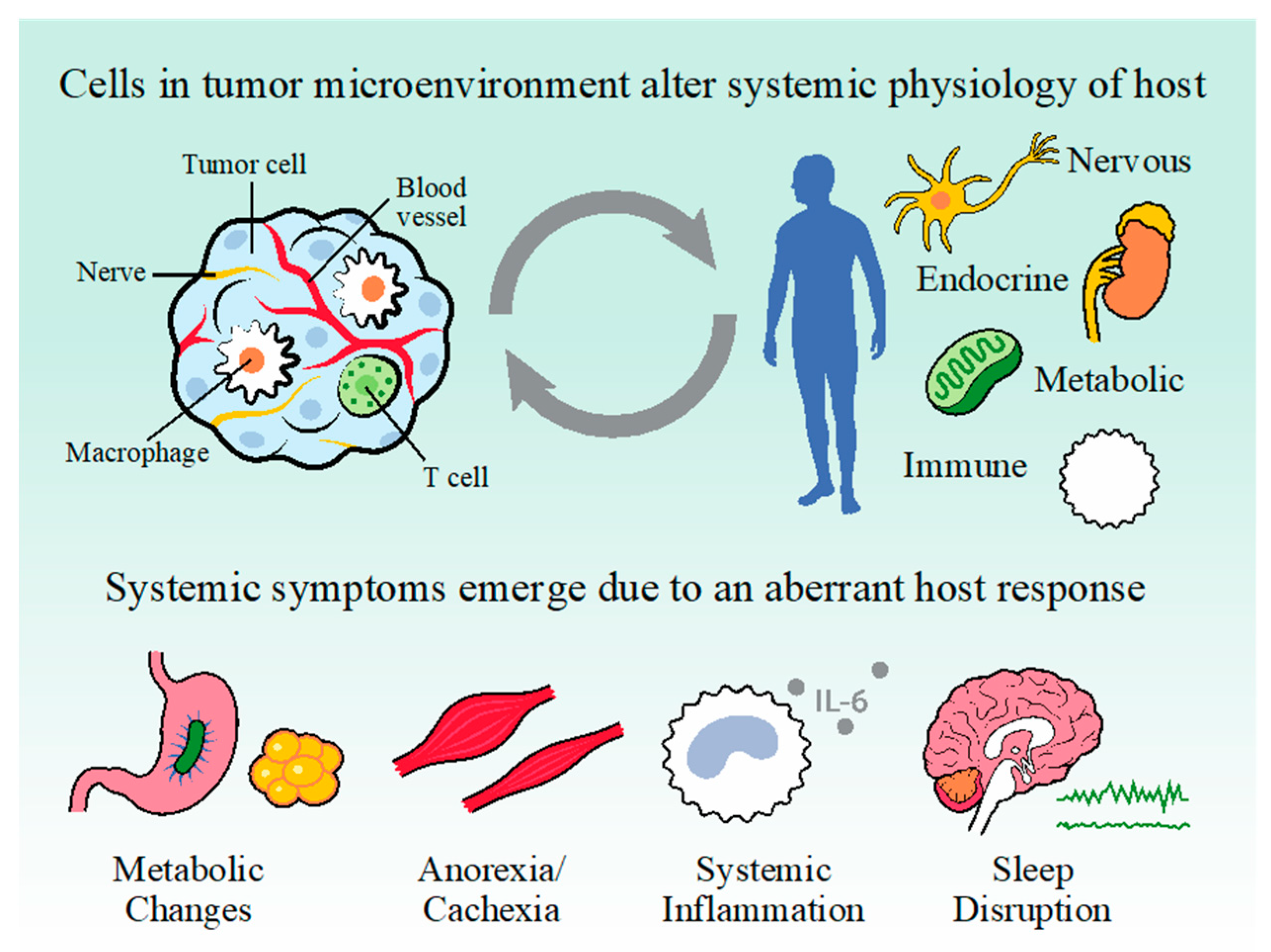
Molecular Mechanisms of Cancer-Induced Sleep Disruption


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sleep Neurocircuitry
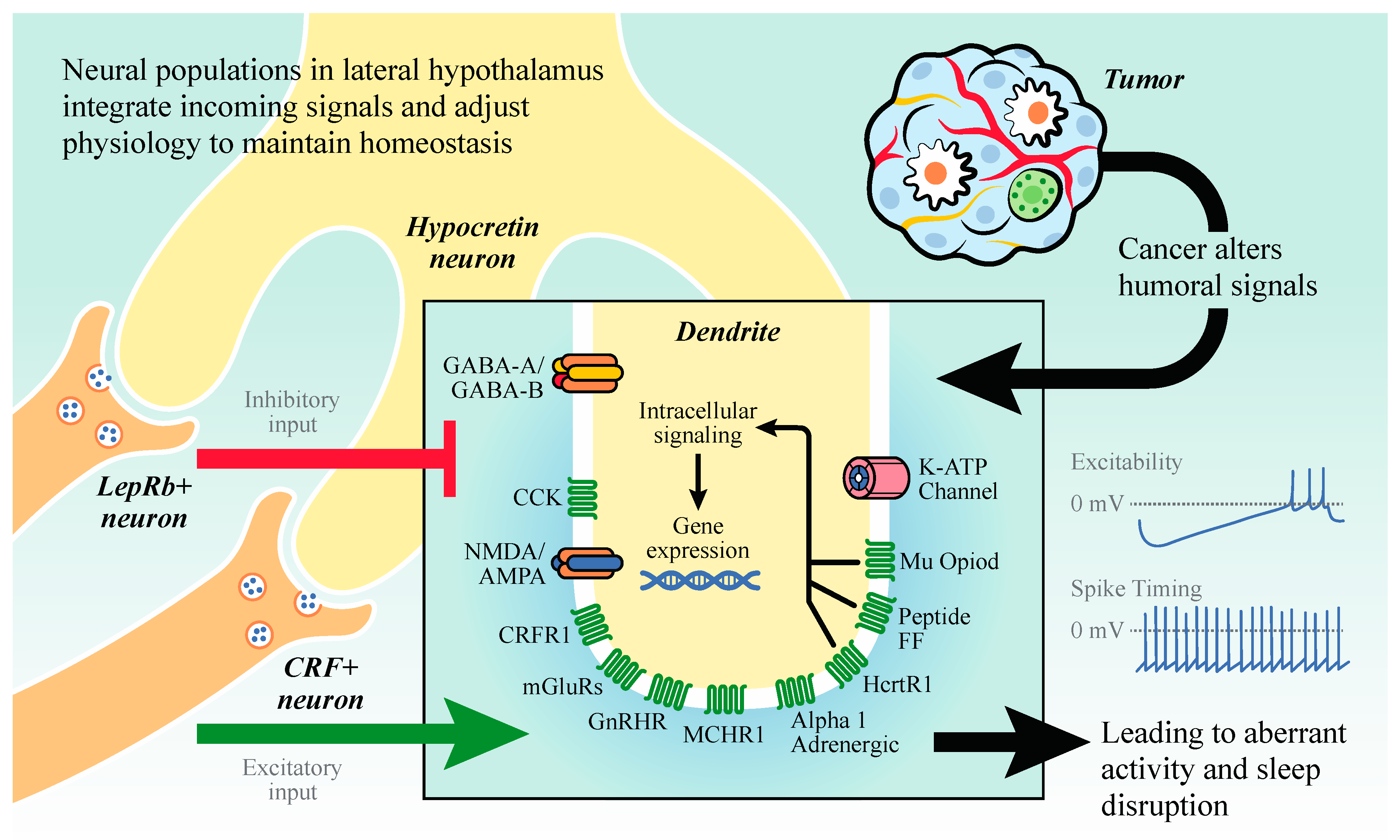
2.1. Hypocretin/Orexin (HO) Neurons
2.2. Melanin Concentrating Hormone (MCH) Neurons
2.3. VLPO GABAergic Neurons
2.4. VTA
2.5. Dorsal Raphe
2.6. LC Noradrenergic Neurons
3. Sleep Disruption in Patients with Cancer and Cancer Survivors
4. Immune Pathways Deregulated by Cancer that Influence Sleep
5. Interleukin-1
6. Interleukin-6
7. Tumor Necrosis Factor
8. Transforming Growth Factor Beta, Interleukin-4, and Interleukin-10
9. Cancer, Energy Balance, and Sleep
10. Preclinical Research
11. Conclusions and Unanswered Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5HT | Serotonin |
| AKT | Protein kinase B |
| BBB | Blood brain barrier |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra large |
| bFGF | Basic fibroblast growth factor |
| CCK | Cholecystokinin |
| CCL | C-C motif chemokine ligand |
| COX2 | Cyclooxygenase-2 |
| CRF | Corticotropin releasing factor |
| CRFR1 | Corticotropin releasing factor receptor 1 |
| DR | Dorsal raphe |
| EEG | Electroencephalogram |
| EGF | Epidermal growth factor |
| EMG | Electromyogram |
| FGF | Fibroblast growth factor |
| GABA | Gamma-Aminobutyric acid |
| GH | Growth Hormone |
| GHRH | Growth hormone-releasing hormone |
| GHSR | Growth hormone secretagogue receptor |
| GnRHR | Gonadotropin releasing hormone receptor |
| HcrtR1 | Hypocretin receptor 1 |
| HGF | Hepatocyte growth factor |
| HO | Hypocretin/Orexin |
| IGF | Insulin-like growth factor |
| IL-1 | Interleukin-1 |
| IL-10 | Interleukin-10 |
| IL-1R | Interleukin-1 receptor |
| IL-2 | Interleukin-2 |
| IL-4 | Interleukin-4 |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| K-ATP | ATP sensitive potassium channel |
| LC | Locus coeruleus |
| LepRb | Long-form leptin receptor |
| LH | Lateral Hypothalamus |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinases |
| MCH | Melanin concentrating hormone |
| MCHR1 | Melanin concentrating hormone receptor 1 |
| Mcl-1 | Induced myeloid leukemia cell differentiation protein |
| mGluR | Metabotropic glutamate receptor |
| MMP | Matrix metalloproteinases |
| NE | Norepinephrine |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NO | Nitric oxide |
| NREM | Non-rapid eye movement |
| NREM | Non rapid eye movement |
| Ob-R | Leptin receptor |
| PDGF | Platelet-derived growth factor |
| PGE-2 | Prostaglandin E2 |
| PI-3K | Phosphoinositide 3-kinase |
| REM | Rapid eye movement |
| REM | Rapid eye movement |
| sIL-6R | Soluble interleukin-6 receptor |
| STAT3 | Signal transducer and activator of transcription 3 |
| TGF-b | Transforming growth factor beta |
| TNF-a | Tumor necrosis factor alpha |
| TNFR2 | Tumor necrosis factor receptor 2 |
| VEGF | Vascular endothelial growth factor |
| VLPO | Ventrolateral preoptic area |
| VTA | Ventral tegmental area |
| WASO | Wake after sleep onset |
References
- Borniger, J.C. Central Regulation of Breast Cancer Growth and Metastasis. J. Cancer Metast. Treat 2019, 5, 23. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Cash, E.; Sephton, S.E.; Chagpar, A.B.; Spiegel, D.; Rebholz, W.N.; Zimmaro, L.A.; Tillie, J.M.; Dhabhar, F.S. Circadian disruption and biomarkers of tumor progression in breast cancer patients awaiting surgery. Brain Behav. Immun. 2015, 48, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Sephton, S.E.; Sapolsky, R.M.; Kraemer, H.C.; Spiegel, D. Diurnal Cortisol Rhythm as a Predictor of Breast Cancer Survival. JNCI J. Natl. Cancer Inst. 2000, 92, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, J.; Heiligensetzer, M.; Krakowski-Roosen, H.; Büchler, M.W.; Friess, H.; Martignoni, M.E. Cachexia Worsens Prognosis in Patients with Resectable Pancreatic Cancer. J. Gastrointest. Surg. 2008, 12, 1193. [Google Scholar] [CrossRef] [PubMed]
- Sephton, S.E.; Lush, E.; Dedert, E.A.; Floyd, A.R.; Rebholz, W.N.; Dhabhar, F.S.; Spiegel, D.; Salmon, P. Diurnal cortisol rhythm as a predictor of lung cancer survival. Brain Behav. Immun. 2013, 30 (Suppl.), S163–S170. [Google Scholar] [CrossRef]
- Ancoli-Israel, S.; Liu, L.; Marler, M.R.; Parker, B.A.; Jones, V.; Sadler, G.R.; Dimsdale, J.; Cohen-Zion, M.; Fiorentino, L. Fatigue, sleep, and circadian rhythms prior to chemotherapy for breast cancer. Support. Care Cancer 2006, 14, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Palesh, O.G.; Roscoe, J.A.; Mustian, K.M.; Roth, T.; Savard, J.; Ancoli-Israel, S.; Heckler, C.; Purnell, J.Q.; Janelsins, M.C.; Morrow, G.R. Prevalence, Demographics, and Psychological Associations of Sleep Disruption in Patients With Cancer: University of Rochester Cancer Center–Community Clinical Oncology Program. J. Clin. Oncol. 2010, 28, 292–298. [Google Scholar] [CrossRef]
- Palesh, O.; Aldridge-Gerry, A.; Zeitzer, J.M.; Koopman, C.; Neri, E.; Giese-Davis, J.; Jo, B.; Kraemer, H.; Nouriani, B.; Spiegel, D. Actigraphy-Measured Sleep Disruption as a Predictor of Survival among Women with Advanced Breast Cancer. Sleep 2014, 37, 837–842. [Google Scholar] [CrossRef]
- Borniger, J.C.; Walker, W.H., II; Surbhi; Emmer, K.M.; Zhang, N.; Zalenski, A.A.; Muscarella, S.L.; Fitzgerald, J.A.; Smith, A.N.; Braam, C.J.; et al. A Role for Hypocretin/Orexin in Metabolic and Sleep Abnormalities in a Mouse Model of Non-metastatic Breast Cancer. Cell Metab. 2018, 28, 118–129.e5. [Google Scholar] [CrossRef]
- Hakim, F.; Wang, Y.; Zhang, S.X.; Zheng, J.; Yolcu, E.S.; Carreras, A.; Khalyfa, A.; Shirwan, H.; Almendros, I.; Gozal, D. Fragmented sleep accelerates tumor growth and progression through recruitment of tumor-associated macrophages and TLR4 signaling. Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Loomis, A.L.; Harvey, E.N.; Hobart, G. Potential rhythms of the cerebral cortex during sleep. Science 1935, 81, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.M. The Neurobiology of Sleep. Semin. Neurol. 2009, 29, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Szymusiak, R.; Gvilia, I.; McGinty, D. Hypothalamic control of sleep. Sleep Med. 2007, 8, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Crunelli, V.; Hughes, S.W. The slow (<1 Hz) rhythm of non-REM sleep: A dialogue between three cardinal oscillators. Nat. Neurosci. 2010, 13, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Contreras, D.; Steriade, M. State-dependent fluctuations of low-frequency rhythms in corticothalamic networks. Neuroscience 1997, 76, 25–38. [Google Scholar] [CrossRef]
