1. Introduction
Lung cancer is the leading cause of cancer death globally, representing 18% of all cancer deaths [
1]. Non-small cell lung cancer (NSCLC) is the most prevalent subtype of all lung cancers (approximately 80–85%) [
2]. Adjuvant chemotherapy after surgical resection is the prioritized strategy against NSCLC to prolong survival. Although the investigation and development of targeted drugs and immunotherapy gained much progress in cancer therapy, the five-year survival rate is only 16%, indicating a poor outcome of prognosis [
3].
Activation of apoptosis is one of the major mechanisms of chemotherapeutic actions. As mitochondria play a pivotal role on triggering apoptosis [
4], currently many clinically-used anti-tumor drugs, including the first-line therapies such as cisplatin and paclitaxel, exert a killing effect on cancer cells at least in part by mitochondrial apoptotic priming, or direct impairment of mitochondrial respiratory chain complexes. With this strategy, high doses of chemotherapy are normally required for an effective anti-tumor effect. However, treatment with high dosages of non-targeted drugs is indeed a double-edged sword that the cytotoxicity affects tumor and healthy cells simultaneously.
It is well known that mitochondria in tumor cells are vulnerable due to its frequent membrane “fission”, and as a result generates excessive reactive oxygen species (ROS) like hydrogen peroxide (H
2O
2)—the predominant one, superoxide anions (O
2−), and hydroxyl free radicals (HO) [
5,
6]. When ROS overwhelms the cellular antioxidant defense system, the MAPK signaling pathways are activated and eventually result in the programmed cell death [
7]. To reduce oxidative damage and maintain cellular homeostasis, tumor cells tend to upregulate autophagy to eliminate defective mitochondria [
8,
9]. Autophagy acts as a conserved degradative process of misfolded proteins and damaged organelles. In this process, autophagic cargo is engulfed by autophagosome, which is then fused with lysosome to form autolysosome. Subsequently, the cargo is broken down by various cathepsins in the autolysosome for molecules recycling. This entire catabolic process of autophagy, also known as autophagic flux, is crucial for maintaining cellular homeostasis and organelle quality control [
10].
Recently, we screened the small molecule chemical library presented by Gu et al. [
11] and discovered that
N-methylparoxetine (NMP) was able to inhibit autophagy flux in NSCLC cells (data not shown). Hypothetically, inhibiting autophagy of cancer cells may undermine cellular redox state by excessive ROS accumulation and, consequently, apoptosis induction. Through indirect mitochondria impairment, NMP was expected to exert more striking killing effects in NSCLC cells with relatively low dosage. NMP is the precursor of paroxetine, a commonly used anti-depressant drug. Several studies have reported that paroxetine can exert a good anticancer activity in human cancer cells, including colon, oral, and breast cancer cells [
12,
13,
14]. The structural resemblance of NMP and paroxetine implies that they may have similar physical, chemical, pharmacological, and adsorption, distribution, metabolism, excretion, and toxicological (ADMET) properties, with predictable clinical application potential (
Figure 1A) [
15].
In the present study, the anti-cancer effect of NMP on NSCLC cells was evaluated. Further, we revealed underlining mechanisms whereby NMP suppressed autophagy and induced apoptosis. The possible relationship between autophagy inhibition and apoptosis through ROS-MAPK signaling pathways was also examined.
