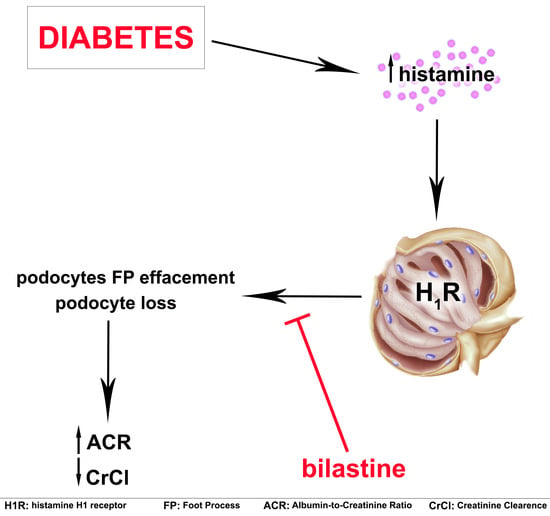
Effect of Bilastine on Diabetic Nephropathy in DBA2/J Mice

 ,
,  ,
,  ,
,
Abstract
:
1. Introduction
2. Results
2.1. Bilastine Effects on Renal Function
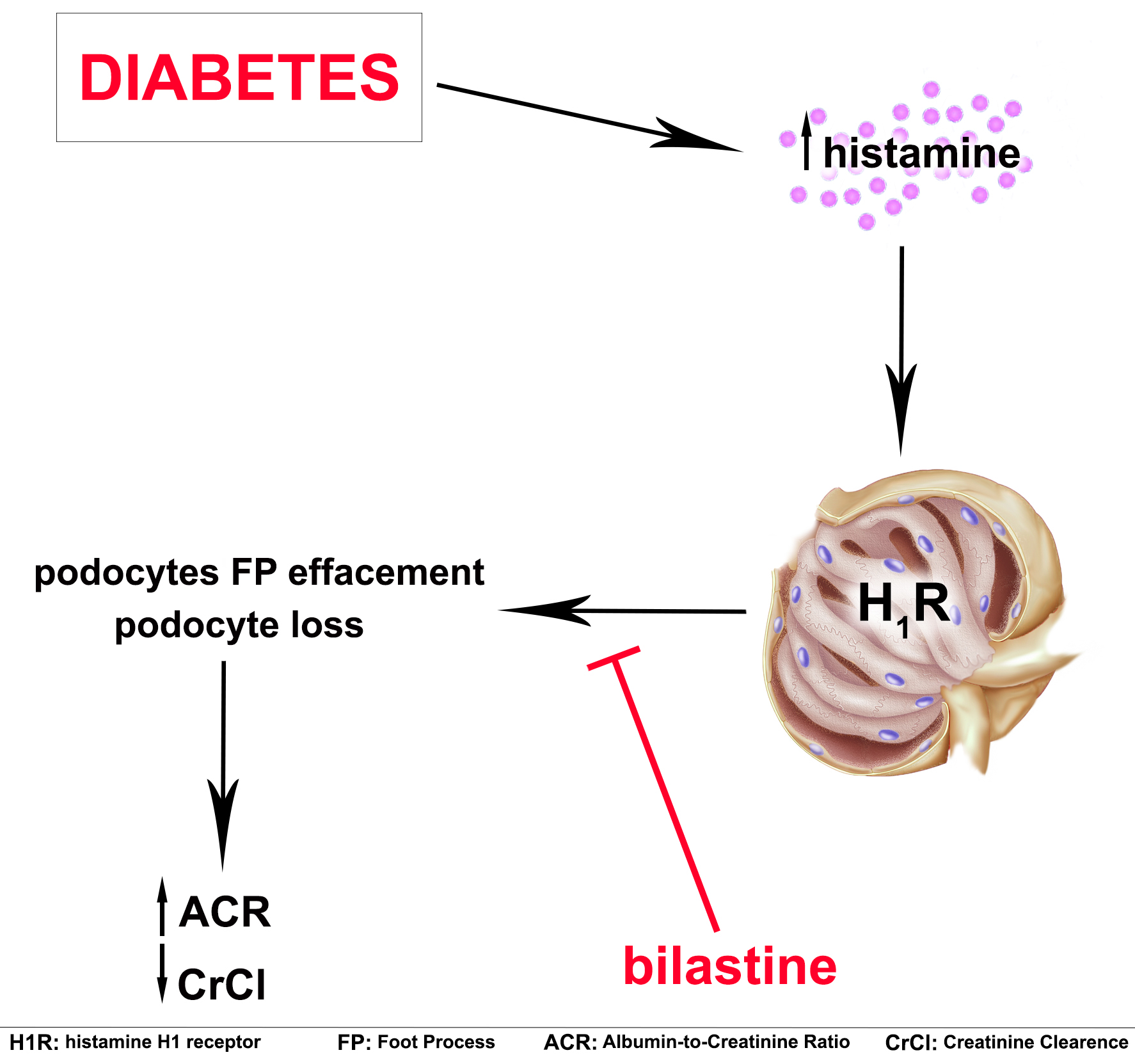
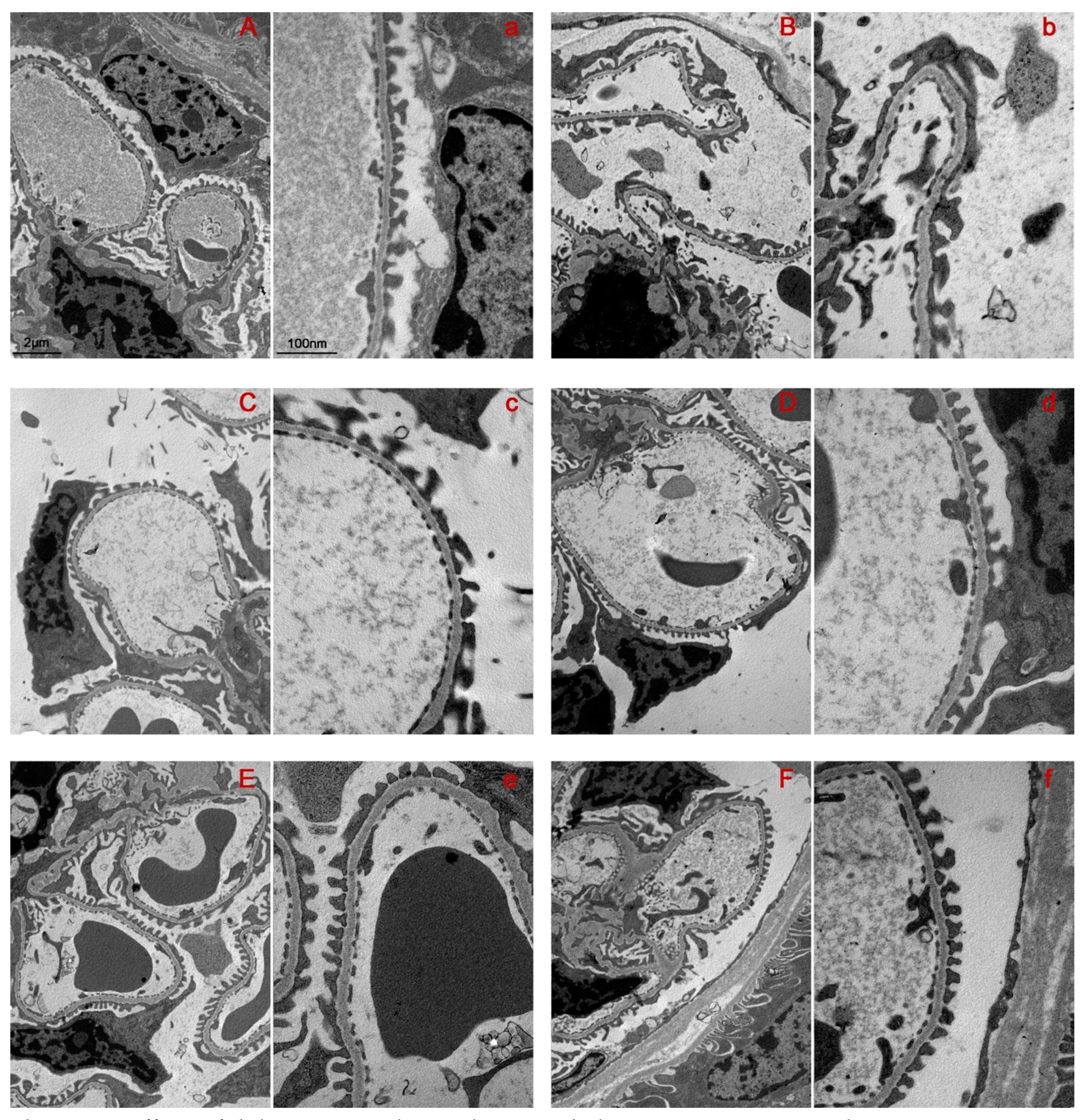
2.2. Bilastine Effect on Glomerular Structure Alterations
2.3. Bilastine Effect on Slit Diaphragm and Cytoarchitecture Protein Expression
2.4. Bilastine Effect on NHE3 Expression
2.5. Bilastine Effect on Tubular Infiltration and Renal Fibrosis
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Care and Ethics Statement
4.3. Experiment Protocol
4.4. Renal Function Evaluations
4.5. Morphological Analysis
4.6. Transmission Electron Microscopy
4.7. Immunofluorescence Analysis
4.8. Immunoblotting
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ESRD | End-Stage Renal Disease |
| ACR | Albumin-to-Creatinine Ratio |
| GBM | Glomerular Basement Membrane |
| IP3 | inositol 1,4,5-trisphosphate |
| NHE | Sodium-Hydrogen Exchanger |
| PAS | Periodic Acid-Schiff |
| PKC | Protein Kinases C |
| STZ | Streptozotocin |
| UPE | Urinary Protein Excretion |
| CrCl | Creatinine Clearance |
| DN | Diabetic Nephropathy |
| FP | Foot Processes |
| SD | Slit Diaphragm |
| ZO | Zonula Occludens |
References
- Fouli, G.E.; Gnudi, L. The Future: Experimental Therapies for Renal Disease in Diabetes. Nephron 2018, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Koye, D.N.; Magliano, D.J.; Nelson, R.G.; Pavkov, M.E. The Global Epidemiology of Diabetes and Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Palsson, R.; Patel, U.D. Cardiovascular complications of diabetic kidney disease. Adv. Chronic Kidney Dis. 2014, 21, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Tangri, N.; Pintilie, M.; Levey, A.S.; Naimark, D. Risk of end-stage renal disease and death after cardiovascular events in chronic kidney disease. Circulation 2014, 130, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.M.; Nee, R.; Ceckowski, K.A.; Knight, K.R.; Abbott, K.C. Diabetic nephropathy as the cause of end-stage kidney disease reported on the medical evidence form CMS2728 at a single center. Clin. Kidney J. 2017, 10, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Neumiller, J.J.; Alicic, R.Z.; Tuttle, K.R. Therapeutic Considerations for Antihyperglycemic Agents in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2263–2274. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.P.; Jandeleit-Dahm, K. The pathobiology of diabetic vascular complications--cardiovascular and kidney disease. J. Mol. Med. 2014, 92, 441–452. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Grange, C.; Gurrieri, M.; Verta, R.; Fantozzi, R.; Pini, A.; Rosa, A.C. Histamine in the kidneys: What is its role in renal pathophysiology? Br. J. Pharm. 2019. [Google Scholar] [CrossRef]
- Pini, A.; Verta, R.; Grange, C.; Gurrieri, M.; Rosa, A.C. Histamine and diabetic nephropathy: An up-to-date overview. Clin. Sci. (Lond.) 2019, 133, 41–54. [Google Scholar] [CrossRef]
- Gill, D.S.; Barradas, M.A.; Fonseca, V.A.; Dandona, P. Plasma histamine concentrations are elevated in patients with diabetes mellitus and peripheral vascular disease. Metabolism 1989, 38, 243–247. [Google Scholar] [CrossRef]
- Alkan, M.; Machavoine, F.; Rignault, R.; Dam, J.; Dy, M.; Thieblemont, N. Histidine Decarboxylase Deficiency Prevents Autoimmune Diabetes in NOD Mice. J. Diabetes Res. 2015, 2015, 965056. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.S.; Thompson, C.S.; Dandona, P. Increased histamine in plasma and tissues in diabetic rats. Diabetes Res. 1988, 7, 31–34. [Google Scholar] [PubMed]
- Gill, D.S.; Thompson, C.S.; Dandona, P. Histamine synthesis and catabolism in various tissues in diabetic rats. Metabolism 1990, 39, 815–818. [Google Scholar] [CrossRef]
- Pini, A.; Grange, C.; Veglia, E.; Argenziano, M.; Cavalli, R.; Guasti, D.; Calosi, L.; Ghe, C.; Solarino, R.; Thurmond, R.L.; et al. Histamine H4 receptor antagonism prevents the progression of diabetic nephropathy in male DBA2/J mice. Pharm. Res 2018, 128, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Sedor, J.R.; Abboud, H.E. Actions and metabolism of histamine in glomeruli and tubules of the human kidney. Kidney Int. 1984, 26, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Sedor, J.R.; Abboud, H.E. Histamine modulates contraction and cyclic nucleotides in cultured rat mesangial cells. Differential effects mediated by histamine H1 and H2 receptors. J. Clin. Investig. 1985, 75, 1679–1689. [Google Scholar] [CrossRef]