- Contreras, D.; Destexhe, A.; Sejnowski, T.J.; Steriade, M. Control of spatiotemporal coherence of a thalamic oscillation by corticothalamic feedback. Science 1996, 274, 771–774. [Google Scholar] [CrossRef]
- Borbély, A.A. A two process model of sleep regulation. Hum. Neurobiol. 1982, 1, 195–204. [Google Scholar]
- Borbély, A.A.; Daan, S.; Wirz-Justice, A.; Deboer, T. The two-process model of sleep regulation: A reappraisal. J. Sleep Res. 2016, 25, 131–143. [Google Scholar] [CrossRef]
- Scammell, T.E.; Arrigoni, E.; Lipton, J.O. Neural Circuitry of Wakefulness and Sleep. Neuron 2017, 93, 747–765. [Google Scholar] [CrossRef]
- De Lecea, L.; Kilduff, T.S.; Peyron, C.; Gao, X.-B.; Foye, P.E.; Danielson, P.E.; Fukuhara, C.; Battenberg, E.L.F.; Gautvik, V.T.; Bartlett, F.S.; et al. The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. USA 1998, 95, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Saper, C.B.; Fuller, P.M.; Pedersen, N.P.; Lu, J.; Scammell, T.E. Sleep State Switching. Neuron 2010, 68, 1023–1042. [Google Scholar] [CrossRef] [PubMed]
- Bonnavion, P.; Mickelsen, L.E.; Fujita, A.; de Lecea, L.; Jackson, A.C. Hubs and spokes of the lateral hypothalamus: Cell types, circuits and behaviour. J. Physiol. 2016, 594, 6443–6462. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Amemiya, A.; Ishii, M.; Matsuzaki, I.; Chemelli, R.M.; Tanaka, H.; Williams, S.C.; Richardson, J.A.; Kozlowski, G.P.; Wilson, S.; et al. Orexins and Orexin Receptors: A Family of Hypothalamic Neuropeptides and G Protein-Coupled Receptors that Regulate Feeding Behavior. Cell 1998, 92, 573–585. [Google Scholar] [CrossRef]
- Adamantidis, A.R.; Zhang, F.; Aravanis, A.M.; Deisseroth, K.; de Lecea, L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 2007, 450, 420–424. [Google Scholar] [CrossRef]
- Tsunematsu, T.; Tabuchi, S.; Tanaka, K.F.; Boyden, E.S.; Tominaga, M.; Yamanaka, A. Long-lasting silencing of orexin/hypocretin neurons using archaerhodopsin induces slow-wave sleep in mice. Behav. Brain Res. 2013, 255, 64–74. [Google Scholar] [CrossRef]
- Chemelli, R.M.; Willie, J.T.; Sinton, C.M.; Elmquist, J.K.; Scammell, T.; Lee, C.; Richardson, J.A.; Williams, S.C.; Xiong, Y.; Kisanuki, Y.; et al. Narcolepsy in orexin knockout mice: Molecular genetics of sleep regulation. Cell 1999, 98, 437–451. [Google Scholar] [CrossRef]
- Hara, J.; Beuckmann, C.T.; Nambu, T.; Willie, J.T.; Chemelli, R.M.; Sinton, C.M.; Sugiyama, F.; Yagami, K.; Goto, K.; Yanagisawa, M.; et al. Genetic Ablation of Orexin Neurons in Mice Results in Narcolepsy, Hypophagia, and Obesity. Neuron 2001, 30, 345–354. [Google Scholar] [CrossRef]
- Crocker, A.; España, R.A.; Papadopoulou, M.; Saper, C.B.; Faraco, J.; Sakurai, T.; Honda, M.; Mignot, E.; Scammell, T.E. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology 2005, 65, 1184–1188. [Google Scholar] [CrossRef]
- Latorre, D.; Kallweit, U.; Armentani, E.; Foglierini, M.; Mele, F.; Cassotta, A.; Jovic, S.; Jarrossay, D.; Mathis, J.; Zellini, F.; et al. T cells in patients with narcolepsy target self-antigens of hypocretin neurons. Nature 2018, 562, 63. [Google Scholar] [CrossRef]
- Luo, G.; Ambati, A.; Lin, L.; Bonvalet, M.; Partinen, M.; Ji, X.; Maecker, H.T.; Mignot, E.J.-M. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc. Natl. Acad. Sci. USA 2018, 115, E12323–E12332. [Google Scholar] [CrossRef] [PubMed]
- Borniger, J.C.; de Lecea, L. The Hypocretin Arousal Network. In Oxford Research Encyclopedia of Neuroscience; Oxford University Press: Oxford, UK, 2019; ISBN 978-0-19-026408-6. [Google Scholar]
- Yoshida, K.; McCormack, S.; España, R.A.; Crocker, A.; Scammell, T.E. Afferents to the orexin neurons of the rat brain. J. Comp. Neurol. 2006, 494, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T. The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nat. Rev. Neurosci. 2007, 8, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Mieda, M. Connectomics of orexin-producing neurons: Interface of systems of emotion, energy homeostasis and arousal. Trends Pharmacol. Sci. 2011, 32, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T. Roles of orexin/hypocretin in regulation of sleep/wakefulness and energy homeostasis. Sleep Med. Rev. 2005, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.E.; Brill, J.; Bonnavion, P.; Huguenard, J.R.; Huerta, R.; Lecea, L. de Mechanism for Hypocretin-mediated sleep-to-wake transitions. Proc. Natl. Acad. Sci. USA 2012, 109, E2635–E2644. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.E.; de Lecea, L. Optogenetic investigation of neural circuits in vivo. Trends Mol. Med. 2011, 17, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Bonnavion, P.; Jackson, A.C.; Carter, M.E.; Lecea, L. de Antagonistic interplay between hypocretin and leptin in the lateral hypothalamus regulates stress responses. Nat. Commun. 2015, 6, 6266. [Google Scholar] [CrossRef]
- Giardino, W.J.; Eban-Rothschild, A.; Christoffel, D.J.; Li, S.-B.; Malenka, R.C.; Lecea, L. de Parallel circuits from the bed nuclei of stria terminalis to the lateral hypothalamus drive opposing emotional states. Nat. Neurosci. 2018, 21, 1084. [Google Scholar] [CrossRef]
- Barnes, P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. Lond. Engl. 1979 1998, 94, 557–572. [Google Scholar] [CrossRef]
- Geerling, J.C.; Mettenleiter, T.C.; Loewy, A.D. Orexin neurons project to diverse sympathetic outflow systems. Neuroscience 2003, 122, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.-X.; Serlie, M.J.; Ackermans, M.T.; Foppen, E.; Buijs, R.M.; Sauerwein, H.P.; Fliers, E.; Kalsbeek, A. A Major Role for Perifornical Orexin Neurons in the Control of Glucose Metabolism in Rats. Diabetes 2009, 58, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, J.C.; Presse, F.; Arias, C.; Peto, C.; Vaughan, J.; Nahon, J.-L.; Vale, W.; Sawchenko, P.E. The melanin-concentrating hormone system of the rat brain: An immuno- and hybridization histochemical characterization. J. Comp. Neurol. 1992, 319, 218–245. [Google Scholar] [CrossRef] [PubMed]
- Monti, J.M.; Torterolo, P.; Lagos, P. Melanin-concentrating hormone control of sleep-wake behavior. Sleep Med. Rev. 2013, 17, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Hassani, O.K.; Lee, M.G.; Jones, B.E. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep–wake cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 2418–2422. [Google Scholar] [CrossRef] [PubMed]
