Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy


Abstract
:1. Introduction
2. Cellular Systems Related to AD
2.1. Microglia
2.2. Glial Cells
2.2.1. Astrocytes
2.2.2. Oligodendrocytes
2.2.3. NG2-Glia
2.3. Neurons
3. Organelles and Organelle Related Processes
3.1. Mitochondria
3.2. Autophagy (Mitophagy)
- Macroautophagy—engulfment of cytoplasmic components by autophagy vesicles and fusion with lysosomes regulated by autophagy-related proteins and specific autophagy receptors.
- Microautophagy—direct engulfment of cytoplasmic components by lysosomes [92].
3.3. Endocytic Processes
4. Stem Cell Therapies
4.1. Endogenous Regeneration
4.2. Engrafted Regeneration
4.1.1. ESCs
4.1.2. NSCs
4.1.3. iPSCs
4.1.4. MSCs
4.3. Translation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APP | Amyloid Precursor Protein |
| APOE | Apolipoprotein E |
| CIE | Clathrin-independent endocytosis |
| CME | Clathrin-mediated endocytosis |
| HC | Hippocampus |
| SGZ | Sub-granular zone |
| SVZ | Subventricular zone |
| ESCs | Embryonic stem cells |
| NSCs | Neural stem cells |
| iPSCs | Induced Pluripotent stem cells |
| MSCs | Mesenchymal stem cells |
References
- Yankner, B.A.; Lu, T. Amyloid beta-protein toxicity and the pathogenesis of alzheimer disease. J. Biol. Chem. 2009, 284, 4755–4759. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin e. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.; Tremblay, M.E. Chronic stress as a risk factor for alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol. Stress 2018, 9, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, A.; Takeuchi, H.; Takahashi, K.; Tanaka, F. Microglia in alzheimer’s disease: Risk factors and inflammation. Front. Neurol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Rezaie, P.; Male, D. Mesoglia & microglia--a historical review of the concept of mononuclear phagocytes within the central nervous system. J. Hist. Neurosci. 2002, 11, 325–374. [Google Scholar]
- Chan, W.Y.; Kohsaka, S.; Rezaie, P. The origin and cell lineage of microglia: New concepts. Brain Res. Rev. 2007, 53, 344–354. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Tgf-beta pathway as a potential target in neurodegeneration and alzheimer’s. Curr. Alzheimer Res. 2006, 3, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Xue, Q.S. Life and death of microglia. J. Neuroimmune. Pharm. 2009, 4, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Zinn, R.; Vissel, B. Microglia: A new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol. Learn. Mem. 2013, 105, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial p2y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C. Early and late cns inflammation in alzheimer’s disease: Two extremes of a continuum? Trends Pharm. Sci. 2017, 38, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. Nlrp3 is activated in alzheimer’s disease and contributes to pathology in app/ps1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in alzheimer’s disease. J. Clin. Invest. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in alzheimer’s disease. J. Cell. Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. Trem2 binds to apolipoproteins, including apoe and clu/apoj, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. Trem2, microglia, and neurodegenerative diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. Trem2 lipid sensing sustains the microglial response in an alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The trem2-apoe pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581 e569. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; De Roeck, A.; Van den Bossche, T.; Van Cauwenberghe, C.; Bettens, K.; Vermeulen, S.; Mattheijssens, M.; Peeters, K.; Engelborghs, S.; Vandenbulcke, M.; et al. Mutations in abca7 in a belgian cohort of alzheimer’s disease patients: A targeted resequencing study. Lancet Neurol. 2015, 14, 814–822. [Google Scholar] [CrossRef]
- Kim, W.S.; Li, H.; Ruberu, K.; Chan, S.; Elliott, D.A.; Low, J.K.; Cheng, D.; Karl, T.; Garner, B. Deletion of abca7 increases cerebral amyloid-beta accumulation in the j20 mouse model of alzheimer’s disease. J. Neurosci. 2013, 33, 4387–4394. [Google Scholar] [CrossRef]
- Jehle, A.W.; Gardai, S.J.; Li, S.; Linsel-Nitschke, P.; Morimoto, K.; Janssen, W.J.; Vandivier, R.W.; Wang, N.; Greenberg, S.; Dale, B.M.; et al. Atp-binding cassette transporter a7 enhances phagocytosis of apoptotic cells and associated erk signaling in macrophages. J. Cell. Biol. 2006, 174, 547–556. [Google Scholar] [CrossRef]
- Abe-Dohmae, S.; Ikeda, Y.; Matsuo, M.; Hayashi, M.; Okuhira, K.; Ueda, K.; Yokoyama, S. Human abca7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 2004, 279, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Volonte, C.; Apolloni, S.; Skaper, S.D.; Burnstock, G. P2x7 receptors: Channels, pores and more. CNS Neurol. Disord. Drug Targets 2012, 11, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Monif, M.; Reid, C.A.; Powell, K.L.; Smart, M.L.; Williams, D.A. The p2x7 receptor drives microglial activation and proliferation: A trophic role for p2x7r pore. J. Neurosci. 2009, 29, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, P.; Zhang, J.; Chen, W.; Gu, L. Silencing of the p2x(7) receptor enhances amyloid-beta phagocytosis by microglia. Biochem. Biophys. Res. Commun. 2013, 434, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Numakawa, T.; Shimazu, K.; Koshimizu, H.; Hara, T.; Hatanaka, H.; Mei, L.; Lu, B.; Kojima, M. Bdnf-induced recruitment of trkb receptor into neuronal lipid rafts: Roles in synaptic modulation. J. Cell. Biol. 2004, 167, 1205–1215. [Google Scholar] [CrossRef]
