Increased mtDNA Abundance and Improved Function in Human Barth Syndrome Patient Fibroblasts Following AAV-TAZ Gene Delivery

Abstract
:1. Introduction
2. Results
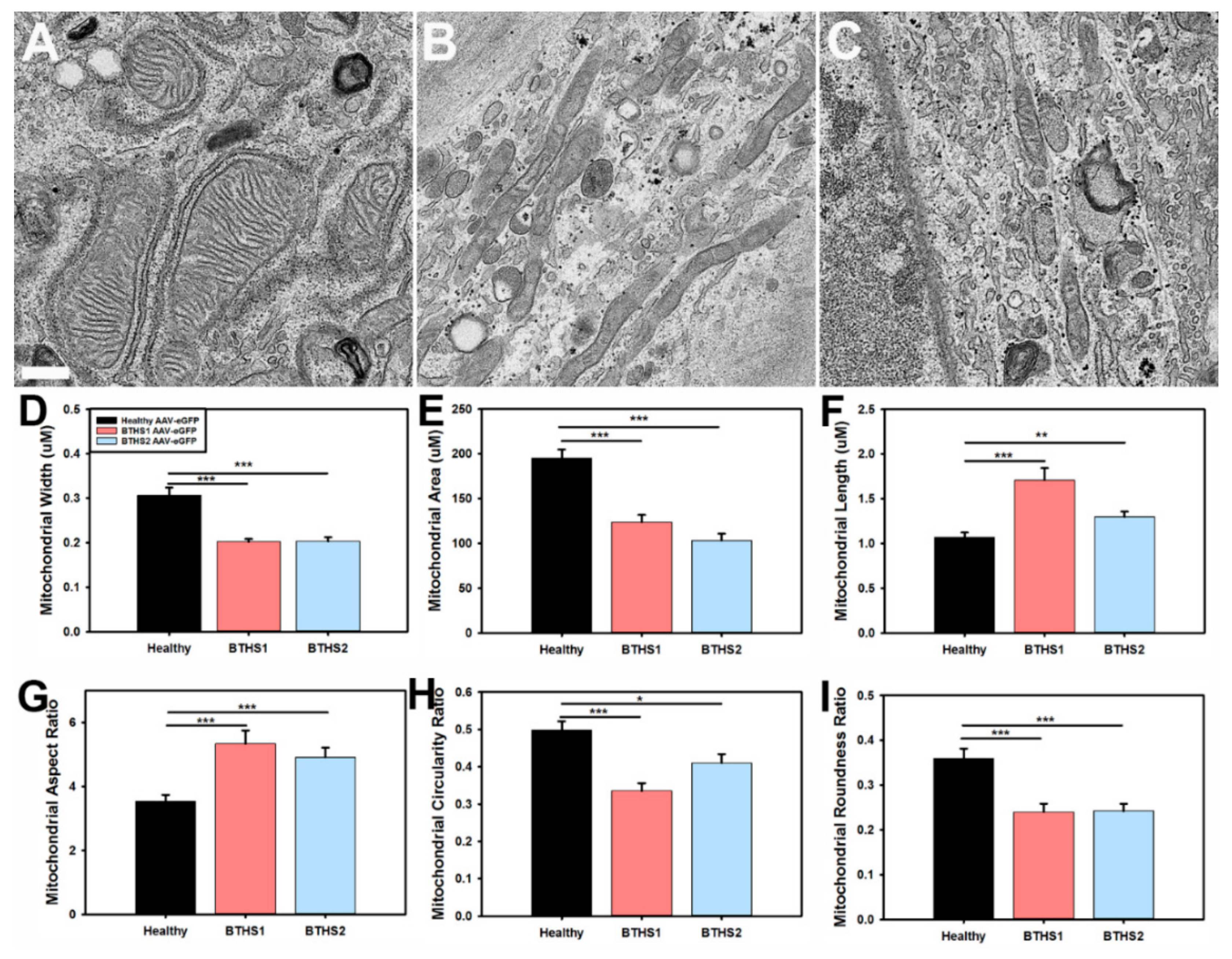
2.1. BTHS Fibroblast Mitochondria Display Structural Alterations
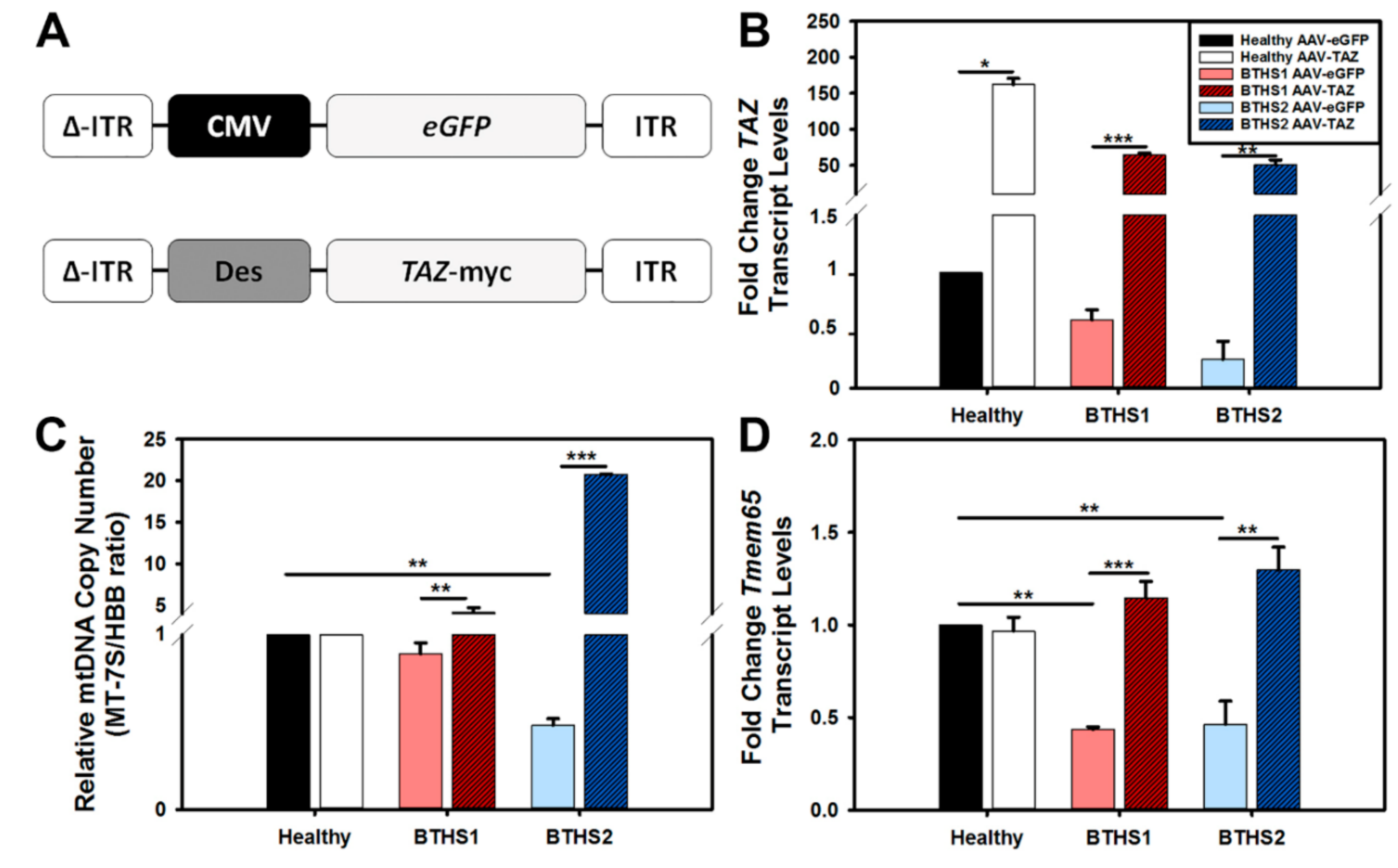
2.2. AAV-TAZ Administration Increases mtDNA Copy Numbers
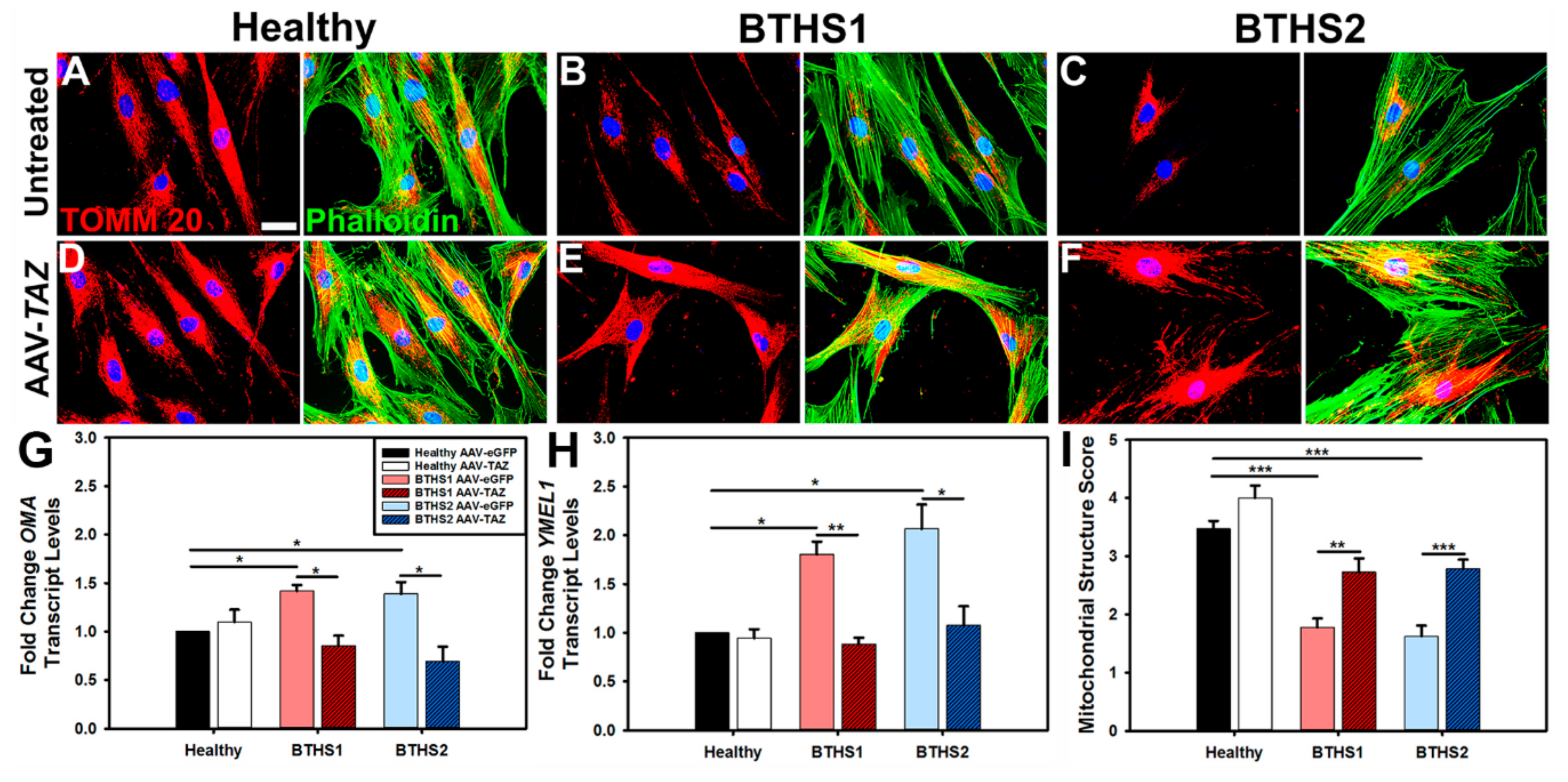
2.3. Mitochondrial Fragmentation in BTHS
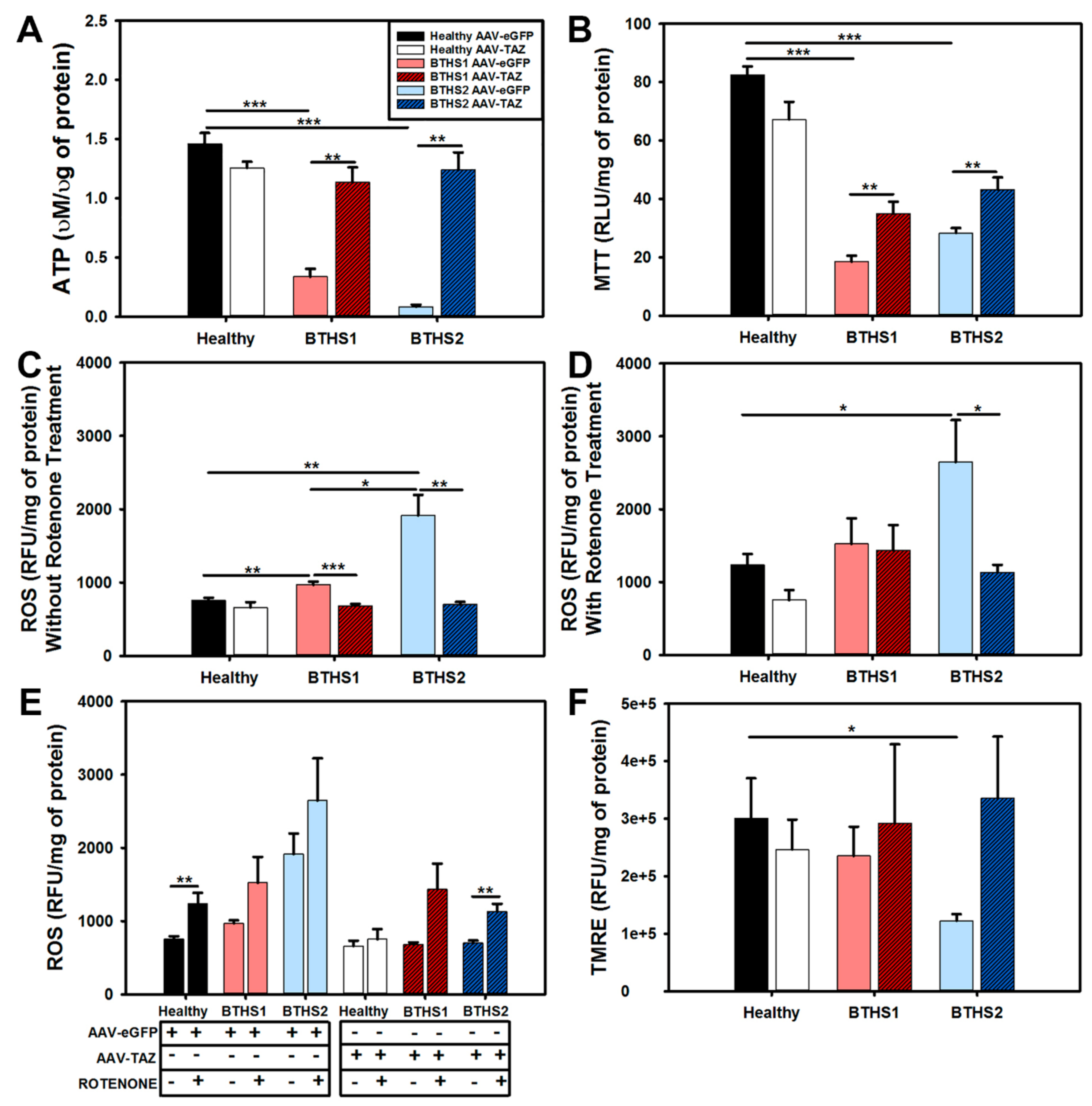
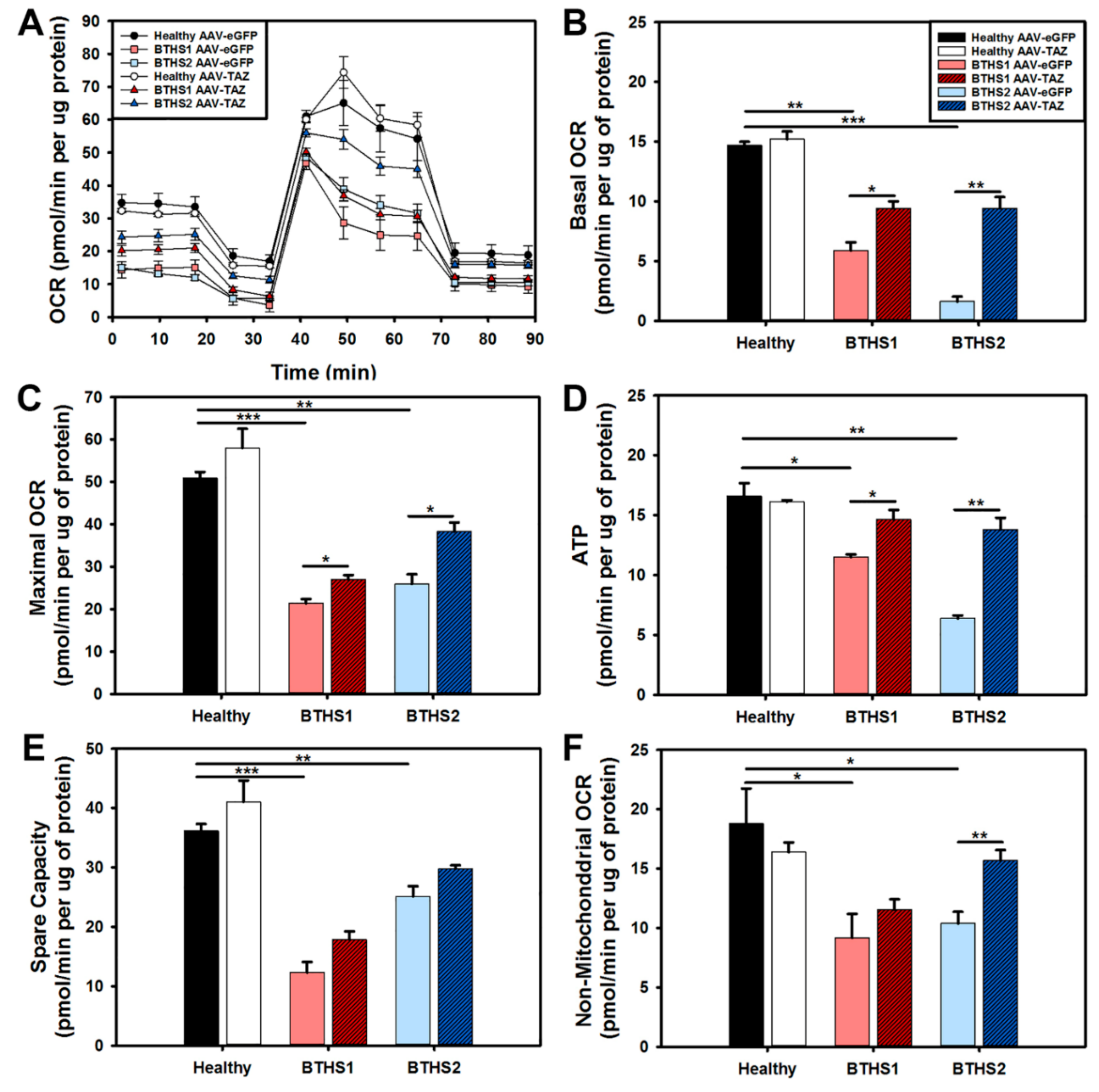
2.4. Impact of AAV-TAZ Treatment on Mitochondrial Function
3. Discussion
4. Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Houtkooper, R.H.; Turkenburg, M.; Poll-The, B.T.; Karall, D.; Perez-Cerda, C.; Morrone, A.; Malvagia, S.; Wanders, R.J.; Kulik, W.; Vaz, F.M. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta 2009, 1788, 2003–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini-Chohan, H.K.; Holmes, M.G.; Chicco, A.J.; Taylor, W.A.; Moore, R.L.; McCune, S.A.; Hickson-Bick, D.L.; Hatch, G.M.; Sparagna, G.C. Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J. Lipid Res. 2009, 50, 1600–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlame, M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 2008, 49, 1607–1620. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Ren, M.; Xu, Y.; Greenberg, M.L.; Haller, I. Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 2005, 138, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kelley, R.I.; Blanck, T.J.; Schlame, M. Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 2003, 278, 51380–51385. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Kelley, R.I.; Feigenbaum, A.; Towbin, J.A.; Heerdt, P.M.; Schieble, T.; Wanders, R.J.; DiMauro, S.; Blanck, T.J. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 2003, 42, 1994–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valianpour, F.; Mitsakos, V.; Schlemmer, D.; Towbin, J.A.; Taylor, J.M.; Ekert, P.G.; Thorburn, D.R.; Munnich, A.; Wanders, R.J.; Barth, P.G.; et al. Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis. J. Lipid Res. 2005, 46, 1182–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Neuwald, A.F. Barth syndrome may be due to an acyltransferase deficiency. Curr. Biol. 1997, 7, R465–R466. [Google Scholar] [CrossRef]
- Spencer, C.T.; Bryant, R.M.; Day, J.; Gonzalez, I.L.; Colan, S.D.; Thompson, W.R.; Berthy, J.; Redfearn, S.P.; Byrne, B.J. Cardiac and clinical phenotype in Barth syndrome. Pediatrics 2006, 118, e337–e346. [Google Scholar] [CrossRef]
- Bashir, A.; Bohnert, K.L.; Reeds, D.N.; Peterson, L.R.; Bittel, A.J.; de Las Fuentes, L.; Pacak, C.A.; Byrne, B.J.; Cade, W.T. Impaired cardiac and skeletal muscle bioenergetics in children, adolescents, and young adults with Barth syndrome. Physiol. Rep. 2017, 5, e13130. [Google Scholar] [CrossRef] [PubMed]
- Cade, W.T.; Spencer, C.T.; Reeds, D.N.; Waggoner, A.D.; O’Connor, R.; Maisenbacher, M.; Crowley, J.R.; Byrne, B.J.; Peterson, L.R. Substrate metabolism during basal and hyperinsulinemic conditions in adolescents and young-adults with Barth syndrome. J. Inherit. Metab. Dis. 2013, 36, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.T.; Byrne, B.J.; Bryant, R.M.; Margossian, R.; Maisenbacher, M.; Breitenger, P.; Benni, P.B.; Redfearn, S.; Marcus, E.; Cade, W.T. Impaired Cardiac Reserve and Severely Diminished Skeletal Muscle Oxygen Utilization Mediate Exercise Intolerance in Barth Syndrome. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2122–H2129. [Google Scholar] [CrossRef] [PubMed]