- Willie, J.T.; Sinton, C.M.; Maratos-Flier, E.; Yanagisawa, M. Abnormal response of melanin-concentrating hormone deficient mice to fasting: Hyperactivity and rapid eye movement sleep suppression. Neuroscience 2008, 156, 819–829. [Google Scholar] [CrossRef]
- Apergis-Schoute, J.; Iordanidou, P.; Faure, C.; Jego, S.; Schöne, C.; Aitta-Aho, T.; Adamantidis, A.; Burdakov, D. Optogenetic Evidence for Inhibitory Signaling from Orexin to MCH Neurons via Local Microcircuits. J. Neurosci. 2015, 35, 5435–5441. [Google Scholar] [CrossRef] [PubMed]
- Palesh, O.; Scheiber, C.; Kesler, S.; Mustian, K.; Koopman, C.; Schapira, L. Management of side effects during and post-treatment in breast cancer survivors. Breast J. 2018, 24, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Kosse, C.; Burdakov, D. Natural hypothalamic circuit dynamics underlying object memorization. bioRxiv 2019, 603936. [Google Scholar] [CrossRef]
- Sherin, J.E.; Shiromani, P.J.; McCarley, R.W.; Saper, C.B. Activation of Ventrolateral Preoptic Neurons During Sleep. Science 1996, 271, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Sherin, J.E.; Elmquist, J.K.; Torrealba, F.; Saper, C.B. Innervation of Histaminergic Tuberomammillary Neurons by GABAergic and Galaninergic Neurons in the Ventrolateral Preoptic Nucleus of the Rat. J. Neurosci. 1998, 18, 4705–4721. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Greco, M.A.; Shiromani, P.; Saper, C.B. Effect of Lesions of the Ventrolateral Preoptic Nucleus on NREM and REM Sleep. J. Neurosci. 2000, 20, 3830–3842. [Google Scholar] [CrossRef] [PubMed]
- Szymusiak, R.; Alam, N.; Steininger, T.L.; McGinty, D. Sleep–waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 1998, 803, 178–188. [Google Scholar] [CrossRef]
- Varin, C.; Rancillac, A.; Geoffroy, H.; Arthaud, S.; Fort, P.; Gallopin, T. Glucose Induces Slow-Wave Sleep by Exciting the Sleep-Promoting Neurons in the Ventrolateral Preoptic Nucleus: A New Link between Sleep and Metabolism. J. Neurosci. 2015, 35, 9900–9911. [Google Scholar] [CrossRef] [PubMed]
- Dahan, L.; Astier, B.; Vautrelle, N.; Urbain, N.; Kocsis, B.; Chouvet, G. Prominent burst firing of dopaminergic neurons in the ventral tegmental area during paradoxical sleep. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Steffensen, S.C.; Henriksen, S.J. Discharge profiles of ventral tegmental area GABA neurons during movement, anesthesia, and the sleep-wake cycle. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 1757–1766. [Google Scholar] [CrossRef]
- Eban-Rothschild, A.; Rothschild, G.; Giardino, W.J.; Jones, J.R.; de Lecea, L. VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat. Neurosci. 2016, 19, 1356–1366. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Ma, Y.; Tossell, K.; Harris, J.J.; Harding, E.C.; Ba, W.; Miracca, G.; Wang, D.; Li, L.; et al. GABA and glutamate neurons in the VTA regulate sleep and wakefulness. Nat. Neurosci. 2019, 22, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Matsubara, T.; Miyazaki, T.; Ono, D.; Abe, M.; Sakimura, K.; Yamanaka, A. GABA neurons in the ventral tegmental area regulate non-rapid eye movement sleep in mice. bioRxiv 2019. [Google Scholar] [CrossRef]
- Eban-Rothschild, A.; Appelbaum, L.; de Lecea, L. Neuronal Mechanisms for Sleep/Wake Regulation and Modulatory Drive. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 937–952. [Google Scholar] [CrossRef]
- Ancoli-Israel, S.; Moore, P.J.; Jones, V. The relationship between fatigue and sleep in cancer patients: A review. Eur. J. Cancer Care (Engl.) 2001, 10, 245–255. [Google Scholar] [CrossRef]
- Bower, J.E.; Ganz, P.A.; Desmond, K.A.; Rowland, J.H.; Meyerowitz, B.E.; Belin, T.R. Fatigue in breast cancer survivors: Occurrence, correlates, and impact on quality of life. J. Clin. Oncol. 2000, 18, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; Meagher, M.W.; Cleeland, C.S. Translational approaches to treatment-induced symptoms in cancer patients. Nat. Rev. Clin. Oncol. 2012, 9, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shaanan, T.L.; Azulay-Debby, H.; Dubovik, T.; Starosvetsky, E.; Korin, B.; Schiller, M.; Green, N.L.; Admon, Y.; Hakim, F.; Shen-Orr, S.S.; et al. Activation of the reward system boosts innate and adaptive immunity. Nat. Med. 2016, 22, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shaanan, T.L.; Schiller, M.; Azulay-Debby, H.; Korin, B.; Boshnak, N.; Koren, T.; Krot, M.; Shakya, J.; Rahat, M.A.; Hakim, F.; et al. Modulation of anti-tumor immunity by the brain’s reward system. Nat. Commun. 2018, 9, 2723. [Google Scholar] [CrossRef] [PubMed]
- Teunis, M.A.T.; Kavelaars, A.; Voest, E.; Bakker, J.M.; Ellenbroek, B.A.; Cools, A.R.; Heijnen, C.J. Reduced tumor growth, experimental metastasis formation, and angiogenesis in rats with a hyperreactive dopaminergic system. FASEB J. 2002, 16, 1465–1467. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.M.; Buchanan, G.F.; Richerson, G.B. Insomnia Caused by Serotonin Depletion is Due to Hypothermia. Sleep 2015, 38, 1985–1993. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Yanase, M.; Yamashita, A.; Kitabatake, C.; Hamada, A.; Suhara, Y.; Narita, M.; Ikegami, D.; Sakai, H.; Yamazaki, M.; et al. Analysis of sleep disorders under pain using an optogenetic tool: Possible involvement of the activation of dorsal raphe nucleus-serotonergic neurons. Mol. Brain 2013, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, J.; Li, Y.; Hu, F.; Lu, Y.; Ma, M.; Feng, Q.; Zhang, J.; Wang, D.; Zeng, J.; et al. Dorsal Raphe Neurons Signal Reward through 5-HT and Glutamate. Neuron 2014, 81, 1360–1374. [Google Scholar] [CrossRef]
- Cho, J.R.; Treweek, J.B.; Robinson, J.E.; Xiao, C.; Bremner, L.R.; Greenbaum, A.; Gradinaru, V. Dorsal Raphe Dopamine Neurons Modulate Arousal and Promote Wakefulness by Salient Stimuli. Neuron 2017, 94, 1205–1219.e8. [Google Scholar] [CrossRef]
- Opp, M.R. Cytokines and sleep. Sleep Med. Rev. 2005, 9, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Del Gallo, F.; Opp, M.R.; Imeri, L. The reciprocal link between sleep and immune responses. Arch. Ital. Biol. 2014, 152, 93–102. [Google Scholar] [PubMed]
- Imeri, L.; Opp, M.R. How (and why) the immune system makes us sleep. Nat. Rev. Neurosci. 2009, 10, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, D.; Franciosi, S.; Opp, M.R.; Imeri, L. Interleukin-1 inhibits firing of serotonergic neurons in the dorsal raphe nucleus and enhances GABAergic inhibitory post-synaptic potentials. Eur. J. Neurosci. 2007, 26, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.G.; Galpern, W.R.; Dunlap, K.; Dinarello, C.A.; Turner, T.J. Interleukin-1 augments gamma-aminobutyric acidA receptor function in brain. Mol. Pharmacol. 1991, 39, 105–108. [Google Scholar] [PubMed]