- Shieh, C.H.; Heinrich, A.; Serchov, T.; van Calker, D.; Biber, K. P2x7-dependent, but differentially regulated release of il-6, ccl2, and tnf-alpha in cultured mouse microglia. Glia 2014, 62, 592–607. [Google Scholar] [CrossRef]
- Murphy, N.; Lynch, M.A. Activation of the p2x(7) receptor induces migration of glial cells by inducing cathepsin b degradation of tissue inhibitor of metalloproteinase 1. J. Neurochem. 2012, 123, 761–770. [Google Scholar] [CrossRef]
- Bartlett, R.; Yerbury, J.J.; Sluyter, R. P2x7 receptor activation induces reactive oxygen species formation and cell death in murine eoc13 microglia. Mediat. Inflamm. 2013, 2013, 271813. [Google Scholar] [CrossRef]
- He, Y.; Taylor, N.; Fourgeaud, L.; Bhattacharya, A. The role of microglial p2x7: Modulation of cell death and cytokine release. J. Neuroinflamm. 2017, 14, 135. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Jones, D.N.C. Emerging role of the p2x7-nlrp3-il1beta pathway in mood disorders. Psychoneuroendocrinology 2018, 98, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. Liaisons dangereuses: P2x(7) and the inflammasome. Trends Pharm. Sci. 2007, 28, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Thawkar, B.S.; Kaur, G. Inhibitors of nf-kappab and p2x7/nlrp3/caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef]
- Fields, R.D.; Araque, A.; Johansen-Berg, H.; Lim, S.S.; Lynch, G.; Nave, K.A.; Nedergaard, M.; Perez, R.; Sejnowski, T.; Wake, H. Glial biology in learning and cognition. Neuroscientist 2014, 20, 426–431. [Google Scholar] [CrossRef]
- Jourdain, P.; Becq, F.; Lengacher, S.; Boinot, C.; Magistretti, P.J.; Marquet, P. The human cftr protein expressed in cho cells activates aquaporin-3 in a camp-dependent pathway: Study by digital holographic microscopy. J. Cell. Sci. 2014, 127, 546–556. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein e promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Mohamed, A.; Posse de Chaves, E. Abeta internalization by neurons and glia. Int. J. Alzheimers Dis. 2011, 2011, 127984. [Google Scholar] [PubMed]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate a beta 42 and give rise to astrocytic amyloid plaques in alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Leal, M.C.; Dorfman, V.B.; Gamba, A.F.; Frangione, B.; Wisniewski, T.; Castano, E.M.; Sigurdsson, E.M.; Morelli, L. Plaque-associated overexpression of insulin-degrading enzyme in the cerebral cortex of aged transgenic tg2576 mice with alzheimer pathology. J. Neuropathol. Exp. Neurol. 2006, 65, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Mulder, S.D.; Veerhuis, R.; Blankenstein, M.A.; Nielsen, H.M. The effect of amyloid associated proteins on the expression of genes involved in amyloid-beta clearance by adult human astrocytes. Exp. Neurol. 2012, 233, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Pihlaja, R.; Koistinaho, J.; Kauppinen, R.; Sandholm, J.; Tanila, H.; Koistinaho, M. Multiple cellular and molecular mechanisms are involved in human abeta clearance by transplanted adult astrocytes. Glia 2011, 59, 1643–1657. [Google Scholar] [CrossRef]
- Delekate, A.; Fuchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic p2y1 receptor signalling mediates astrocytic hyperactivity in vivo in an alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.Z.; Lipski, J. Involvement of trpv4 channels in abeta(40)-induced hippocampal cell death and astrocytic ca(2+) signalling. Neurotoxicology 2014, 41, 64–72. [Google Scholar] [CrossRef]
- Xiu, J.; Nordberg, A.; Zhang, J.T.; Guan, Z.Z. Expression of nicotinic receptors on primary cultures of rat astrocytes and up-regulation of the alpha7, alpha4 and beta2 subunits in response to nanomolar concentrations of the beta-amyloid peptide(1-42). Neurochem. Int. 2005, 47, 281–290. [Google Scholar] [CrossRef]
- Lee, L.; Kosuri, P.; Arancio, O. Picomolar amyloid-beta peptides enhance spontaneous astrocyte calcium transients. J. Alzheimers Dis. 2014, 38, 49–62. [Google Scholar] [CrossRef]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-beta and alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell. Death Dis. 2013, 4, e623. [Google Scholar] [CrossRef]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Lim, D. Differential deregulation of astrocytic calcium signalling by amyloid-beta, tnfalpha, Il-1beta and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Augusto, E.; Machado, N.J.; dos Santos-Rodrigues, A.; Cunha, R.A.; Agostinho, P. Astrocytic adenosine a2a receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J. Alzheimers Dis. 2012, 31, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of abeta1-40 and abeta1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Malone, M.J.; Szoke, M.C. Neurochemical changes in white matter: Aged human brain and alzheimer’s disease. Arch. Neurol. 1985, 42, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.; Scheltens, P.; Gholkar, A.; Ballard, C.; McKeith, I.; Ince, P.; Perry, R.; O’brien, J. White matter lesions on magnetic resonance imaging in dementia with lewy bodies, alzheimer’s disease, vascular dementia, and normal aging. J. Neurol. Neurosurg. Psychiatry 1999, 67, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Cummings, J.L.; Sultzer, D.; Henderson, V.W.; Nuechterlein, K.H.; Mintz, J. White matter structural integrity in healthy aging adults and patients with alzheimer disease: A magnetic resonance imaging study. Arch. Neurol. 2003, 60, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.E.; Chen, F.; Chalk, J.B.; Zelaya, F.O.; Strugnell, W.E.; Benson, M.; Semple, J.; Doddrell, D.M. Loss of connectivity in alzheimer’s disease: An evaluation of white matter tract integrity with colour coded mr diffusion tensor imaging. J. Neurol. Neurosurg. Psychiatry 2000, 69, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-K.; Kim, J.H.; Lin, S.-J.; Brendza, R.P.; Holtzman, D.M. Diffusion tensor imaging detects age-dependent white matter changes in a transgenic mouse model with amyloid deposition. Neurobiol. Dis. 2004, 15, 640–647. [Google Scholar] [CrossRef]