- Veglia, E.; Pini, A.; Moggio, A.; Grange, C.; Premoselli, F.; Miglio, G.; Tiligada, K.; Fantozzi, R.; Chazot, P.L.; Rosa, A.C. Histamine type 1-receptor activation by low dose of histamine undermines human glomerular slit diaphragm integrity. Pharm. Res. 2016, 114, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, E.; Grange, C.; Pini, A.; Moggio, A.; Lanzi, C.; Camussi, G.; Chazot, P.L.; Rosa, A.C. Histamine receptor expression in human renal tubules: A comparative pharmacological evaluation. Inflamm. Res. 2015, 64, 261–270. [Google Scholar] [CrossRef]
- Rosa, A.C.; Grange, C.; Pini, A.; Katebe, M.A.; Benetti, E.; Collino, M.; Miglio, G.; Bani, D.; Camussi, G.; Chazot, P.L.; et al. Overexpression of histamine H(4) receptors in the kidney of diabetic rat. Inflamm. Res. 2013, 62, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Pini, A.; Chazot, P.L.; Veglia, E.; Moggio, A.; Rosa, A.C. H3 receptor renal expression in normal and diabetic rats. Inflamm. Res. 2015, 64, 271–273. [Google Scholar] [CrossRef]
- Gardner, T.W.; Lesher, T.; Khin, S.; Vu, C.; Barber, A.J.; Brennan, W.A., Jr. Histamine reduces ZO-1 tight-junction protein expression in cultured retinal microvascular endothelial cells. Biochem. J. 1996, 320, 717–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichikawa, I.; Brenner, B.M. Mechanisms of action of hisamine and histamine antagonists on the glomerular microcirculation in the rat. Circ. Res. 1979, 45, 737–745. [Google Scholar] [CrossRef]
- Bolton, W.K.; Spargo, B.A.; Lewis, E.J. Chronic autologous immune complex glomerulopathy: Effect of cyproheptadine. J. Lab. Clin. Med. 1974, 83, 695–704. [Google Scholar] [PubMed]
- Anbar, H.S.; Shehatou, G.S.; Suddek, G.M.; Gameil, N.M. Comparison of the effects of levocetirizine and losartan on diabetic nephropathy and vascular dysfunction in streptozotocin-induced diabetic rats. Eur. J. Pharm. 2016, 780, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Alpers, C.E.; Hudkins, K.L. Mouse models of diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2011, 20, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Lucero, M.L.; Patterson, A.B. Whole-body tissue distribution of total radioactivity in rats after oral administration of [(1)(4)C]-bilastine. Drug Chem. Toxicol. 2012, 35, 1–7. [Google Scholar] [CrossRef]