- Manfridi, A.; Brambilla, D.; Bianchi, S.; Mariotti, M.; Opp, M.R.; Imeri, L. Interleukin-1beta enhances non-rapid eye movement sleep when microinjected into the dorsal raphe nucleus and inhibits serotonergic neurons in vitro. Eur. J. Neurosci. 2003, 18, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Serantes, R.; Arnalich, F.; Figueroa, M.; Salinas, M.; Andrés-Mateos, E.; Codoceo, R.; Renart, J.; Matute, C.; Cavada, C.; Cuadrado, A.; et al. Interleukin-1beta enhances GABAA receptor cell-surface expression by a phosphatidylinositol 3-kinase/Akt pathway: Relevance to sepsis-associated encephalopathy. J. Biol. Chem. 2006, 281, 14632–14643. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hopf, F.W.; Li, S.-B.; Lecea, L. de In vivo cell type-specific CRISPR knockdown of dopamine beta hydroxylase reduces locus coeruleus evoked wakefulness. Nat. Commun. 2018, 9, 5211. [Google Scholar] [CrossRef]
- Carter, M.E.; Yizhar, O.; Chikahisa, S.; Nguyen, H.; Adamantidis, A.; Nishino, S.; Deisseroth, K.; de Lecea, L. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat. Neurosci. 2010, 13, 1526–1533. [Google Scholar] [CrossRef]
- Elam, M.; Svensson, T.H.; Thoren, P. Differentiated cardiovascular afferent regulation of locus coeruleus neurons and sympathetic nerves. Brain Res. 1985, 358, 77–84. [Google Scholar] [CrossRef]
- Elam, M.; Svensson, T.H.; Thorén, P. Locus coeruleus neurons and sympathetic nerves: Activation by cutaneous sensory afferents. Brain Res. 1986, 366, 254–261. [Google Scholar] [CrossRef]
- Elam, M.; Yoa, T.; Svensson, T.H.; Thoren, P. Regulation of locus coeruleus neurons and splanchnic, sympathetic nerves by cardiovascular afferents. Brain Res. 1984, 290, 281–287. [Google Scholar] [CrossRef]
- Svensson, T.H. Peripheral, autonomic regulation of locus coeruleus noradrenergic neurons in brain: putative implications for psychiatry and psychopharmacology. Psychopharmacology 1987, 92, 1–7. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Cohen, J.D. An integrative theory of locus coeruleus-norepinephrine function: Adaptive gain and optimal performance. Annu. Rev. Neurosci. 2005, 28, 403–450. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.R.; Szabadi, E. Functional Neuroanatomy of the Noradrenergic Locus Coeruleus: Its Roles in the Regulation of Arousal and Autonomic Function Part I: Principles of Functional Organisation. Curr. Neuropharmacol. 2008, 6, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.R.; MacLean, A.W.; Brundage, M.D.; Schulze, K. Sleep disturbance in cancer patients. Soc. Sci. Med. 2002, 54, 1309–1321. [Google Scholar] [CrossRef]
- Budhrani, P.H.; Lengacher, C.A.; Kip, K.; Tofthagen, C.; Jim, H. An Integrative Review of Subjective and Objective Measures of Sleep Disturbances in Breast Cancer Survivors. Clin. J. Oncol. Nurs. 2015, 19, 185–191. [Google Scholar] [CrossRef]
- Fiorentino, L.; Rissling, M.; Liu, L.; Ancoli-Israel, S. The symptom cluster of sleep, fatigue and depressive symptoms in breast cancer patients: Severity of the problem and treatment options. Drug Discov. Today Dis. Models 2011, 8, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Erren, T.C.; Morfeld, P.; Foster, R.G.; Reiter, R.J.; Groß, J.V.; Westermann, I.K. Sleep and cancer: Synthesis of experimental data and meta-analyses of cancer incidence among some 1,500,000 study individuals in 13 countries. Chronobiol. Int. 2016, 33, 325–350. [Google Scholar] [CrossRef]
- Segrin, C.; Badger, T.A. Psychological and physical distress are interdependent in breast cancer survivors and their partners. Psychol. Health Med. 2014, 19, 716–723. [Google Scholar] [CrossRef]
- Costa, A.R.; Fontes, F.; Pereira, S.; Gonçalves, M.; Azevedo, A.; Lunet, N. Impact of breast cancer treatments on sleep disturbances—A systematic review. Breast 2014, 23, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.; Sartor, M.-L.; Young, A.H.; Cleare, A.J. Is chronic fatigue syndrome an inflammatory disorder? J. Psychosom. Res. 2016, 85, 82. [Google Scholar] [CrossRef][Green Version]
- Shen, J.; Barbera, J.; Shapiro, C.M. Distinguishing sleepiness and fatigue: Focus on definition and measurement. Sleep Med. Rev. 2006, 10, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.E.; Ganz, P.A.; Irwin, M.R.; Kwan, L.; Breen, E.C.; Cole, S.W. Inflammation and Behavioral Symptoms After Breast Cancer Treatment: Do Fatigue, Depression, and Sleep Disturbance Share a Common Underlying Mechanism? J. Clin. Oncol. 2011, 29, 3517–3522. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.E.; Ganz, P.A.; Aziz, N.; Fahey, J.L. Fatigue and proinflammatory cytokine activity in breast cancer survivors. Psychosom. Med. 2002, 64, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.E.; Lamkin, D.M. Inflammation and cancer-related fatigue: Mechanisms, contributing factors, and treatment implications. Brain Behav. Immun. 2013, 30, S48–S57. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Mills, P.J.; Rissling, M.; Fiorentino, L.; Natarajan, L.; Dimsdale, J.E.; Sadler, G.R.; Parker, B.A.; Ancoli-Israel, S. Fatigue and sleep quality are associated with changes in inflammatory markers in breast cancer patients undergoing chemotherapy. Brain Behav. Immun. 2012, 26, 706–713. [Google Scholar] [CrossRef]
- Krueger, J.M.; Obál, J.; Fang, J.; Kubota, T.; Taishi, P. The role of cytokines in physiological sleep regulation. Ann. N. Y. Acad. Sci. 2001, 933, 211–221. [Google Scholar] [CrossRef]
- Krueger, J.M.; Frank, M.G.; Wisor, J.P.; Roy, S. Sleep function: Toward elucidating an enigma. Sleep Med. Rev. 2016, 28, 46–54. [Google Scholar] [CrossRef]
- Schmidt, E.-M.; Linz, B.; Diekelmann, S.; Besedovsky, L.; Lange, T.; Born, J. Effects of an interleukin-1 receptor antagonist on human sleep, sleep-associated memory consolidation, and blood monocytes. Brain Behav. Immun. 2015, 47, 178–185. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar] [PubMed]
- Ben-Baruch, A. Host microenvironment in breast cancer development: Inflammatory cells, cytokines and chemokines in breast cancer progression: Reciprocal tumor–microenvironment interactions. Breast Cancer Res. 2003, 5, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Role of Chemokines In Tumor Growth. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2065851/ (accessed on 30 April 2019).