- Desai, M.K.; Sudol, K.L.; Janelsins, M.C.; Mastrangelo, M.A.; Frazer, M.E.; Bowers, W.J. Triple-transgenic alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 2009, 57, 54–65. [Google Scholar] [CrossRef]
- Desai, M.K.; Mastrangelo, M.A.; Ryan, D.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Early oligodendrocyte/myelin pathology in alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 2010, 177, 1422–1435. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hayashi, M.; Nakano, H.; Fukutani, Y.; Sasaki, K.; Shimazaki, M.; Koshino, Y. Apoptosis of astrocytes with enhanced lysosomal activity and oligodendrocytes in white matter lesions in alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2002, 28, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Sjöbeck, M.; Englund, E. Glial levels determine severity of white matter disease in alzheimer’s disease: A neuropathological study of glial changes. Neuropathol. Appl. Neurobiol. 2003, 29, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, S.; Ahmed, S.H.; Chen, H.; Ku, G.; Goldberg, M.P.; Hsu, C.Y. Amyloid-β peptides are cytotoxic to oligodendrocytes. J. Neurosci. 2001, 21, RC118. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (ng2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Salins, P.; Shawesh, S.; He, Y.; Dibrov, A.; Kashour, T.; Arthur, G.; Amara, F. Lovastatin protects human neurons against abeta-induced toxicity and causes activation of beta-catenin-tcf/lef signaling. Neurosci. Lett. 2007, 412, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.P.; Zhao, J.; Li, S. Roles of ng2 glial cells in diseases of the central nervous system. Neurosci. Bull. 2011, 27, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar] [CrossRef] [Green Version]
- Nukina, N.; Ihara, Y. One of the antigenic determinants of paired helical filaments is related to tau protein. J. Biochem. 1986, 99, 1541–1544. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Basurto-Islas, G.; Santa-Maria, I.; Mena, R.; Binder, L.I.; Avila, J.; Smith, M.A.; Perry, G.; Garcia-Sierra, F. Cleavage and conformational changes of tau protein follow phosphorylation during alzheimer’s disease. Int. J. Exp. Pathol. 2008, 89, 81–90. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Basurto-Islas, G.; Lee, H.G.; Perry, G.; Zhu, X.; Castellani, R.J.; Smith, M.A. Causes versus effects: The increasing complexities of alzheimer’s disease pathogenesis. Expert. Rev. Neurother. 2010, 10, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.I. Coupled reductions in brain oxidative phosphorylation and synaptic function can be quantified and staged in the course of alzheimer disease. Neurotox. Res. 2003, 5, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Molecular machinery of mitochondrial fusion and fission. J. Biol. Chem. 2008, 283, 13501–13505. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. Alzheimer’s disease: Diverse aspects of mitochondrial malfunctioning. Int. J. Clin. Exp. Pathol. 2010, 3, 570–581. [Google Scholar]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in alzheimer’s disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F.; Terlecky, S.R.; Chiang, H.L.; Olson, T.S.; Isenman, L.D.; Short-Russell, S.R.; Freundlieb, S.; Terlecky, L.J. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin. Cell. Biol. 1990, 1, 449–455. [Google Scholar] [PubMed]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell. Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, E.; Cuervo, A.M. Chaperone-mediated autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Bandyopadhyay, U.; Sridhar, S.; Kiffin, R.; Martinez-Vicente, M.; Kon, M.; Orenstein, S.J.; Wong, E.; Cuervo, A.M. Chaperone-mediated autophagy at a glance. J. Cell. Sci. 2011, 124, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.A. Neuronal autophagy: A housekeeper or a fighter in neuronal cell survival? Exp. Neurobiol. 2012, 21, 1–8. [Google Scholar] [CrossRef]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural. Regen. Res. 2013, 8, 2275–2283. [Google Scholar]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Invest. 2015, 125, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Liu, Y.; Sun, M. Autophagy and alzheimer’s disease. Cell. Mol. Neurobiol. 2017, 37, 377–388. [Google Scholar] [CrossRef]
- Whyte, L.S.; Lau, A.A.; Hemsley, K.M.; Hopwood, J.J.; Sargeant, T.J. Endo-lysosomal and autophagic dysfunction: A driving factor in alzheimer’s disease? J. Neurochem. 2017, 140, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Perry, G.; Moreira, P.I. Mitochondrial traffic jams in alzheimer’s disease—Pinpointing the roadblocks. Biochim. Biophys. Acta 2016, 1862, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Caberlotto, L.; Nguyen, T.P.; Lauria, M.; Priami, C.; Rimondini, R.; Maioli, S.; Cedazo-Minguez, A.; Sita, G.; Morroni, F.; Corsi, M.; et al. Cross-disease analysis of alzheimer’s disease and type-2 diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases. Sci. Rep. 2019, 9, 3965. [Google Scholar] [CrossRef] [PubMed]
- Bitsikas, V.; Riento, K.; Howe, J.D.; Barry, N.P.; Nichols, B.J. The role of flotillins in regulating abeta production, investigated using flotillin 1-/-, flotillin 2-/- double knockout mice. PLoS ONE 2014, 9, e85217. [Google Scholar] [CrossRef] [PubMed]