3. Discussion
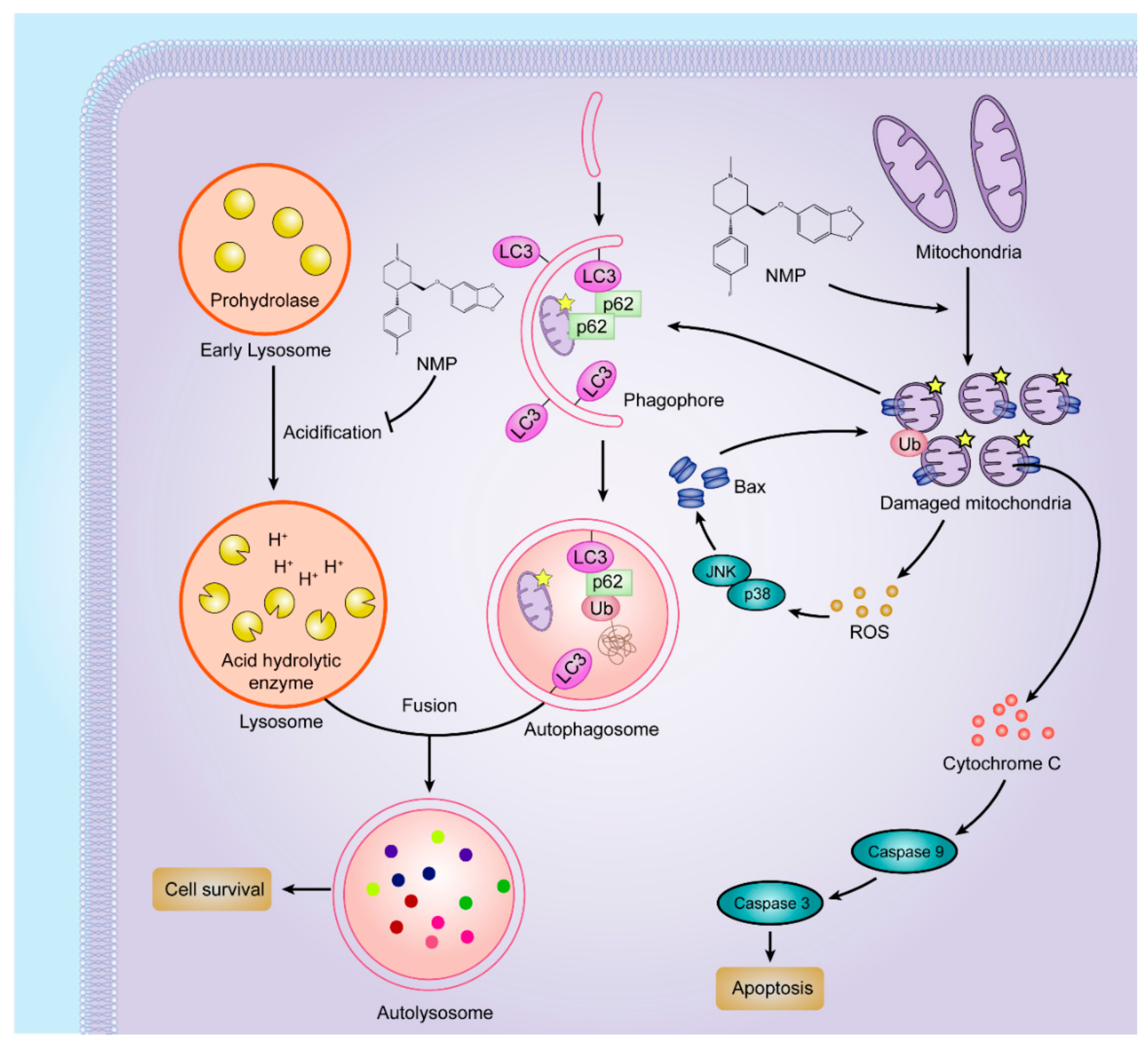
In this study, we proposed that NMP was an effective inducer of apoptosis in NSCLC cells by dual pathways (
Figure 9). On one hand, NMP stimulated mitochondrial fission and fragmentation. Fragmented mitochondria showed round morphology (i.e., depolarization) with MOMP leading to the leakage of ROS and apoptosis factors, such as cytochrome c. In favor of homeostasis maintenance, cells can eliminate impaired mitochondria to avoid excessive accumulation of ROS by autophagy (“self-eating”). However, exclusively striking mitochondria may not effectively kill cancer cells due to the concurrent upregulated autophagic pathway. Recently, it has been confirmed that inhibition of autophagy can be a new strategy to improve the efficacy of mitochondrial disrupting agents [
19]. In this study, we demonstrated that NMP inhibited autophagy in NSCLC cells. NMP disrupted the ROS removal via blockade of the late-staged autophagy flux. As an excessive accumulation of ROS is a risk factor damaging mitochondria [
20], NMP can exert more efficient apoptotic effects on cancer cells with this positive feedback loop from ROS to mitochondria. This reassures the future development of NMP as a promising drug for clinical chemotherapeutic application.