- Qian, B.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Mizia-Malarz, A.; Sobol, G.; Woś, H. Proangiogenic factors: vascular-endothelial growth factor (VEGF) and basic fibroblast growth factor--the characteristics and function. Przegl. Lek. 2008, 65, 353–357. [Google Scholar]
- Baldwin, A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J. Clin. Investig. 2001, 107, 241–246. [Google Scholar] [CrossRef]
- Quan, N.; Banks, W.A. Brain-immune communication pathways. Brain Behav. Immun. 2007, 21, 727–735. [Google Scholar] [CrossRef]
- Quan, N. Immune-To-Brain Signaling: How Important are the Blood–Brain Barrier-independent Pathways? Mol. Neurobiol. 2008, 37, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.M.; Nagaraja, S.; Ocampo, A.; Tam, L.T.; Wood, L.S.; Pallegar, P.N.; Greene, J.J.; Geraghty, A.C.; Goldstein, A.K.; Ni, L.; et al. Methotrexate Chemotherapy Induces Persistent Tri-glial Dysregulation that Underlies Chemotherapy-Related Cognitive Impairment. Cell 2019, 176, 43–55.e13. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Lewis, A.M.; Varghese, S.; Xu, H.; Alexander, H.R. Interleukin-1 and cancer progression: The emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med. 2006, 4, 48. [Google Scholar] [CrossRef]
- Elaraj, D.M.; Weinreich, D.M.; Varghese, S.; Puhlmann, M.; Hewitt, S.M.; Carroll, N.M.; Feldman, E.D.; Turner, E.M.; Alexander, H.R. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1088–1096. [Google Scholar] [CrossRef]
- Gemma, A.; Takenaka, K.; Hosoya, Y.; Matuda, K.; Seike, M.; Kurimoto, F.; Ono, Y.; Uematsu, K.; Takeda, Y.; Hibino, S.; et al. Altered expression of several genes in highly metastatic subpopulations of a human pulmonary adenocarcinoma cell line. Eur. J. Cancer Oxf. Engl. 1990 2001, 37, 1554–1561. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Goehler, L.E.; Relton, J.K.; Dripps, D.; Kiechle, R.; Tartaglia, N.; Maier, S.F.; Watkins, L.R. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: A possible mechanism for immune-to-brain communication. Brain Res. Bull. 1997, 43, 357–364. [Google Scholar] [CrossRef]
- Liu, X.; Nemeth, D.P.; McKim, D.B.; Zhu, L.; DiSabato, D.J.; Berdysz, O.; Gorantla, G.; Oliver, B.; Witcher, K.G.; Wang, Y.; et al. Cell-Type-Specific Interleukin 1 Receptor 1 Signaling in the Brain Regulates Distinct Neuroimmune Activities. Immunity 2019, 50, 317–333.e6. [Google Scholar] [CrossRef] [PubMed]
- Imeri, L.; Opp, M.R.; Krueger, J.M. An IL-1 receptor and an IL-1 receptor antagonist attenuate muramyl dipeptide- and IL-1-induced sleep and fever. Am. J. Physiol. 1993, 265, R907–R913. [Google Scholar] [CrossRef] [PubMed]
- Opp, M.R.; Krueger, J.M. Anti-interleukin-1 beta reduces sleep and sleep rebound after sleep deprivation in rats. Am. J. Physiol. 1994, 266, R688–R695. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Kapás, L.; Fang, J.; Krueger, J.M. Somnogenic relationships between tumor necrosis factor and interleukin-1. Am. J. Physiol. 1999, 276, R1132–R1140. [Google Scholar] [CrossRef]
- Nisticò, G.; De Sarro, G. Behavioral and electrocortical spectrum power effects after microinfusion of lymphokines in several areas of the rat brain. Ann. N. Y. Acad. Sci. 1991, 621, 119–134. [Google Scholar] [CrossRef]
- Alam, M.N.; McGinty, D.; Bashir, T.; Kumar, S.; Imeri, L.; Opp, M.R.; Szymusiak, R. Interleukin-1beta modulates state-dependent discharge activity of preoptic area and basal forebrain neurons: role in sleep regulation. Eur. J. Neurosci. 2004, 20, 207–216. [Google Scholar] [CrossRef]
- Kapás, L.; Shibata, M.; Kimura, M.; Krueger, J.M. Inhibition of nitric oxide synthesis suppresses sleep in rabbits. Am. J. Physiol. 1994, 266, R151–R157. [Google Scholar] [CrossRef]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer Oxf. Engl. 1990 2005, 41, 2502–2512. [Google Scholar] [CrossRef]
- Wang, Q.; Horiatis, D.; Pinski, J. Interleukin-6 inhibits the growth of prostate cancer xenografts in mice by the process of neuroendocrine differentiation. Int. J. Cancer 2004, 111, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Michalaki, V.; Syrigos, K.; Charles, P.; Waxman, J. Serum levels of IL-6 and TNF-α correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 2004, 90, 2312–2316. [Google Scholar] [CrossRef] [PubMed]
- Knüpfer, H.; Preiss, R. Significance of interleukin-6 (IL-6) in breast cancer (review). Breast Cancer Res. Treat. 2007, 102, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Songür, N.; Kuru, B.; Kalkan, F.; Ozdilekcan, C.; Cakmak, H.; Hizel, N. Serum interleukin-6 levels correlate with malnutrition and survival in patients with advanced non-small cell lung cancer. Tumori 2004, 90, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Knüpfer, H.; Preiss, R. Serum interleukin-6 levels in colorectal cancer patients—A summary of published results. Int. J. Colorectal Dis. 2010, 25, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 2004, 21, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef]
- Rose-John, S. IL-6 Trans-Signaling via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Reynaud, D.; Pietras, E.; Barry-Holson, K.; Mir, A.; Binnewies, M.; Jeanne, M.; Sala-Torra, O.; Radich, J.P.; Passegué, E. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 2011, 20, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Denson, J.L.; Brüning, J.C. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015, 36, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, X.L.; Qu, Y.; Wu, J.; Zhu, Y.Q.; Sun, W.J.; Li, L.Z. Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett. 2010, 295, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Hemann, M.T. DNA damage-mediated induction of a chemoresistant niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Konsman, J.P.; Luheshi, G.N.; Bluthé, R.M.; Dantzer, R. The vagus nerve mediates behavioural depression, but not fever, in response to peripheral immune signals; a functional anatomical analysis. Eur. J. Neurosci. 2000, 12, 4434–4446. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.F.; Goehler, L.E.; Fleshner, M.; Watkins, L.R. The role of the vagus nerve in cytokine-to-brain communication. Ann. N. Y. Acad. Sci. 1998, 840, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Hohagen, F.; Ebert, T.; Timmer, J.; Ganter, U.; Krieger, S.; Lis, S.; Postler, E.; Voderholzer, U.; Berger, M. Interleukin-6 serum levels in healthy persons correspond to the sleep-wake cycle. Clin. Investig. 1994, 72, 315. [Google Scholar] [CrossRef] [PubMed]
- Vgontzas, A.N.; Papanicolaou, D.A.; Bixler, E.O.; Lotsikas, A.; Zachman, K.; Kales, A.; Prolo, P.; Wong, M.-L.; Licinio, J.; Gold, P.W.; et al. Circadian Interleukin-6 Secretion and Quantity and Depth of Sleep. J. Clin. Endocrinol. Metab. 1999, 84, 2603–2607. [Google Scholar] [CrossRef] [PubMed]
- Redwine, L.; Hauger, R.L.; Gillin, J.C.; Irwin, M. Effects of sleep and sleep deprivation on interleukin-6, growth hormone, cortisol, and melatonin levels in humans. J. Clin. Endocrinol. Metab. 2000, 85, 3597–3603. [Google Scholar] [CrossRef] [PubMed]
- Späth-Schwalbe, E.; Hansen, K.; Schmidt, F.; Schrezenmeier, H.; Marshall, L.; Burger, K.; Fehm, H.L.; Born, J. Acute Effects of Recombinant Human Interleukin-6 on Endocrine and Central Nervous Sleep Functions in Healthy Men. J. Clin. Endocrinol. Metab. 1998, 83, 1573–1579. [Google Scholar] [CrossRef]
- Opp, M.; Obal, F.; Cady, A.B.; Johannsen, L.; Krueger, J.M. Interleukin-6 is pyrogenic but not somnogenic. Physiol. Behav. 1989, 45, 1069–1072. [Google Scholar] [CrossRef]
- Hogan, D.; Morrow, J.D.; Smith, E.M.; Opp, M.R. Interleukin-6 alters sleep of rats. J. Neuroimmunol. 2003, 137, 59–66. [Google Scholar] [CrossRef]
- Dimitrov, S.; Lange, T.; Benedict, C.; Nowell, M.A.; Jones, S.A.; Scheller, J.; Rose-John, S.; Born, J. Sleep enhances IL-6 trans-signaling in humans. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 2174–2176. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P.A.; Trinder, J.; Curtis, N. Sick and tired: Does sleep have a vital role in the immune system? Nat. Rev. Immunol. 2004, 4, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M. Effects of sleep and sleep loss on immunity and cytokines. Brain Behav. Immun. 2002, 16, 503–512. [Google Scholar] [CrossRef]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef]