- Tate, B.A.; Mathews, P.M. Targeting the role of the endosome in the pathophysiology of alzheimer’s disease: A strategy for treatment. Sci. Aging Knowl. Env. 2006, 2006, re2. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, S.B.; Chabot, C.; Gratton, J.P.; Poirier, J. The caveolin scaffolding domain modifies 2-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor binding properties by inhibiting phospholipase a2 activity. J. Biol. Chem. 2004, 279, 356–362. [Google Scholar] [CrossRef]
- Okamoto, C.T. Endocytosis and transcytosis. Adv. Drug Deliv. Rev. 1998, 29, 215–228. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R.; Pun, S.; Stallmach, L.; Sadasivam, G.; Santos, A.F.; Caroni, P.; Fuhrer, C. Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 2006, 25, 4050–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakadate, K. Developmental changes in the flotillin-1 expression pattern of the rat visual cortex. Neuroscience 2015, 292, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.M.; Torrado, A.I.; Arocho, L.C.; Rosas, O.R.; Rodriguez, A.E.; Toro, F.K.; Salgado, I.K.; Torres, Y.A.; Silva, W.I.; Miranda, J.D. Expression profile of flotillin-2 and its pathophysiological role after spinal cord injury. J. Mol. Neurosci. 2013, 49, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.; Simons, K. Caveolae, digs, and the dynamics of sphingolipid-cholesterol microdomains. Curr. Opin. Cell. Biol. 1997, 9, 534–542. [Google Scholar] [CrossRef]
- Fra, A.M.; Williamson, E.; Simons, K.; Parton, R.G. De novo formation of caveolae in lymphocytes by expression of vip21-caveolin. Proc. Natl. Acad. Sci. USA 1995, 92, 8655–8659. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, T.; Ueda, H.; Trapp, B.D.; Nishiyama, K.; Sha, J.F.; Volonte, D.; Galbiati, F.; Byrd, A.L.; Bassell, G.; Serizawa, H.; et al. Affinity-purification and characterization of caveolins from the brain: Differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain Res. 1998, 804, 177–192. [Google Scholar] [CrossRef]
- Bilderback, T.R.; Gazula, V.R.; Lisanti, M.P.; Dobrowsky, R.T. Caveolin interacts with trk a and p75(ntr) and regulates neurotrophin signaling pathways. J. Biol. Chem. 1999, 274, 257–263. [Google Scholar] [CrossRef]
- Hibbert, A.P.; Kramer, B.M.; Miller, F.D.; Kaplan, D.R. The localization, trafficking and retrograde transport of bdnf bound to p75ntr in sympathetic neurons. Mol. Cell. Neurosci. 2006, 32, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Head, B.P.; Patel, H.H.; Tsutsumi, Y.M.; Hu, Y.; Mejia, T.; Mora, R.C.; Insel, P.A.; Roth, D.M.; Drummond, J.C.; Patel, P.M. Caveolin-1 expression is essential for n-methyl-d-aspartate receptor-mediated src and extracellular signal-regulated kinase 1/2 activation and protection of primary neurons from ischemic cell death. FASEB J. 2008, 22, 828–840. [Google Scholar] [CrossRef]
- Gaudreault, S.B.; Blain, J.F.; Gratton, J.P.; Poirier, J. A role for caveolin-1 in post-injury reactive neuronal plasticity. J. Neurochem. 2005, 92, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, T.; Trapp, B.D.; Song, K.S.; Schlegel, A.; Lisanti, M.P.; Okamoto, T. Caveolae, plasma membrane microdomains for alpha-secretase-mediated processing of the amyloid precursor protein. J. Biol. Chem. 1998, 273, 10485–10495. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, K.; Trapp, B.D.; Ikezu, T.; Ransohoff, R.M.; Tomita, T.; Iwatsubo, T.; Kanazawa, I.; Hsiao, K.K.; Lisanti, M.P.; Okamoto, T. Caveolin-3 upregulation activates beta-secretase-mediated cleavage of the amyloid precursor protein in alzheimer’s disease. J. Neurosci. 1999, 19, 6538–6548. [Google Scholar] [CrossRef] [PubMed]
- Alsaqati, M.; Thomas, R.S.; Kidd, E.J. Proteins involved in endocytosis are upregulated by ageing in the normal human brain: Implications for the development of alzheimer’s disease. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Girardot, N.; Allinquant, B.; Langui, D.; Laquerriere, A.; Dubois, B.; Hauw, J.J.; Duyckaerts, C. Accumulation of flotillin-1 in tangle-bearing neurones of alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2003, 29, 451–461. [Google Scholar] [CrossRef]
- Head, B.P.; Peart, J.N.; Panneerselvam, M.; Yokoyama, T.; Pearn, M.L.; Niesman, I.R.; Bonds, J.A.; Schilling, J.M.; Miyanohara, A.; Headrick, J.; et al. Loss of caveolin-1 accelerates neurodegeneration and aging. PLoS ONE 2010, 5, e15697. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic alzheimer’s disease and down syndrome: Differential effects of apoe genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Thomas, R.S.; Lelos, M.J.; Good, M.A.; Kidd, E.J. Clathrin-mediated endocytic proteins are upregulated in the cortex of the tg2576 mouse model of alzheimer’s disease-like amyloid pathology. Biochem. Biophys. Res. Commun. 2011, 415, 656–661. [Google Scholar] [CrossRef]
- van Helmond, Z.K.; Miners, J.S.; Bednall, E.; Chalmers, K.A.; Zhang, Y.; Wilcock, G.K.; Love, S.; Kehoe, P.G. Caveolin-1 and -2 and their relationship to cerebral amyloid angiopathy in alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2007, 33, 317–327. [Google Scholar] [CrossRef]
- Trushina, E.; Du Charme, J.; Parisi, J.; McMurray, C.T. Neurological abnormalities in caveolin-1 knock out mice. Behav. Brain Res. 2006, 172, 24–32. [Google Scholar] [CrossRef]