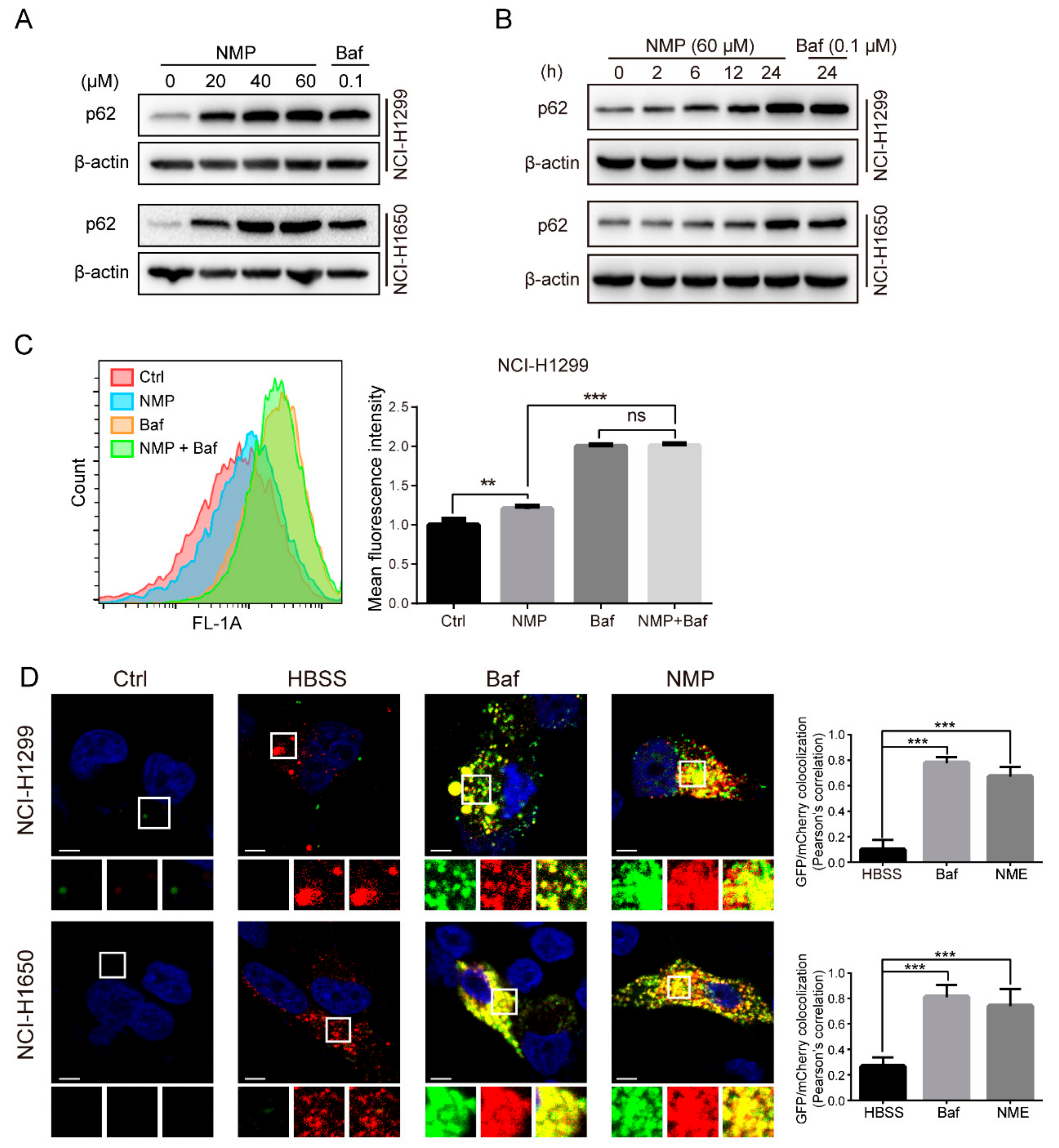
To delineate the actions of NMP on autophagy, we firstly quantified autophagosome formation in NSCLC cells. Autophagosome, a double-membraned structure, was initiated from the endoplasmic reticulum. Cargos, such as unfolded proteins and damaged organelles, were mediated by receptors and capsuled in autophagosome, fused with lysosome to form, and degraded in autolysosome. Accordingly, autophagosome is a central structure during autophagosome flux. Secondly, we examined LC3-II expression. LC3-II, a well-established marker of autophagosome, is generated by cleavage of its precursor LC3 and conjugation to lipid phosphatidylethanolamine (PE) in the autophagosome, and eventually degraded alongside the cargo. Although the expression of LC3-II was upregulated in NSCLC cells following NMP treatment, the data did not indicate if NMP inhibited or promoted autophagy. Therefore, we then measured the expression levels of p62, which is responsible for recruiting ubiquitinated-substrate (i.e., the cargo) to the LC3-II autophagosomal receptor, preceding degradation. Autophagy-dependent pathway remains the prevailing degradation pathway of p62 although it can also be degraded by proteasome. The dose-dependent upregulation of p62 in our data suggested that NMP inhibited autophagy of NSCLC cells. Further, we demonstrated that NMP inhibited late staged autophagy flux. Specifically, we treated GFP-LC3-expressing NSCLC cells with NMP in combination with a widely accepted late autophagy inhibitor, bafilomycin A1 and