- Cade, W.T.; Bohnert, K.L.; Peterson, L.R.; Patterson, B.W.; Bittel, A.J.; Okunade, A.L.; de Las Fuentes, L.; Steger-May, K.; Bashir, A.; Schweitzer, G.G.; et al. Blunted fat oxidation upon submaximal exercise is partially compensated by enhanced glucose metabolism in children, adolescents, and young adults with Barth syndrome. J. Inherit. Metab. Dis. 2019, 42, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Cade, W.T.; Bohnert, K.L.; Reeds, D.N.; Peterson, L.R.; Bittel, A.J.; Bashir, A.; Byrne, B.J.; Taylor, C.L. Peak oxygen uptake (VO2peak) across childhood, adolescence and young adulthood in Barth syndrome: Data from cross-sectional and longitudinal studies. PLoS ONE 2018, 13, e0197776. [Google Scholar] [CrossRef]
- Cade, W.T.; Reeds, D.N.; Peterson, L.R.; Bohnert, K.L.; Tinius, R.A.; Benni, P.B.; Byrne, B.J.; Taylor, C.L. Endurance Exercise Training in Young Adults with Barth Syndrome: A Pilot Study. Jimd Rep. 2017, 32, 15–24. [Google Scholar] [PubMed]
- Bowron, A.; Honeychurch, J.; Williams, M.; Tsai-Goodman, B.; Clayton, N.; Jones, L.; Shortland, G.J.; Qureshi, S.A.; Heales, S.J.; Steward, C.G. Barth syndrome without tetralinoleoyl cardiolipin deficiency: A possible ameliorated phenotype. J. Inherit. Metab. Dis. 2015, 38, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; Maianski, N.A.; Tool, A.T.; Becker, K.; Plecko, B.; Valianpour, F.; Wanders, R.J.; Pereira, R.; Van Hove, J.; Verhoeven, A.J.; et al. Neutrophils in Barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood 2004, 103, 3915–3923. [Google Scholar] [CrossRef] [Green Version]
- Suzuki-Hatano, S.; Saha, M.; Rizzo, S.A.; Witko, R.L.; Gosiker, B.J.; Ramanathan, M.; Soustek, M.S.; Jones, M.D.; Kang, P.B.; Byrne, B.J.; et al. AAV-Mediated TAZ Gene Replacement Restores Mitochondrial and Cardioskeletal Function in Barth Syndrome. Hum. Gene 2019, 30, 139–154. [Google Scholar] [CrossRef]
- Suzuki-Hatano, S.; Saha, M.; Soustek, M.S.; Kang, P.B.; Byrne, B.J.; Cade, W.T.; Pacak, C.A. AAV9-TAZ Gene Replacement Ameliorates Cardiac TMT Proteomic Profiles in a Mouse Model of Barth Syndrome. Mol. Methods Clin. Dev. 2019, 13, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Jayandharan, G.R.; Li, B.; Ling, C.; Ma, W.; Srivastava, A.; Zhong, L. High-efficiency transduction of fibroblasts and mesenchymal stem cells by tyrosine-mutant AAV2 vectors for their potential use in cellular therapy. Hum. Gene Ther. 2010, 21, 1527–1543. [Google Scholar] [CrossRef] [PubMed]