- Gioiosa, L.; Raggi, C.; Ricceri, L.; Jasmin, J.F.; Frank, P.G.; Capozza, F.; Lisanti, M.P.; Alleva, E.; Sargiacomo, M.; Laviola, G. Altered emotionality, spatial memory and cholinergic function in caveolin-1 knock-out mice. Behav. Brain Res. 2008, 188, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Jasmin, J.F.; Malhotra, S.; Singh Dhallu, M.; Mercier, I.; Rosenbaum, D.M.; Lisanti, M.P. Caveolin-1 deficiency increases cerebral ischemic injury. Circ. Res. 2007, 100, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Wais, P.E.; Wixted, J.T.; Hopkins, R.O.; Squire, L.R. The hippocampus supports both the recollection and the familiarity components of recognition memory. Neuron 2006, 49, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Manns, J.R.; Hopkins, R.O.; Reed, J.M.; Kitchener, E.G.; Squire, L.R. Recognition memory and the human hippocampus. Neuron 2003, 37, 171–180. [Google Scholar] [CrossRef]
- Vargha-Khadem, F.; Gadian, D.G.; Watkins, K.E.; Connelly, A.; Van Paesschen, W.; Mishkin, M. Differential effects of early hippocampal pathology on episodic and semantic memory. Science 1997, 277, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, A.D.; Kahana, M.J.; Caplan, J.B.; Fields, T.A.; Isham, E.A.; Newman, E.L.; Fried, I. Cellular networks underlying human spatial navigation. Nature 2003, 425, 184. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Nakamura, K.; Fukuda, M.; Tamura, R. Place recognition responses of neurons in monkey hippocampus. Neurosci. Lett. 1991, 121, 194–198. [Google Scholar] [CrossRef]
- West, M.J.; Coleman, P.D.; Flood, D.G.; Troncoso, J.C. Differences in the pattern of hippocampal neuronal loss in normal ageing and alzheimer’s disease. Lancet 1994, 344, 769–772. [Google Scholar] [CrossRef]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef]
- Lazarov, O.; Marr, R.A. Neurogenesis and alzheimer’s disease: At the crossroads. Exp. Neurol. 2010, 223, 267–281. [Google Scholar] [CrossRef]
- Marlatt, M.W.; Lucassen, P.J. Neurogenesis and alzheimer’s disease: Biology and pathophysiology in mice and men. Curr. Alzheimer Res. 2010, 7, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Hassouna, I.; Ott, C.; Wüstefeld, L.; Offen, N.; Neher, R.A.; Mitkovski, M.; Winkler, D.; Sperling, S.; Fries, L.; Goebbels, S. Revisiting adult neurogenesis and the role of erythropoietin for neuronal and oligodendroglial differentiation in the hippocampus. Mol. Psychiatry 2016, 21, 1752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fang, X.; Huang, D.; Luo, Q.; Zheng, M.; Wang, K.; Cao, L.; Yin, Z. Erythropoietin signaling increases neurogenesis and oligodendrogenesis of endogenous neural stem cells following spinal cord injury both in vivo and in vitro. Mol. Med. Rep. 2018, 17, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Cevik, B.; Solmaz, V.; Yigitturk, G.; Cavusoğlu, T.; Peker, G.; Erbas, O. Neuroprotective effects of erythropoietin on alzheimer’s dementia model in rats. Adv. Clin. Exp. Med. 2017, 26, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Sachs, B.D.; Caron, M.G. Chronic fluoxetine increases extra-hippocampal neurogenesis in adult mice. Int. J. Neuropsychopharmacol. 2015, 18. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Lee, J.K.; Lee, J.E.; Min, W.K.; Schuchman, E.H.; Jin, H.K.; Bae, J.s. Combined effects of hematopoietic progenitor cell mobilization from bone marrow by granulocyte colony stimulating factor and amd3100 and chemotaxis into the brain using stromal cell-derived factor-1α in an alzheimer’s disease mouse model. Stem Cells 2011, 29, 1075–1089. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Müller, F.-J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cells improve cognition via bdnf in a transgenic model of alzheimer disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C. Combined adult neurogenesis and bdnf mimic exercise effects on cognition in an alzheimer’s mouse model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef]
- Liu, P.Z.; Nusslock, R. Exercise-mediated neurogenesis in the hippocampus via bdnf. Front. Neurosci. 2018, 12, 52. [Google Scholar] [CrossRef]
- McGinley, L.M.; Sims, E.; Lunn, J.S.; Kashlan, O.N.; Chen, K.S.; Bruno, E.S.; Pacut, C.M.; Hazel, T.; Johe, K.; Sakowski, S.A. Human cortical neural stem cells expressing insulin-like growth factor-i: A novel cellular therapy for alzheimer’s disease. Stem Cells Transl. Med. 2016, 5, 379–391. [Google Scholar] [CrossRef]
- Capsoni, S.; Marinelli, S.; Ceci, M.; Vignone, D.; Amato, G.; Malerba, F.; Paoletti, F.; Meli, G.; Viegi, A.; Pavone, F. Intranasal “painless” human nerve growth factors slows amyloid neurodegeneration and prevents memory deficits in app x ps1 mice. PLoS ONE 2012, 7, e37555. [Google Scholar] [CrossRef]
- Lee, H.J.; Lim, I.J.; Park, S.W.; Kim, Y.B.; Ko, Y.; Kim, S.U. Human neural stem cells genetically modified to express human nerve growth factor (ngf) gene restore cognition in the mouse with ibotenic acid-induced cognitive dysfunction. Cell Transplant. 2012, 21, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Herrán, E.; Pérez-González, R.; Igartua, M.; Pedraz, J.L.; Carro, E.; Hernández, R.M. Vegf-releasing biodegradable nanospheres administered by craniotomy: A novel therapeutic approach in the app/ps1 mouse model of alzheimer’s disease. J. Control. Release 2013, 170, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Yousef, H.; Conboy, M.J.; Morgenthaler, A.; Schlesinger, C.; Bugaj, L.; Paliwal, P.; Greer, C.; Conboy, I.M.; Schaffer, D. Systemic attenuation of the tgf-β pathway by a single drug simultaneously rejuvenates hippocampal neurogenesis and myogenesis in the same old mammal. Oncotarget 2015, 6, 11959. [Google Scholar] [CrossRef] [PubMed]
- Speisman, R.B.; Kumar, A.; Rani, A.; Pastoriza, J.M.; Severance, J.E.; Foster, T.C.; Ormerod, B.K. Environmental enrichment restores neurogenesis and rapid acquisition in aged rats. Neurobiol. Aging 2013, 34, 263–274. [Google Scholar] [CrossRef]