quantified the GFP-LC3 signals. If NMP inhibited early autophagy, the combined treatment would lead to the low intensity of GFP-fluorescence. Our data showed a similar intensity of GFP-LC3 fluorescence intensities between the combined treatment and the bafilomycin A1-standalone group. These results suggested that NMP inhibited late autophagy as bafilomycin A1 did. In addition, NSCLC cells were transiently transfected with mCherry-GFP-LC3 constructs to trace autophagy flux. The tandem fluorescence system labels autophagosome with yellow fluorescence (i.e., merging of green and red) and autolysosome with a red signal due to quenching of GFP in an acidic environment. A large proportion of yellow fluorescence was detected in the NMP group, similar to that in bafilomycin A1 group. We speculated that NMP might suppress lysosomal acidification. This was confirmed by decreased AO and lysotracker stainings, and the impeded maturation of lysosomal cathepsins. Of note, here lies another uncertainty—the fusion of autophagosome and lysosome may be obstructed. To clarify the action of NMP on it, we specifically labeled lysosome in GFP-LC3-expressing NSCLC cells with LysoBrite
TM Red. In contrast to the positive control group treated with chloroquine (CQ) group, a fusion inhibitor, NMP group showed a larger percentage of colocalizing autophagosome and lysosome (yellow fluorescence,
Supplementary Figure S1), suggesting treatment of NMP did not inhibit the fusion process to form autolysosome.
The accumulation of autophagosomes was reported to be stress leading to autophagic cell death [