- Wolthers, O.D. Bilastine: A new nonsedating oral H1 antihistamine for treatment of allergic rhinoconjunctivitis and urticaria. Biomed. Res. Int. 2013, 2013, 626837. [Google Scholar] [CrossRef] [PubMed]
- Biemesderfer, D.; Pizzonia, J.; Abu-Alfa, A.; Exner, M.; Reilly, R.; Igarashi, P.; Aronson, P.S. NHE3: A Na+/H+ exchanger isoform of renal brush border. Am. J. Physiol. 1993, 265, F736–F742. [Google Scholar] [CrossRef]
- Banks, R.O.; Inscho, E.W.; Jacobson, E.D. Histamine H1 receptor antagonists inhibit autoregulation of renal blood flow in the dog. Circ. Res. 1984, 54, 527–535. [Google Scholar] [CrossRef]
- Schulte, K.; Berger, K.; Boor, P.; Jirak, P.; Gelman, I.H.; Arkill, K.P.; Neal, C.R.; Kriz, W.; Floege, J.; Smeets, B.; et al. Origin of parietal podocytes in atubular glomeruli mapped by lineage tracing. J. Am. Soc. Nephrol. 2014, 25, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Teng, B.; Duong, M.; Tossidou, I.; Yu, X.; Schiffer, M. Role of protein kinase C in podocytes and development of glomerular damage in diabetic nephropathy. Front. Endocrinol. 2014, 5, 179. [Google Scholar] [CrossRef]
- Xu, Z.G.; Ryu, D.R.; Yoo, T.H.; Jung, D.S.; Kim, J.J.; Kim, H.J.; Choi, H.Y.; Kim, J.S.; Adler, S.G.; Natarajan, R.; et al. P-Cadherin is decreased in diabetic glomeruli and in glucose-stimulated podocytes in vivo and in vitro studies. Nephrol. Dial. Transplant. 2005, 20, 524–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miethe, S.; Guarino, M.; Alhamdan, F.; Simon, H.U.; Renz, H.; Dufour, J.F.; Potaczek, D.P.; Garn, H. Effects of obesity on asthma: Immunometabolic links. Pol. Arch. Intern. Med. 2018, 128, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Klamt, S.; Vogel, M.; Kapellen, T.M.; Hiemisch, A.; Prenzel, F.; Zachariae, S.; Ceglarek, U.; Thiery, J.; Kiess, W. Association between IgE-mediated allergies and diabetes mellitus type 1 in children and adolescents. Pediatr. Diabetes 2015, 16, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.P. Links between allergy and cardiovascular or hemostatic system. Int. J. Cardiol. 2014, 170, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Girardi, A.C.; Di Sole, F. Deciphering the mechanisms of the Na+/H+ exchanger-3 regulation in organ dysfunction. Am. J. Physiol. Cell Physiol. 2012, 302, C1569–C1587. [Google Scholar] [CrossRef]