- Monteiro, B.M.; Moreira, F.A.; Massensini, A.R.; Moraes, M.F.; Pereira, G.S. Enriched environment increases neurogenesis and improves social memory persistence in socially isolated adult mice. Hippocampus 2014, 24, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H.; Kempermann, G.; Gage, F.H. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999, 2, 266. [Google Scholar] [CrossRef]
- Van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise enhances learning and hippocampal neurogenesis in aged mice. J. Neurosci. 2005, 25, 8680–8685. [Google Scholar] [CrossRef]
- Hüttenrauch, M.; Brauss, A.; Kurdakova, A.; Borgers, H.; Klinker, F.; Liebetanz, D.; Salinas-Riester, G.; Wiltfang, J.; Klafki, H.; Wirths, O. Physical activity delays hippocampal neurodegeneration and rescues memory deficits in an alzheimer disease mouse model. Transl. Psychiatry 2016, 6, e800. [Google Scholar] [CrossRef]
- Alipour, M.; Nabavi, S.M.; Arab, L.; Vosough, M.; Pakdaman, H.; Ehsani, E.; Shahpasand, K. Stem cell therapy in alzheimer’s disease: Possible benefits and limiting drawbacks. Mol. Biol. Rep. 2019, 46, 1425–1446. [Google Scholar] [CrossRef]
- Pera, M.F.; Trounson, A.O. Human embryonic stem cells: Prospects for development. Development 2004, 131, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, T.; Oh, S.-H.; Pi, L.; Hatch, H.M.; Shupe, T.; Petersen, B.E. Teratoma formation leads to failure of treatment for type i diabetes using embryonic stem cell-derived insulin-producing cells. Am. J. Pathol. 2005, 166, 1781–1791. [Google Scholar] [CrossRef]
- Richards, M.; Fong, C.-Y.; Chan, W.-K.; Wong, P.-C.; Bongso, A. Human feeders support prolonged undifferentiated growth of human inner cell masses and embryonic stem cells. Nat. Biotechnol. 2002, 20, 933. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, F.H.; Alaie, H.; Karbalaie, K.; Tanhaei, S.; Esfahani, M.H.N.; Baharvand, H. Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in alzheimerian rats. Differentiation 2009, 78, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, C.J.; Lyass, L.; Bhattacharyya, B.J.; Belmadani, A.; Miller, R.J.; Kessler, J.A. The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells 2011, 29, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Li, Y.; Zhang, T.; Jiang, M.; Qian, Y.; Zhang, M.; Sheng, N.; Feng, S.; Tang, K.; Yu, X. Esc-derived basal forebrain cholinergic neurons ameliorate the cognitive symptoms associated with alzheimer’s disease in mouse models. Stem Cell Rep. 2015, 5, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Weick, J.P.; Liu, H.; Krencik, R.; Zhang, X.; Ma, L.; Zhou, G.-m.; Ayala, M.; Zhang, S.-C. Medial ganglionic eminence–like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 2013, 31, 440. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.S.; LeComte, M.D.; Granger, J.C.; Quinlan, N.J.; Spees, J.L. Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J. Neurosci. 2012, 32, 7926–7940. [Google Scholar] [CrossRef]
- Liang, Y.; Ågren, L.; Lyczek, A.; Walczak, P.; Bulte, J.W. Neural progenitor cell survival in mouse brain can be improved by co-transplantation of helper cells expressing bfgf under doxycycline control. Exp. Neurol. 2013, 247, 73–79. [Google Scholar] [CrossRef]
- Nakaji-Hirabayashi, T.; Kato, K.; Iwata, H. In vivo study on the survival of neural stem cells transplanted into the rat brain with a collagen hydrogel that incorporates laminin-derived polypeptides. Bioconjugate Chem. 2013, 24, 1798–1804. [Google Scholar] [CrossRef]
- Lee, I.-S.; Jung, K.; Kim, I.-S.; Lee, H.; Kim, M.; Yun, S.; Hwang, K.; Shin, J.E.; Park, K.I. Human neural stem cells alleviate alzheimer-like pathology in a mouse model. Mol. Neurodegener. 2015, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of alzheimer’s disease and neuronal loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, H.; Sun, X.; Zuo, F.; Lei, J.; Wang, Z.; Bao, X.; Wang, R. Human neural stem cell transplantation rescues cognitive defects in app/ps1 model of alzheimer’s disease by enhancing neuronal connectivity and metabolic activity. Front. Aging Neurosci. 2016, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.; Luo, M.; Ji, W.; Long, D. Effects of engrafted neural stem cells in alzheimer’s disease rats. Neurosci. Lett. 2009, 450, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.; Long, D.; Gu, H.; Yang, D.; Hong, L.; Leng, S. Bdnf improves the effects of neural stem cells on the rat model of alzheimer’s disease with unilateral lesion of fimbria-fornix. Neurosci. Lett. 2008, 440, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Yang, G.; Bae, D.K.; Lee, S.H.; Yang, Y.H.; Kyung, J.; Kim, D.; Choi, E.K.; Choi, K.C.; Kim, S.U. Human adipose tissue-derived mesenchymal stem cells improve cognitive function and physical activity in ageing mice. J. Neurosci. Res. 2013, 91, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Lilja, A.M.; Malmsten, L.; Röjdner, J.; Voytenko, L.; Verkhratsky, A.; Ögren, S.O.; Nordberg, A.; Marutle, A. Neural stem cell transplant-induced effect on neurogenesis and cognition in alzheimer tg2576 mice is inhibited by concomitant treatment with amyloid-lowering or cholinergic 7 nicotinic receptor drugs. Neural Plast. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Desplats, P.; Ubhi, K.; Mante, M.; Florio, J.; Adame, A.; Winter, S.; Brandstaetter, H.; Meier, D.; Masliah, E. Neuro-peptide treatment with cerebrolysin improves the survival of neural stem cell grafts in an app transgenic model of alzheimer disease. Stem Cell Res. 2015, 15, 54–67. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, H.h.; Wang, Y.; Gu, G.j.; Zhang, W.; Xia, R. Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of alzheimer’s disease. J. Neurochem. 2016, 136, 815–825. [Google Scholar] [CrossRef]