18,
19]. To test whether NMP induced cell death is due to autophagic cell death, 3-methyladenine (3-MA), an inhibitor of early autophagy targeting PI3K (also termed Vps34), was used to block autophagosome formation [
16]. The combination of NMP and 3-MA did not show any decreased, but indeed increased inhibition rate of lung cancer cells proliferation compared with the NMP treatment alone (
Supplementary Figure S2A,B). This result was then confirmed by apoptotic analysis with PI/Annexin V staining (
Supplementary Figure S2C). Since 3-MA treatment was not able to reverse the inhibitory effect of NMP on NSCLC cells proliferation, NMP-induced cell death could not be explained by autophagic cell death.
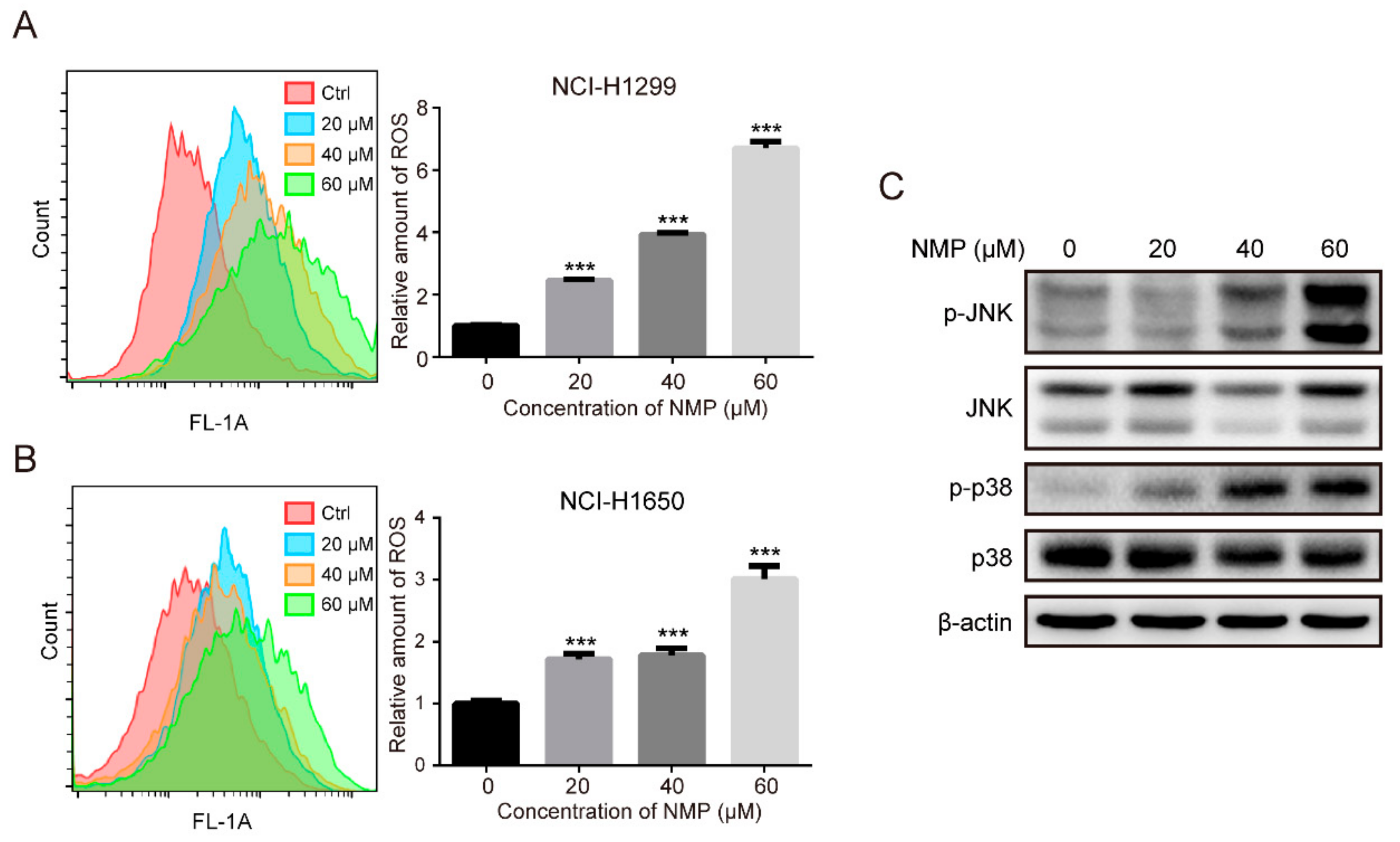
Excessive ROS accumulation is well known to activate MAPK pathways, resulting in cell death. For example, palmitate could induce H9c2 cell death through activation of ROS/MAPK signaling pathways [
21]. Also, isoliensinine could induce apoptosis in human breast cancer cells through ROS generation and p38/JNK activation [
22]. Our data showed that NMP treatment resulted in a significant increase of intracellular ROS (
Figure 4A,B) and activation of p38/JNK (
Figure 4C). To test whether ROS is involved in regulating MAPK pathways, we treated NSCLC cells with a ROS scavenger, NAC, and found a significant reduction in NMP-induced apoptosis (
Figure 5B) with abolished activation of p38 and JNK (
Figure 5C). These data indicated that ROS might induce apoptosis by activating p38/JNK.