- Brouckaert, P.G.; Leroux-Roels, G.G.; Guisez, Y.; Tavernier, J.; Fiers, W. In vivo anti-tumour activity of recombinant human and murine TNF, alone and in combination with murine IFN-gamma, on a syngeneic murine melanoma. Int. J. Cancer 1986, 38, 763–769. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Lee, A.; Aldam, G.; Moodie, E.; Thomas, J.A.; Tavernier, J.; Fiers, W. Human tumor xenografts treated with recombinant human tumor necrosis factor alone or in combination with interferons. Cancer Res. 1986, 46, 3990–3993. [Google Scholar]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J. Exp. Med. 1988, 167, 1067–1085. [CrossRef]
- Wallach, D. Preparations of lymphotoxin induce resistance to their own cytotoxic effect. J. Immunol. Baltim. Md. 1950 1984, 132, 2464–2469. [Google Scholar]
- Moore, R.J.; Owens, D.M.; Stamp, G.; Arnott, C.; Burke, F.; East, N.; Holdsworth, H.; Turner, L.; Rollins, B.; Pasparakis, M.; et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 1999, 5, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Szlosarek, P.W.; Grimshaw, M.J.; Kulbe, H.; Wilson, J.L.; Wilbanks, G.D.; Burke, F.; Balkwill, F.R. Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium. Mol. Cancer Ther. 2006, 5, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bäumel, M.; Männel, D.N.; Howard, O.M.Z.; Oppenheim, J.J. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol. Baltim. Md. 1950 2007, 179, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF–TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Kastin, A.J.; Daniel, J.; Yu, C.; Baryshnikova, L.M.; von Bartheld, C.S. TNFα trafficking in cerebral vascular endothelial cells. J. Neuroimmunol. 2007, 185, 47–56. [Google Scholar] [CrossRef][Green Version]
- Pan, W.; Stone, K.P.; Hsuchou, H.; Manda, V.K.; Zhang, Y.; Kastin, A.J. Cytokine signaling modulates blood-brain barrier function. Curr. Pharm. Des. 2011, 17, 3729–3740. [Google Scholar] [CrossRef]
- Opp, M.R.; Toth, L.A. Neural-immune interactions in the regulation of sleep. Front. Biosci. J. Virtual Libr. 2003, 8, d768–d779. [Google Scholar] [CrossRef]
- Bredow, S.; Guha-Thakurta, N.; Taishi, P.; Obál, F.; Krueger, J.M. Diurnal variations of tumor necrosis factor alpha mRNA and alpha-tubulin mRNA in rat brain. Neuroimmunomodulation 1997, 4, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Krueger, J.M. Diurnal variation of TNF alpha in the rat brain. Neuroreport 1997, 8, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Shoham, S.; Davenne, D.; Cady, A.B.; Dinarello, C.A.; Krueger, J.M. Recombinant tumor necrosis factor and interleukin 1 enhance slow-wave sleep. Am. J. Physiol. 1987, 253, R142–R149. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wang, Y.; Krueger, J.M. Mice lacking the TNF 55 kDa receptor fail to sleep more after TNFalpha treatment. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 5949–5955. [Google Scholar] [CrossRef]
- Kubota, T.; Li, N.; Guan, Z.; Brown, R.A.; Krueger, J.M. Intrapreoptic microinjection of TNF-alpha enhances non-REM sleep in rats. Brain Res. 2002, 932, 37–44. [Google Scholar] [CrossRef]
- Zhan, S.; Cai, G.-Q.; Zheng, A.; Wang, Y.; Jia, J.; Fang, H.; Yang, Y.; Hu, M.; Ding, Q. Tumor necrosis factor-alpha regulates the Hypocretin system via mRNA degradation and ubiquitination. Biochim. Biophys. Acta 2011, 1812, 565–571. [Google Scholar] [CrossRef] [PubMed]
- De Sarro, G.; Gareri, P.; Sinopoli, V.A.; David, E.; Rotiroti, D. Comparative, behavioural and electrocortical effects of tumor necrosis factor-alpha and interleukin-1 microinjected into the locus coeruleus of rat. Life Sci. 1997, 60, 555–564. [Google Scholar] [CrossRef]
- Terao, A.; Matsumura, H.; Yoneda, H.; Saito, M. Enhancement of slow-wave sleep by tumor necrosis factor-alpha is mediated by cyclooxygenase-2 in rats. Neuroreport 1998, 9, 3791–3796. [Google Scholar] [CrossRef] [PubMed]
- Obal, F.; Krueger, J.M. Biochemical regulation of non-rapid-eye-movement sleep. Front. Biosci. J. Virtual Libr. 2003, 8, d520–d550. [Google Scholar]
- Engle, S.J.; Hoying, J.B.; Boivin, G.P.; Ormsby, I.; Gartside, P.S.; Doetschman, T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999, 59, 3379–3386. [Google Scholar]
- Padua, D.; Massagué, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Chau, G.-Y.; Wu, C.-W.; Lui, W.-Y.; Chang, T.-J.; Kao, H.-L.; Wu, L.-H.; King, K.-L.; Loong, C.-C.; Hsia, C.-Y.; Chi, C.-W. Serum Interleukin-10 But Not Interleukin-6 Is Related to Clinical Outcome in Patients With Resectable Hepatocellular Carcinoma. Ann. Surg. 2000, 231, 552–558. [Google Scholar] [CrossRef] [PubMed]
- König, A.; Vilsmaier, T.; Rack, B.; Friese, K.; Janni, W.; Jeschke, U.; Andergassen, U.; Trapp, E.; Jückstock, J.; Jäger, B.; et al. Determination of Interleukin-4, -5, -6, -8 and -13 in Serum of Patients with Breast Cancer Before Treatment and its Correlation to Circulating Tumor Cells. Anticancer Res. 2016, 36, 3123–3130. [Google Scholar] [PubMed]
- Goldstein, R.; Hanley, C.; Morris, J.; Cahill, D.; Chandra, A.; Harper, P.; Chowdhury, S.; Maher, J.; Burbridge, S. Clinical Investigation of the Role of Interleukin-4 and Interleukin-13 in the Evolution of Prostate Cancer. Cancers 2011, 3, 4281–4293. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S.; Marincola, F.M.; Young, H.A. Interleukin-10 and the immune response against cancer: A counterpoint. J. Leukoc. Biol. 2005, 78, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Joyce, J.A. Alternative activation of tumor-associated macrophages by IL-4. Cell Cycle 2010, 9, 4824–4835. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the brain: A cytokine to remember. J. Immunol. Baltim. Md. 1950 2012, 189, 4213–4219. [Google Scholar] [CrossRef]
- Kushikata, T.; Fang, J.; Krueger, J.M. Interleukin-10 inhibits spontaneous sleep in rabbits. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 1999, 19, 1025–1030. [Google Scholar] [CrossRef]
- Kushikata, T.; Fang, J.; Wang, Y.; Krueger, J.M. Interleukin-4 inhibits spontaneous sleep in rabbits. Am. J. Physiol. 1998, 275, R1185–R1191. [Google Scholar] [CrossRef] [PubMed]
- Opp, M.R.; Smith, E.M.; Hughes, T.K. Interleukin-10 (cytokine synthesis inhibitory factor) acts in the central nervous system of rats to reduce sleep. J. Neuroimmunol. 1995, 60, 165–168. [Google Scholar] [CrossRef]
- Kubota, T.; Fang, J.; Kushikata, T.; Krueger, J.M. Interleukin-13 and transforming growth factor-beta1 inhibit spontaneous sleep in rabbits. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R786–R792. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-C.; Hsiao, M. Ghrelin and cancer progression. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Kangawa, K. Ghrelin: Structure and function. Physiol. Rev. 2005, 85, 495–522. [Google Scholar] [CrossRef]
- Cassoni, P.; Ghé, C.; Marrocco, T.; Tarabra, E.; Allia, E.; Catapano, F.; Deghenghi, R.; Ghigo, E.; Papotti, M.; Muccioli, G. Expression of ghrelin and biological activity of specific receptors for ghrelin and des-acyl ghrelin in human prostate neoplasms and related cell lines. Eur. J. Endocrinol. 2004, 150, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Toshinai, K.; Yamaguchi, H.; Sun, Y.; Smith, R.G.; Yamanaka, A.; Sakurai, T.; Date, Y.; Mondal, M.S.; Shimbara, T.; Kawagoe, T.; et al. Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology 2006, 147, 2306–2314. [Google Scholar] [CrossRef]
- Delhanty, P.J.D.; Neggers, S.J.; van der Lely, A.J. Mechanisms in endocrinology: Ghrelin: The differences between acyl- and des-acyl ghrelin. Eur. J. Endocrinol. 2012, 167, 601–608. [Google Scholar] [CrossRef]
- Au, C.C.; Furness, J.B.; Brown, K.A. Ghrelin and Breast Cancer: Emerging Roles in Obesity, Estrogen Regulation, and Cancer. Front. Oncol. 2017, 6. [Google Scholar] [CrossRef]