- Ross, C.A.; Akimov, S.S. Human-induced pluripotent stem cells: Potential for neurodegenerative diseases. Hum. Mol. Genet. 2014, 23, R17–R26. [Google Scholar] [CrossRef]
- Hossini, A.M.; Megges, M.; Prigione, A.; Lichtner, B.; Toliat, M.R.; Wruck, W.; Schröter, F.; Nuernberg, P.; Kroll, H.; Makrantonaki, E. Induced pluripotent stem cell-derived neuronal cells from a sporadic alzheimer’s disease donor as a model for investigating ad-associated gene regulatory networks. BMC Genom. 2015, 16, 84. [Google Scholar]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial alzheimer’s disease appv717i mutation alters app processing and tau expression in ipsc-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.-e.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A. Gain of toxic apolipoprotein e4 effects in human ipsc-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Kwon, Y.W.; Ahn, H.S.; Jeong, H.; Lee, Y.Y.; Moon, M.; Baik, S.H.; Kim, D.K.; Song, H.; Yi, E.C. Protein-induced pluripotent stem cells ameliorate cognitive dysfunction and reduce aβ deposition in a mouse model of alzheimer’s disease. Stem Cells Transl. Med. 2017, 6, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Shimizu, J.; Takai, K.; Arimitsu, N.; Saito, A.; Kono, T.; Umehara, T.; Ueda, Y.; Wakisaka, S.; Suzuki, T. Restoration of spatial memory dysfunction of human app transgenic mice by transplantation of neuronal precursors derived from human ips cells. Neurosci. Lett. 2013, 557, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Hallett, P.J.; Deleidi, M.; Astradsson, A.; Smith, G.A.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.-L. Successful function of autologous ipsc-derived dopamine neurons following transplantation in a non-human primate model of parkinson’s disease. Cell Stem Cell 2015, 16, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.-H.; Ko, J.-Y.; Chang, M.-Y.; Yi, S.-H.; Kim, D.; Kim, C.-H.; Shim, J.-W.; Jo, A.-Y.; Kim, B.-W.; Lee, H. Protein-based human ips cells efficiently generate functional dopamine neurons and can treat a rat model of parkinson disease. J. Clin. Investig. 2011, 121, 2326–2335. [Google Scholar] [CrossRef]
- Kikuchi, T.; Morizane, A.; Onoe, H.; Hayashi, T.; Kawasaki, T.; Saiki, H.; Miyamoto, S.; Takahashi, J. Survival of human induced pluripotent stem cell–derived midbrain dopaminergic neurons in the brain of a primate model of parkinson’s disease. J. Parkinson’s Dis. 2011, 1, 395–412. [Google Scholar]
- Eckert, A.; Huang, L.; Gonzalez, R.; Kim, H.-S.; Hamblin, M.H.; Lee, J.-P. Bystander effect fuels human induced pluripotent stem cell-derived neural stem cells to quickly attenuate early stage neurological deficits after stroke. Stem Cells Transl. Med. 2015, 4, 841–851. [Google Scholar] [CrossRef]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef]
- Lee, J.; Kuroda, S.; Shichinohe, H.; Ikeda, J.; Seki, T.; Hida, K.; Tada, M.; Sawada, K.i.; Iwasaki, Y. Migration and differentiation of nuclear fluorescence-labeled bone marrow stromal cells after transplantation into cerebral infarct and spinal cord injury in mice. Neuropathology 2003, 23, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Ra, J.C.; Shin, I.S.; Kim, S.H.; Kang, S.K.; Kang, B.C.; Lee, H.Y.; Kim, Y.J.; Jo, J.Y.; Yoon, E.J.; Choi, H.J. Safety of intravenous infusion of human adipose tissue-derived mesenchymal stem cells in animals and humans. Stem Cells Dev. 2011, 20, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Lykhmus, O.; Koval, L.; Voytenko, L.; Uspenska, K.; Komisarenko, S.; Deryabina, O.; Shuvalova, N.; Kordium, V.; Ustymenko, A.; Kyryk, V. Intravenously injected mesenchymal stem cells penetrate the brain and treat inflammation-induced brain damage and memory impairment in mice. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, Z.H.; Wei, L.F.; Yang, H.N.; Yang, S.N.; Zhu, Z.Y.; Wang, P.; Zhao, C.P.; Bi, J.Z. Human umbilical cord mesenchymal stem cell-derived neuron-like cells rescue memory deficits and reduce amyloid-beta deposition in an aβpp/ps1 transgenic mouse model. Stem Cell Res. Ther. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Kim, H.S.; Park, J.-M.; Kim, H.W.; Park, M.-k.; Lee, H.-S.; Lim, D.S.; Lee, T.H.; Chopp, M.; Moon, J. Long-term immunomodulatory effect of amniotic stem cells in an alzheimer’s disease model. Neurobiol. Aging 2013, 34, 2408–2420. [Google Scholar] [CrossRef]
- Matchynski-Franks, J.J.; Pappas, C.; Rossignol, J.; Reinke, T.; Fink, K.; Crane, A.; Twite, A.; Lowrance, S.A.; Song, C.; Dunbar, G.L. Mesenchymal stem cells as treatment for behavioral deficits and neuropathology in the 5xfad mouse model of alzheimer’s disease. Cell Transplant. 2016, 25, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Kim, H.N.; Park, H.-J.; Shin, J.Y.; Lee, P.H. Mesenchymal stem cells increase hippocampal neurogenesis and neuronal differentiation by enhancing the wnt signaling pathway in an Alzheimer’s disease model. Cell Transplant. 2015, 24, 1097–1109. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Carvalho, M.M.; Neves-Carvalho, A.; Panchalingam, K.M.; Behie, L.A.; Pinto, L.; Sousa, N.; Salgado, A.J. Secretome of mesenchymal progenitors from the umbilical cord acts as modulator of neural/glial proliferation and differentiation. Stem Cell Rev. Rep. 2015, 11, 288–297. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of sirna to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341. [Google Scholar] [CrossRef]
- Katsuda, T.; Tsuchiya, R.; Kosaka, N.; Yoshioka, Y.; Takagaki, K.; Oki, K.; Takeshita, F.; Sakai, Y.; Kuroda, M.; Ochiya, T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013, 3, 1197. [Google Scholar] [CrossRef]