Several lines of evidence have established that phosphorylation of Bcl-2 by p38 enhanced its degradation via a proteasome-dependent pathway. JNK pathway regulates mitochondrial apoptotic cell death via modulation of Bcl-2 and Bax protein expression [
23,
24,
25,
26]. However, our data showed that NMP did alter the expression of Bcl-2. JNK and p38 were reported to mediate the translocation of Bax from the cytoplasm to the mitochondrial outer membrane by Bax phosphorylation [
27]. The Bax protein plays a crucial role in apoptosis induction through the mitochondria-dependent pathway that controls the release of cytochrome c, a well-known apoptotic factor. When the Bax protein is translocated to the mitochrondrial outer membrane, channels for cytochrome c release were formed [
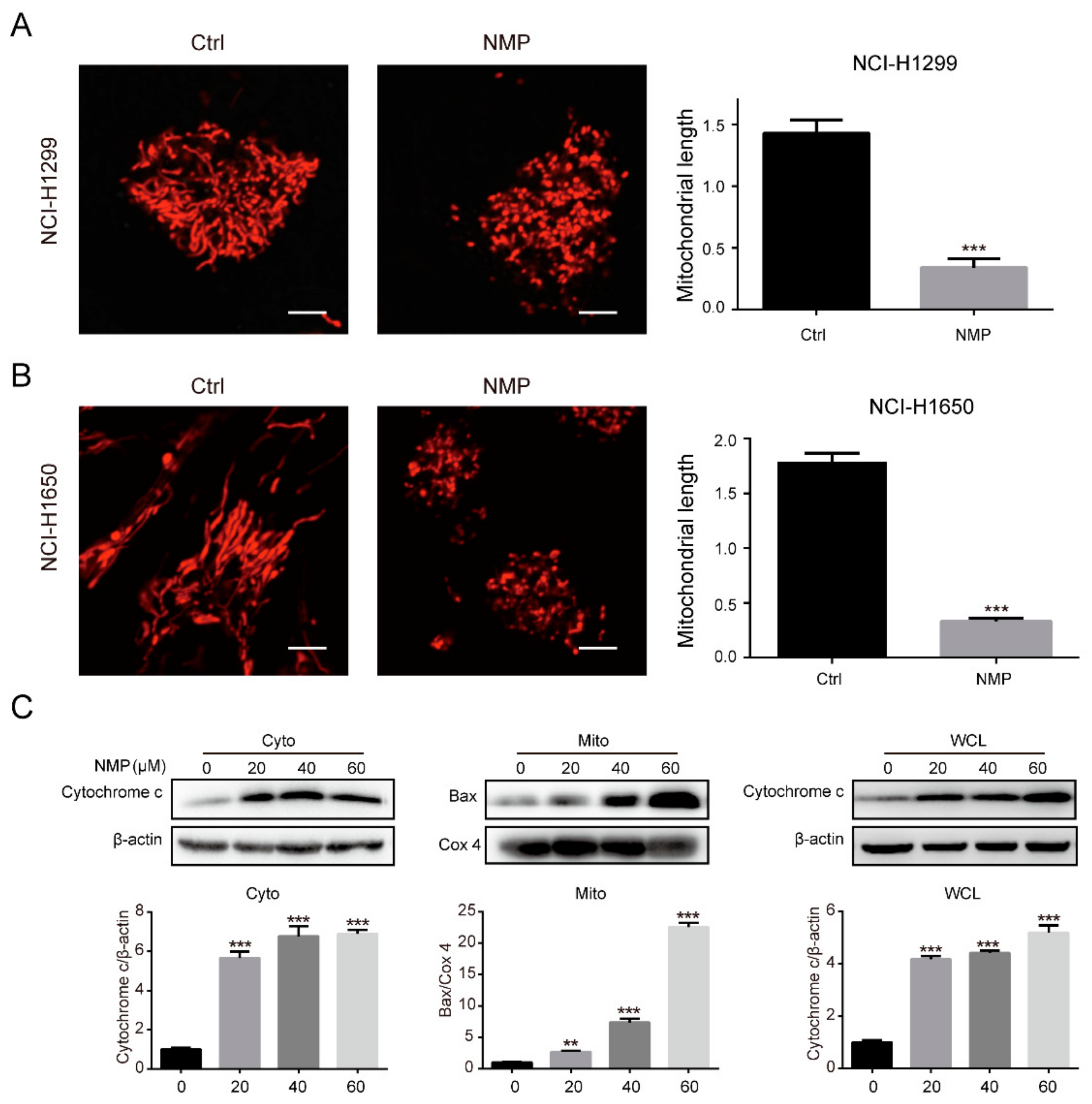
28]. In this study, we found that Bax was accumulated in the mitochrondria fraction while cytochrome c was markedly increased in the cytosolic fraction (
Figure 3C). This suggested that NMP treatment induced Bax translocation to mitochondria, followed by cytochrome c release to cytosol. Collectively, we concluded that NMP induced mitochondria-dependent apoptosis in human NSCLC cells. This was mediated by p38 and JNK activation followed by Bax translocation.