- Grönberg, M.; Fjällskog, M.-L.; Jirström, K.; Janson, E.T. Expression of ghrelin is correlated to a favorable outcome in invasive breast cancer. Acta Oncol. Stockh. Swed. 2012, 51, 386–393. [Google Scholar] [CrossRef]
- Banks, W.A.; Tschöp, M.; Robinson, S.M.; Heiman, M.L. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J. Pharmacol. Exp. Ther. 2002, 302, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Tolle, V.; Bassant, M.-H.; Zizzari, P.; Poindessous-Jazat, F.; Tomasetto, C.; Epelbaum, J.; Bluet-Pajot, M.-T. Ultradian rhythmicity of ghrelin secretion in relation with GH, feeding behavior, and sleep-wake patterns in rats. Endocrinology 2002, 143, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Obal, F.; Alt, J.; Taishi, P.; Gardi, J.; Krueger, J.M. Sleep in mice with nonfunctional growth hormone-releasing hormone receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R131–R139. [Google Scholar] [CrossRef] [PubMed]
- Steiger, A. Ghrelin and sleep-wake regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R573–R574. [Google Scholar] [CrossRef] [PubMed]
- Szentirmai, E.; Hajdu, I.; Obal, F.; Krueger, J.M. Ghrelin-induced sleep responses in ad libitum fed and food-restricted rats. Brain Res. 2006, 1088, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Weikel, J.C.; Wichniak, A.; Ising, M.; Brunner, H.; Friess, E.; Held, K.; Mathias, S.; Schmid, D.A.; Uhr, M.; Steiger, A. Ghrelin promotes slow-wave sleep in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E407–E415. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Lin, L.; Austin, D.; Young, T.; Mignot, E. Short Sleep Duration Is Associated with Reduced Leptin, Elevated Ghrelin, and Increased Body Mass Index. PLoS Med. 2004, 1. [Google Scholar] [CrossRef]
- Schmid, S.M.; Hallschmid, M.; Jauch-Chara, K.; Born, J.; Schultes, B. A single night of sleep deprivation increases ghrelin levels and feelings of hunger in normal-weight healthy men. J. Sleep Res. 2008, 17, 331–334. [Google Scholar] [CrossRef]
- Yamanaka, A.; Beuckmann, C.T.; Willie, J.T.; Hara, J.; Tsujino, N.; Mieda, M.; Tominaga, M.; Yagami, K.; Sugiyama, F.; Goto, K.; et al. Hypothalamic Orexin Neurons Regulate Arousal According to Energy Balance in Mice. Neuron 2003, 38, 701–713. [Google Scholar] [CrossRef]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef]
- Garofalo, C.; Surmacz, E. Leptin and cancer. J. Cell. Physiol. 2006, 207, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Baumann, H.; Morella, K.K.; White, D.W.; Dembski, M.; Bailon, P.S.; Kim, H.; Lai, C.F.; Tartaglia, L.A. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. USA 1996, 93, 8374–8378. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-K.; Ahima, R.S. Leptin signaling. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Somasundar, P.; Frankenberry, K.A.; Skinner, H.; Vedula, G.; McFadden, D.W.; Riggs, D.; Jackson, B.; Vangilder, R.; Hileman, S.M.; Vona-Davis, L.C. Prostate cancer cell proliferation is influenced by leptin. J. Surg. Res. 2004, 118, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Zhang, H.-T.; Du, L.; Liu, X.; Jing, J.; Zhao, X.; Yang, X.; Tian, B. Serum levels of leptin, insulin, and lipids in relation to breast cancer in china. Endocrine 2005, 26, 19–24. [Google Scholar] [CrossRef]
- Chen, D.-C.; Chung, Y.-F.; Yeh, Y.-T.; Chaung, H.-C.; Kuo, F.-C.; Fu, O.-Y.; Chen, H.-Y.; Hou, M.-F.; Yuan, S.-S.F. Serum adiponectin and leptin levels in Taiwanese breast cancer patients. Cancer Lett. 2006, 237, 109–114. [Google Scholar] [CrossRef]
- Stattin, P.; Palmqvist, R.; Söderberg, S.; Biessy, C.; Ardnor, B.; Hallmans, G.; Kaaks, R.; Olsson, T. Plasma leptin and colorectal cancer risk: A prospective study in Northern Sweden. Oncol. Rep. 2003, 10, 2015–2021. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Huang, W.; Jaspan, J.B.; Maness, L.M. Leptin enters the brain by a saturable system independent of insulin. Peptides 1996, 17, 305–311. [Google Scholar] [CrossRef]
- Schoeller, D.A.; Cella, L.K.; Sinha, M.K.; Caro, J.F. Entrainment of the diurnal rhythm of plasma leptin to meal timing. J. Clin. Investig. 1997, 100, 1882–1887. [Google Scholar] [CrossRef]
- Spiegel, K.; Tasali, E.; Penev, P.; Van Cauter, E. Brief communication: Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann. Intern. Med. 2004, 141, 846–850. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J. Leptin: A biomarker for sleep disorders? Sleep Med. Rev. 2014, 18, 283–290. [Google Scholar] [CrossRef]
- Sinton, C.M.; Fitch, T.E.; Gershenfeld, H.K. The effects of leptin on REM sleep and slow wave delta in rats are reversed by food deprivation. J. Sleep Res. 1999, 8, 197–203. [Google Scholar] [CrossRef]
- Laposky, A.D.; Bradley, M.A.; Williams, D.L.; Bass, J.; Turek, F.W. Sleep-wake regulation is altered in leptin-resistant (db/db) genetically obese and diabetic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R2059–R2066. [Google Scholar] [CrossRef]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar] [CrossRef]
- Avril, N.; Menzel, M.; Dose, J.; Schelling, M.; Weber, W.; Jänicke, F.; Nathrath, W.; Schwaiger, M. Glucose metabolism of breast cancer assessed by 18F-FDG PET: Histologic and immunohistochemical tissue analysis. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2001, 42, 9–16. [Google Scholar]
- Poschke, I.; Mao, Y.; Kiessling, R.; de Boniface, J. Tumor-dependent increase of serum amino acid levels in breast cancer patients has diagnostic potential and correlates with molecular tumor subtypes. J. Transl. Med. 2013, 11, 290. [Google Scholar] [CrossRef]
- Lin, X.; Hong, S.; Huang, J.; Chen, Y.; Chen, Y.; Wu, Z. Plasma Apolipoprotein A1 Levels at Diagnosis Are Independent Prognostic Factors in Invasive Ductal Breast Cancer. Discov. Med. 2017, 23, 247–258. [Google Scholar] [PubMed]
- Ryu, T.Y.; Park, J.; Scherer, P.E. Hyperglycemia as a Risk Factor for Cancer Progression. Diabetes Metab. J. 2014, 38, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.M.; Wojtkowiak, J.W.; Hashim, A.I.; Gillies, R.J. Targeting the Metabolic Microenvironment of Tumors. Adv. Pharmacol. San Diego Calif. 2012, 65, 63–107. [Google Scholar]
- Burdakov, D.; Jensen, L.T.; Alexopoulos, H.; Williams, R.H.; Fearon, I.M.; O’Kelly, I.; Gerasimenko, O.; Fugger, L.; Verkhratsky, A. Tandem-Pore K+ Channels Mediate Inhibition of Orexin Neurons by Glucose. Neuron 2006, 50, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Sakurai, T.; Nambu, T.; Yanagisawa, M.; Goto, K. Neurons containing orexin in the lateral hypothalamic area of the adult rat brain are activated by insulin-induced acute hypoglycemia. Neurosci. Lett. 1999, 264, 101–104. [Google Scholar] [CrossRef]
- Kong, D.; Vong, L.; Parton, L.E.; Ye, C.; Tong, Q.; Hu, X.; Choi, B.; Brüning, J.C.; Lowell, B.B. Glucose Stimulation of Hypothalamic MCH Neurons Involves KATP Channels, Is Modulated by UCP2, and Regulates Peripheral Glucose Homeostasis. Cell Metab. 2010, 12, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Karnani, M.M.; Apergis-Schoute, J.; Adamantidis, A.; Jensen, L.T.; de Lecea, L.; Fugger, L.; Burdakov, D. Activation of Central Orexin/Hypocretin Neurons by Dietary Amino Acids. Neuron 2011, 72, 616–629. [Google Scholar] [CrossRef]
- Williams, R.H.; Jensen, L.T.; Verkhratsky, A.; Fugger, L.; Burdakov, D. Control of hypothalamic orexin neurons by acid and CO2. Proc. Natl. Acad. Sci. USA 2007, 104, 10685–10690. [Google Scholar] [CrossRef] [PubMed]
- Masri, S.; Papagiannakopoulos, T.; Kinouchi, K.; Liu, Y.; Cervantes, M.; Baldi, P.; Jacks, T.; Sassone-Corsi, P. Lung Adenocarcinoma Distally Rewires Hepatic Circadian Homeostasis. Cell 2016, 165, 896–909. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, C.S.; Kiss, M.G.; Rattik, S.; He, S.; Vassalli, A.; Valet, C.; Anzai, A.; Chan, C.T.; Mindur, J.E.; Kahles, F.; et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 2019. [Google Scholar] [CrossRef] [PubMed]
- Van Dycke, K.C.G.; Rodenburg, W.; van Oostrom, C.T.M.; van Kerkhof, L.W.M.; Pennings, J.L.A.; Roenneberg, T.; van Steeg, H.; van der Horst, G.T.J. Chronically Alternating Light Cycles Increase Breast Cancer Risk in Mice. Curr. Biol. 2015, 25, 1932–1937. [Google Scholar] [CrossRef]