- Garcia, K.d.O.; Ornellas, F.L.; Matsumoto, P.; Patti, C.d.L.; Mello, L.E.; Frussa-Filho, R.; Han, S.W.; Longo, B.M. Therapeutic effects of the transplantation of vegf overexpressing bone marrow mesenchymal stem cells in the hippocampus of murine model of alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Seo, S.W.; Chang, J.W.; Lee, J.I.; Kim, C.H.; Chin, J.; Choi, S.J.; Kwon, H.; Yun, H.J.; Lee, J.M. Stereotactic brain injection of human umbilical cord blood mesenchymal stem cells in patients with alzheimer’s disease dementia: A phase 1 clinical trial. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2015, 1, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M.H.; Thal, L.; Pay, M.; Salmon, D.P.; Bakay, R.; Patel, P.; Blesch, A.; Vahlsing, H.L.; Ho, G.; Tong, G. A phase 1 clinical trial of nerve growth factor gene therapy for alzheimer disease. Nat. Med. 2005, 11, 551. [Google Scholar] [CrossRef] [PubMed]


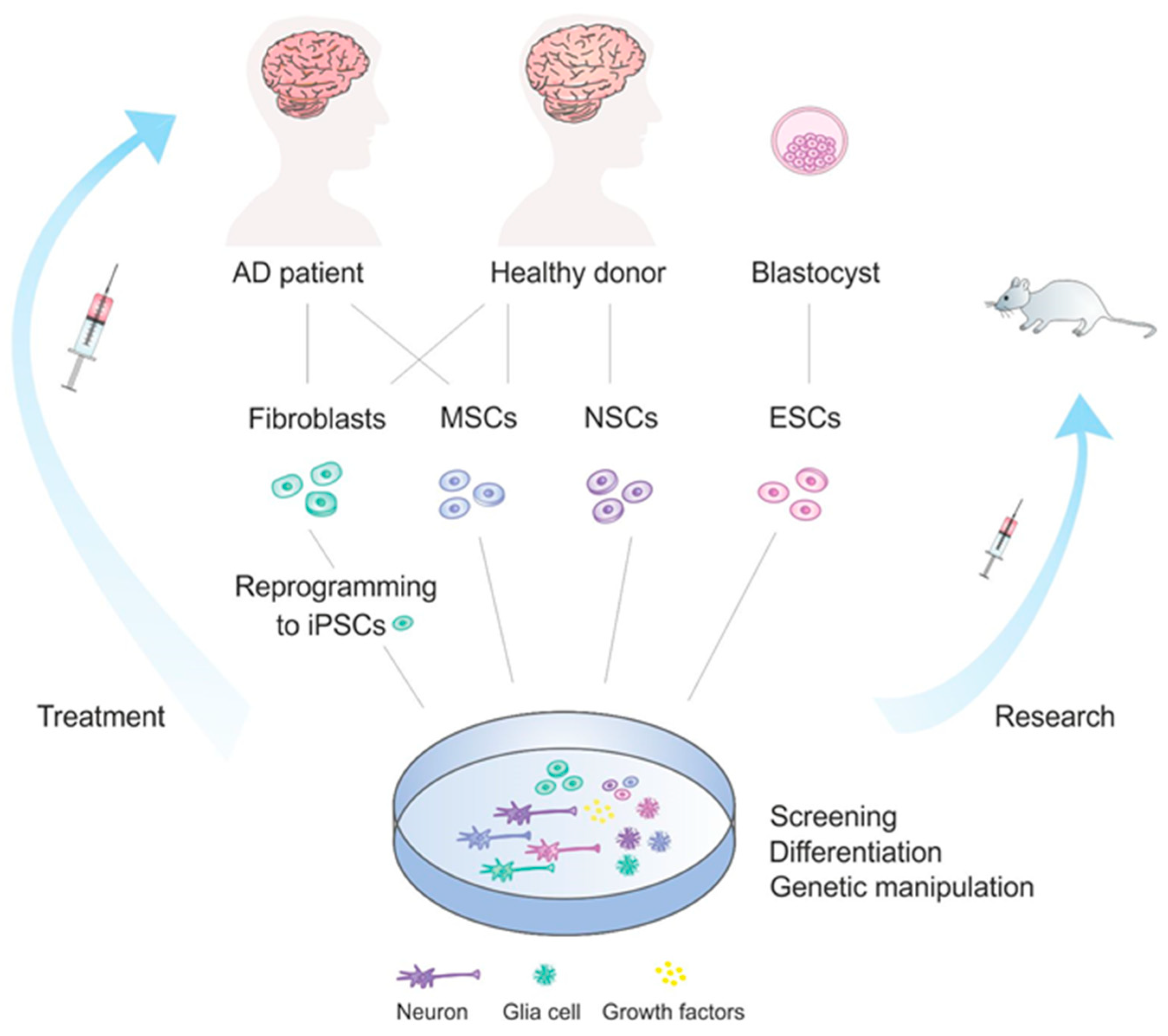{kind=link}
| Cellular System | Physiological Function | Involvement in AD |
|---|---|---|
| Microglia | Component in the determination of cognitive function, preservation of a healthy brain, attack and removal of pathogens and detritus, secretion of tissue rebuilding factors, synaptic remodeling [15,16,17,18,19,20,21,22,24,25,26,27] | Involved in the generation of neuro-inflammation, imbalance of Aβ peptide homeostasis, decrease of phagocytic activity, release of pro-inflammatory neurotoxins and cytokines/chemokines [29,30,31,33,34,35,36,37,38,39,40,41,42,43,44] |
| Astrocytes | Interactions with neurons by releasing and recycling glio-transmitter, control ion homeostasis, energy metabolism, synaptic remodeling and the modulation of oxidative stress leading to control of neurotransmission, synaptic plasticity and the modulation of cognitive functions involved in the degradation of Aβ peptides [8] | Changes in intra- and extracellular degradation of Aβ peptides, release of cytokines and chemokines, expression of ApoE, formation of hyperphosphorylated tau protein [45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63] |
| Oligodendrocytes | Form together with myelin lipid layers the envelope of the neuronal axons [8] | Specific morphological changes during AD progression, deterioration in myelin integrity and axonal destruction, killed by Aβ peptides [64,65,66,67,68,69,73] |
| NG2-glia | Oligodendrocyte precursor cells | Aβ peptides-induced inhibition of Wnt signaling pathway results in an inhibition of the differentiation of NG2-glia [74,76] |
| Neurons | Expression of a large number of molecules for protection against inflammatory attacks and induction of neurological disorders [5] | Formation of intracellular neurofibrillary tangles by hyperphosphorylated tau protein, impaired axonal transport of mitochondria resulting in energy dysfunction, generation of reactive oxygen and nitrogen species [77,78,79,80,81,82,83] |
| Mitochondria | ATP synthesis, reaction to different energy demands by balanced fission and fusion processes and directed transport along axons, protection against ROS damage by elimination of defective constituents | Aggregation of hyperphosphorylated tau in neurofibrillary tangles, perinuclear mis-localization resulting in ATP depletion, synaptic dysfunction, oxidative stress [84,85,86,87,88,89,90,91] |
| Autophagy (Mitophagy) | Degradation of organelles, proteins and lipids mediated by membranes, vesicles and lysosomes, essential for organelle turn-over, synaptic plasticity, anti-inflammatory function in glial cells, oligodendrocyte development, and the myelination process | Dysregulation leading to changes in the expression of several autophagy genes resulting in reduced energy levels, increased ROS production and impaired neuroplasticity [92,93,94,95,96,97,98,99,100,101,102,103,104,105,106] |
| Endocytic processes | Internalization of materials from the cell surface by clathrin-dependent and clathrin-independent pathways, using flotillins or caveolins as the main proteins, as well as the protection against the processing of APP and Aβ toxicity [36] | Only a few data available, volume of total endosomes increases, enhanced levels of several endocytic enzymes [107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125] |
| Stem Cells | Advantages | Disadvantages |
|---|---|---|
| ESCs | Unlimited self-renewal; Pluripotent [161] | Ethical issues; Uncontrolled differentiation [162,163] |
| NSCs | Multipotent [168] | Poor survival [169,170] |
| iPSCs | Autologous transplantation; Pluripotent [180] | Possible pathological phenotype [181,182] |
| MSCs | Easy handling; Multipotent; Intravenous application [190,192,193] | Low rate of neuronal differentiation [191] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasic, V.; Barth, K.; Schmidt, M.H.H. Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy. Int. J. Mol. Sci. 2019, 20, 4272. https://doi.org/10.3390/ijms20174272
Vasic V, Barth K, Schmidt MHH. Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy. International Journal of Molecular Sciences. 2019; 20(17):4272. https://doi.org/10.3390/ijms20174272
Chicago/Turabian StyleVasic, Verica, Kathrin Barth, and Mirko H.H. Schmidt. 2019. "Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy" International Journal of Molecular Sciences 20, no. 17: 4272. https://doi.org/10.3390/ijms20174272
APA StyleVasic, V., Barth, K., & Schmidt, M. H. H. (2019). Neurodegeneration and Neuro-Regeneration—Alzheimer’s Disease and Stem Cell Therapy. International Journal of Molecular Sciences, 20(17), 4272. https://doi.org/10.3390/ijms20174272