4. Materials and Methods
4.1. Cell Culture and Drugs
Human NSCLC cell lines NCI-H1299 (ATCC® CRL-5803™) and NCI-H1650 (ATCC® CRL-5883™) were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). BEAS-2B, an immortalized human bronchial epithelial cell line, was purchased from iCell Bioscience, Inc. (iCell-h023, Shanghai, China). All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin/streptomycin at 37 °C in 5% CO2. All reagents for cell culture were purchased from Gibco Life Technologies (Grand Island, NY, USA) unless otherwise stated.
N-Methylparoxetine (NMP, M2645) was purchased from J and K Chemical (Beijing, China). Bafilomycin A1 (Baf, B1793) was purchased from Sigma Biotechnology (St. Louis, MO, USA). Rapamycin (Rapa, S1039) was purchased from Selleckchem (Houston, TX, USA).
4.2. Cell Viability Assay
Cells were seeded onto 96-well plates and cultured overnight before treatment with different compounds for 24 h. Cell proliferation was assessed using Cell Counting Kit-8 (CCK-8, CK04, Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instruction. Briefly, the medium of each well was removed before adding 100 µL of CCK-8 reagent (10-fold dilution in DMEM). The plates were incubated for 2 h at 37 °C before measuring absorbance at 450 nm.
4.3. Colony Formation Assay
Cells were seeded onto 6-well plates, cultured for two days and treated with drugs for three days. The medium was removed, followed by rinsing colonies with PBS and fixation with 4% paraformaldehyde (PFA) for 20 min. The colonies were then stained with crystal violet and photographed manually.
4.4. CFDA-SE Cell Tracking Assay
Cells were labeled with CFDA-SE Cell Proliferation Assay and Tracking Kit (C0051, Beyotime, Shanghai, China) and seeded in six-well plates with overnight incubation before drug treatment for 24 h. The cells were harvested and washed with PBS before resuspension in HBSS for fluorescence intensity quantification with a BD AccuriTM C6 flow cytometer (BD Pharmingen, San Diego, CA, USA).
4.5. 5-Ethynyl-20-Deoxyuridine (EdU) Cell Proliferation Assay
Cells were seeded on a confocal dish and cultured overnight before treatment for 24 h. 5-Ethynyl-20-deoxyuridine agent (EdU, C0071S, Beyotime, Shanghai, China) was added to each dish and allowed 8 h for incorporation. The cells were fixed in 4% PFA for 20 min and washed three times with PBS containing 3% bovine serum albumin (BSA). The cells were incubated in PBS containing 0.3% TritonX-100 for 15 min, followed by BSA-containing PBS washes three times. The cells were stained with 5 μg/mL Hoechst 33342 at room temperature for 10 min, washed three times with PBS and mounted using Prolong Diamond DAPI (P36966, Karlsruhe, Germany). Images were captured using an LSM 800 confocal microscopy platform (Carl Zeiss, Jena, Germany).
4.6. Cell Apoptosis Detection
Cells were plated onto six-well plates overnight and treated for 24 h. Cell apoptosis detection was performed with FITC Annexin V Apoptosis Detection Kit (556547, BD Pharmingen, San Jose, CA, USA) according to manufacturer’s instruction. Briefly, treated cells were harvested and washed with PBS, followed by suspension in 600 μL annexin V binding buffer. Three microliters of annexin-V-FITC was added to each sample for 20 min at 37 °C in the dark. Then 5 μL PI was added for a further 5 min incubation. For each sample, 10,000 cells were analyzed using FL1 and FL3 channels with an Accuri C6 flow cytometer (BD Pharmingen, San Diego, CA, USA).
4.7. Cellular Fractionation Western Blot Analysis
Cellular fractions from treated cells were extracted with a mitochondrial protein extraction kit (BB-3171) and cytosolic protein extraction kit (BB-3113) from BestBio (Shanghai, China). Treated cells were harvested and washed twice with PBS. Five-hundred μL of mitochondrial or cytoplasmic protein reagent was added to each sample, followed by incubation on ice for 20 min. The samples were vortexed every 5 min. Mitochondrial protein was prepared by centrifugation at 11,000× g for 20 min while cytoplasmic protein was prepared by centrifugation at 16,000× g for 5 min. The mitochondrial protein was lysed in 1× loading buffer. The cytoplasmic protein was quantified using a BCA protein assay kit (MA0082, Meilunbio, Dalian, China). All proteins were stored at −80 °C.