- Schnepp, B.C.; Clark, K.R.; Klemanski, D.L.; Pacak, C.A.; Johnson, P.R. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J. Virol. 2003, 77, 3495–3504. [Google Scholar] [CrossRef] [PubMed]
- Taggart, K.; Estrada, A.; Thompson, P.; Lourenco, F.; Kirmani, S.; Suzuki-Hatano, S.; Pacak, C.A. PDK4 Deficiency Induces Intrinsic Apoptosis in Response to Starvation in Fibroblasts from Doberman Pinschers with Dilated Cardiomyopathy. Biores. Open Access 2017, 6, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Li, B.; Mah, C.S.; Govindasamy, L.; Agbandje-McKenna, M.; Cooper, M.; Herzog, R.W.; Zolotukhin, I.; Warrington, K.H., Jr.; Weigel-Van Aken, K.A.; et al. Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. USA 2008, 105, 7827–7832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazli, A.; Safdar, A.; Saleem, A.; Akhtar, M.; Brady, L.I.; Schwartzentruber, J.; Tarnopolsky, M.A. A mutation in the TMEM65 gene results in mitochondrial myopathy with severe neurological manifestations. Eur. J. Hum. Genet. EJHG 2017, 25, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, N.; Gotoh, T.; Oike, Y.; Yano, M. TMEM65 is a mitochondrial inner-membrane protein. Peer J. 2014, 2, e349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Abbasi, C.; Lazic, S.; Teng, A.C.; Wang, D.; Dubois, N.; Ignatchenko, V.; Wong, V.; Liu, J.; Araki, T.; et