- Ryan, S.; Taylor, C.T.; McNicholas, W.T. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005, 112, 2660–2667. [Google Scholar] [CrossRef]
- Campos-Rodriguez, F.; Martinez-Garcia, M.A.; Martinez, M.; Duran-Cantolla, J.; Peña, M.L.; Masdeu, M.J.; Gonzalez, M.; Campo, F.; Gallego, I.; Marin, J.M.; et al. Association between obstructive sleep apnea and cancer incidence in a large multicenter Spanish cohort. Am. J. Respir. Crit. Care Med. 2013, 187, 99–105. [Google Scholar] [CrossRef]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Bhatty, R.; Kamat, A.A.; Landen, C.N.; Han, L.; Thaker, P.H.; Li, Y.; Gershenson, D.M.; Lutgendorf, S.; Cole, S.W. Stress Hormone–Mediated Invasion of Ovarian Cancer Cells. Clin. Cancer Res. 2006, 12, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Antoni, M.H.; Lutgendorf, S.K.; Cole, S.W.; Dhabhar, F.S.; Sephton, S.E.; McDonald, P.G.; Stefanek, M.; Sood, A.K. The influence of bio-behavioural factors on tumour biology: Pathways and mechanisms. Nat. Rev. Cancer 2006, 6, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Obradović, M.M.S.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Münst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nature 2019, 567, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.Y.; Eichler, V.B. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. 1972, 42, 201–206. [Google Scholar] [CrossRef]
- Oster, H.; Damerow, S.; Kiessling, S.; Jakubcakova, V.; Abraham, D.; Tian, J.; Hoffmann, M.W.; Eichele, G. The circadian rhythm of glucocorticoids is regulated by a gating mechanism residing in the adrenal cortical clock. Cell Metab. 2006, 4, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Vinogradova, I.A.; Panchenko, A.V.; Popovich, I.G.; Zabezhinski, M.A. Light-at-night-induced circadian disruption, cancer and aging. Curr. Aging Sci. 2012, 5, 170–177. [Google Scholar] [CrossRef]
- Cadenas, C.; van de Sandt, L.; Edlund, K.; Lohr, M.; Hellwig, B.; Marchan, R.; Schmidt, M.; Rahnenführer, J.; Oster, H.; Hengstler, J.G. Loss of circadian clock gene expression is associated with tumor progression in breast cancer. Cell Cycle Georget. Tex. 2014, 13, 3282–3291. [Google Scholar] [CrossRef]
- Dauchy, R.T.; Xiang, S.; Mao, L.; Brimer, S.; Wren, M.A.; Yuan, L.; Anbalagan, M.; Hauch, A.; Frasch, T.; Rowan, B.G.; et al. Circadian and Melatonin Disruption by Exposure to Light at Night Drives Intrinsic Resistance to Tamoxifen Therapy in Breast Cancer. Cancer Res. 2014, 74, 4099–4110. [Google Scholar] [CrossRef]
- Dycke, K.C.G.V.; Nijman, R.M.; Wackers, P.F.K.; Jonker, M.J.; Rodenburg, W.; van Oostrom, C.T.M.; Salvatori, D.C.F.; Breit, T.M.; van Steeg, H.; Luijten, M.; et al. A day and night difference in the response of the hepatic transcriptome to cyclophosphamide treatment. Arch. Toxicol. 2015, 89, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Tuveson, D.A. Maximizing mouse cancer models. Nat. Rev. Cancer 2007, 7, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Vander Heiden, M.G.; Jacks, T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Chance, W.T.; Dayal, R.; Friend, L.A.; James, J.H. Elevated blood lactate is not a primary cause of anorexia in tumor-bearing rats. Nutr. Cancer 2004, 48, 174–181. [Google Scholar] [CrossRef]
- Cortes-Campos, C.; Elizondo, R.; Carril, C.; Martínez, F.; Boric, K.; Nualart, F.; Garcia-Robles, M.A. MCT2 Expression and Lactate Influx in Anorexigenic and Orexigenic Neurons of the Arcuate Nucleus. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Kaur, S.; Wang, J.L.; Ferrari, L.; Thankachan, S.; Kroeger, D.; Venner, A.; Lazarus, M.; Wellman, A.; Arrigoni, E.; Fuller, P.M.; et al. A Genetically-Defined Circuit for Arousal from Sleep during Hypercapnia. Neuron 2017, 96, 1153–1167.e5. [Google Scholar] [CrossRef]
- Campos, C.A.; Bowen, A.J.; Han, S.; Wisse, B.E.; Palmiter, R.D.; Schwartz, M.W. Cancer-induced anorexia and malaise are mediated by CGRP neurons in the parabrachial nucleus. Nat. Neurosci. 2017, 20, 934–942. [Google Scholar] [CrossRef]
- Campos, C.A.; Bowen, A.J.; Schwartz, M.W.; Palmiter, R.D. Parabrachial CGRP Neurons Control Meal Termination. Cell Metab. 2016, 23, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Lipp, A.; Tank, J.; Trottenberg, T.; Kupsch, A.; Arnold, G.; Jordan, J. Sympathetic activation due to deep brain stimulation in the region of the STN. Neurology 2005, 65, 774–775. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.E.; Crosswell, A.D.; Stanton, A.L.; Crespi, C.M.; Winston, D.; Arevalo, J.; Ma, J.; Cole, S.W.; Ganz, P.A. Mindfulness meditation for younger breast cancer survivors: A randomized controlled trial. Cancer 2015, 121, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Black, D.S.; Slavich, G.M. Mindfulness meditation and the immune system: a systematic review of randomized controlled trials. Ann. N. Y. Acad. Sci. 2016, 1373, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin Selectively Targets Cancer Stem Cells, and Acts Together with Chemotherapy to Block Tumor Growth and Prolong Remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Mormont, M.-C.; Levi, F. Cancer chronotherapy: principles, applications, and perspectives. Cancer 2003, 97, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Granda, T.G.; Filipski, E.; D’Attino, R.M.; Vrignaud, P.; Anjo, A.; Bissery, M.C.; Lévi, F. Experimental Chronotherapy of Mouse Mammary Adenocarcinoma MA13/C with Docetaxel and Doxorubicin as Single Agents and in Combination. Cancer Res. 2001, 61, 1996–2001. [Google Scholar] [PubMed]
- Borniger, J.C.; Walker II, W.H.; Gaudier-Diaz, M.M.; Stegman, C.J.; Zhang, N.; Hollyfield, J.L.; Nelson, R.J.; DeVries, A.C. Time-of-Day Dictates Transcriptional Inflammatory Responses to Cytotoxic Chemotherapy. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Nohara, K.; Park, N.; Park, Y.-S.; Guillory, B.; Zhao, Z.; Garcia, J.M.; Koike, N.; Lee, C.C.; Takahashi, J.S.; et al. The Small Molecule Nobiletin Targets the Molecular Oscillator to Enhance Circadian Rhythms and Protect against Metabolic Syndrome. Cell Metab. 2016, 23, 610–621. [Google Scholar] [CrossRef]
- Chen, J.; Chen, A.Y.; Huang, H.; Ye, X.; Rollyson, W.D.; Perry, H.E.; Brown, K.C.; Rojanasakul, Y.; Rankin, G.O.; Dasgupta, P.; et al. The flavonoid nobiletin inhibits tumor growth and angiogenesis of ovarian cancers via the Akt pathway. Int. J. Oncol. 2015, 46, 2629–2638. [Google Scholar] [CrossRef]
- Li, N.; Zhang, Z.; Jiang, G.; Sun, H.; Yu, D. Nobiletin sensitizes colorectal cancer cells to oxaliplatin by PI3K/Akt/MTOR pathway. Front. Biosci. Landmark Ed. 2019, 24, 303–312. [Google Scholar] [PubMed]
- Luo, G.; Guan, X.; Zhou, L. Apoptotic effect of citrus fruit extract nobiletin on lung cancer cell line A549 in vitro and in vivo. Cancer Biol. Ther. 2008, 7, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ono, M.; Takeshima, M.; Nakano, S. Antiproliferative and Apoptosis-inducing Activity of Nobiletin Against Three Subtypes of Human Breast Cancer Cell Lines. Anticancer Res. 2014, 34, 1785–1792. [Google Scholar] [PubMed]
- MA, W.; FENG, S.; Yao, X.-J.; YUAN, Z.; Liu, L.; Xie, Y. Use of Nobiletin in Cancer Treatment. US20170143668A1, 25 May 2017. [Google Scholar]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, W.H., II; Borniger, J.C. Molecular Mechanisms of Cancer-Induced Sleep Disruption. Int. J. Mol. Sci. 2019, 20, 2780. https://doi.org/10.3390/ijms20112780
Walker WH II, Borniger JC. Molecular Mechanisms of Cancer-Induced Sleep Disruption. International Journal of Molecular Sciences. 2019; 20(11):2780. https://doi.org/10.3390/ijms20112780
Chicago/Turabian StyleWalker, William H., II, and Jeremy C. Borniger. 2019. "Molecular Mechanisms of Cancer-Induced Sleep Disruption" International Journal of Molecular Sciences 20, no. 11: 2780. https://doi.org/10.3390/ijms20112780
APA StyleWalker, W. H., II, & Borniger, J. C. (2019). Molecular Mechanisms of Cancer-Induced Sleep Disruption. International Journal of Molecular Sciences, 20(11), 2780. https://doi.org/10.3390/ijms20112780