- Segnani, C.; Ippolito, C.; Antonioli, L.; Pellegrini, C.; Blandizzi, C.; Dolfi, A.; Bernardini, N. Histochemical Detection of Collagen Fibers by Sirius Red/Fast Green Is More Sensitive than van Gieson or Sirius Red Alone in Normal and Inflamed Rat Colon. Plos ONE 2015, 10, e0144630. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTRL | CTRL + Bilastine 30 mg/kg | STZ | STZ + Bilastine (mg/kg) | ||||
|---|---|---|---|---|---|---|---|
| 1 | 3 | 10 | 30 | ||||
| Glucosuria ° (mg/dL) | n.d. | n.d. | 1500 ± 224 | 1000 ± 0 | 1188 ± 188 | 1143 ± 143 | 1500 ± 289 |
| Urine volume (mL) | 6.8 ± 0.7 | 6.4 ± 0.7 | 19.8 ± 1.1 * | 22.5 ± 2.9 * | 17.5 ± 3.6 * | 21.6 ± 2.3 * | 32.5 ± 2.5 * |
| Leukocyte | - | - | - | - | - | - | - |
| Urine pH ° | 6.5 ± 0.0 | 6.4 ± 0.1 | 6.0 ± 0.2 | 6.0 ± 0.2 | 6.2 ± 0.1 | 6.0 ± 0.1 | 5.9 ± 0.1 |
| UPE (mg/mL) | 0.8 ± 0.1 | 0.8 ± 0.1 | 3.2 ± 0.2 * | 3.7 ± 0.5 * | 2.0 ± 0.4 | 2.8 ± 0.4 * | 3.6 ± 1.1 * |
| ACR (µg/mg) | 66.9 ± 6.0 | 60.4 ± 9.7 | 257.4 ± 27.2 * | 150.6 ± 33.8 | 101.4 ± 23.1 | 132.2 ± 9.8 | 90.3 ± 40.9 # |
| CrCl (mL/min) | 0.13 ± 0.01 | 0.14 ± 0.01 | 0.05 ± 0.01 * | 0.05 ± 0.01 §* | 0.09 ± 0.03 | 0.14 ± 0.01 §# | 0.15 ± 0.02 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verta, R.; Grange, C.; Gurrieri, M.; Borga, S.; Nardini, P.; Argenziano, M.; Ghè, C.; Cavalli, R.; Benetti, E.; Miglio, G.; et al. Effect of Bilastine on Diabetic Nephropathy in DBA2/J Mice. Int. J. Mol. Sci. 2019, 20, 2554. https://doi.org/10.3390/ijms20102554
Verta R, Grange C, Gurrieri M, Borga S, Nardini P, Argenziano M, Ghè C, Cavalli R, Benetti E, Miglio G, et al. Effect of Bilastine on Diabetic Nephropathy in DBA2/J Mice. International Journal of Molecular Sciences. 2019; 20(10):2554. https://doi.org/10.3390/ijms20102554
Chicago/Turabian StyleVerta, Roberta, Cristina Grange, Maura Gurrieri, Sara Borga, Patrizia Nardini, Monica Argenziano, Corrado Ghè, Roberta Cavalli, Elisa Benetti, Gianluca Miglio, and et al. 2019. "Effect of Bilastine on Diabetic Nephropathy in DBA2/J Mice" International Journal of Molecular Sciences 20, no. 10: 2554. https://doi.org/10.3390/ijms20102554