth Factor 21 Stimulates Pancreatic Islet Autophagy via Inhibition of AMPK-mTOR Signaling. International Journal of Molecular Sciences, 20(10), 2517. https://doi.org/10.3390/ijms20102517