4.8. Western Blot Analysis
The whole cell was lysed in 1× loading buffer. The protein samples were separated with 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes. The membranes were blocked in 5% skim milk powder in TBST for 2 h and incubated overnight with primary antibodies at 4 °C. The membrane was washed three times with TBST (0.05% Tween 20 in Tris-buffered saline) and incubated with secondary antibodies (diluted 1:4000). The immunoreactive bands were visualized with enhanced chemiluminescence (ECL) using an ECL detection system. The band densities were quantified using ImageJ (NIH).
Anti-LC3B antibody (12994S), anti-p62 antibody (5114), anti-PARP antibody (9542), anti-P-JNK antibody (9251), anti-JNK antibody (9252), anti-CytochromeC antibody (11940), and cleaved-caspase 3 antibody (9662) were from Cell Signaling Technology (Boston, MA, USA). CatD antibody (SC-13985) was from Santa Cruz Biotechnology (Dallas, TX, USA). Peroxidase-labeled antibodies anti-mouse IgG (AS003) and anti-rabbit IgG (AS014) were from ABclonal (Wuhan, China).
4.9. Plasmid Transfection Assay
Cells were seeded onto coverslips in 12-well plates and cultured for 24 h. GFP-LC3B or mCherry-GFP-LC3B plasmids (generous gifts from Professor William K. K. Wu, The Chinese University of Hong Kong) were transfected into cells using Lipofectamine 3000 strictly according to the manufacturer’s instructions (Invitrogen). After 6 h, the transfection medium was replaced with fresh culture medium and the cells were incubated for 24 h before drug treatment. The cells were fixed with 4% PFA for 20 min and washed three times with PBS. Five μg/mL Hoechst 33342 was added for 10 min staining at room temperature. Stained cells were mounted on glass slides using Prolong Diamond DAPI and subjected to confocal imaging.
4.10. AO Staining
Cells were seeded onto coverslips in 12-well plates and incubated for 24 h before drug treatment. The medium was removed and the cells were washed three times with PBS. Each sample was incubated with 5 μg/mL Acridine Orange (AO, A6014, Sigma Biotechnology, St. Louis, MO, USA) at 37 °C with 5% CO2 for 20 min and washed three times with PBS. Images were obtained with a laser confocal scanning microscope equipped with an argon laser (excitation wavelength: 488 nm) and a 63× objective lens. In the lysosomal compartments, AO produces red fluorescence (620 nm long pass emission filter) and green fluorescence (emission range: 520–560 nm) in the cytosol and nuclear compartments. The red and green intensities’ ratios in non-nuclear cellular compartments were analyzed using ImageJ.
4.11. LysoTracker Red Staining
Cells were seeded onto coverslips in 12-well plates and cultured for 24 h before drug treatment. The medium was removed and the cells were washed three times with PBS. Each sample was stained with LysoTracker Red DND-99 (L7528, Invitrogen, Carlsbad, CA, USA) for 20 min. Then the samples were washed three times with PBS and imaged under the laser scanning confocal microscope equipped with an argon laser (excitation wavelength: 555 nm) and a 63× objective lens.
4.12. MitoTracker Red CMXRos Staining
Cells were seeded onto coverslips in 12-well plates and cultured for 24 h before drug treatment. The medium was removed and the cells were washed three times with PBS. Each sample was stained with MitoTracker Red CMXRos (9082) from Cell Signaling Technology, Boston, MA, USA) for 20 min and washed three times with PBS. Images were obtained with a laser scanning confocal microscope equipped with an argon laser (excitation wavelength: 555 nm) and a 63× objective lens.
4.13. Intracellular ROS Measurement
Cells were seeded onto 6-well plates and cultured for 24 h before drug treatment. The cells were harvested and washed with PBS, followed by staining with 10 μM DCFH-DA (D399) from Thermo Fisher Scientific (Waltham, MA, USA) for 15 min at 37 °C in dark. Each sample was washed with PBS and resuspended in HBSS. From each sample, 10,000 cells were analyzed using the FL1 channel with a BD Accuri C6 flow cytometer.
4.14. Statistical Analysis
All experiments were repeated at least three times. The statistics were evaluated using one-way analysis of variance (ANOVA) and multiple comparisons. Level of significance was set at *** p < 0.001; ** p < 0.01; and * p < 0.05.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