al. Evolutionarily conserved intercalated disc protein Tmem65 regulates cardiac conduction and connexin 43 function. Nat. Commun. 2015, 6, 8391. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Tan, L.; Shen, R.; Zhang, L.; Zuo, H.; Wang, D.W. Decreased Peripheral Mitochondrial DNA Copy Number is Associated with the Risk of Heart Failure and Long-term Outcomes. Medicine 2016, 95, e3323. [Google Scholar] [CrossRef]
- Yue, P.; Jing, S.; Liu, L.; Ma, F.; Zhang, Y.; Wang, C.; Duan, H.; Zhou, K.; Hua, Y.; Wu, G. Association between mitochondrial DNA copy number and cardiovascular disease: Current evidence based on a systematic review and meta-analysis. PLoS ONE 2018, 13, e0206003. [Google Scholar] [CrossRef]
- Johnson-Cadwell, L.I.; Jekabsons, M.B.; Wang, A.; Polster, B.M.; Nicholls, D.G. ‘Mild Uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J. Neurochem. 2007, 101, 1619–1631. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Mutation: | Exon: | Amino Acid: | Mutation Type: |
|---|---|---|---|
| c.170G > T | Exon 2 | p.Arg57Leu | Missense |
| c.140-152del13 | Exon 2 | p.Arg47Thr fs*32 | Frameshift |
| Gene | Catalog | Company | |
|---|---|---|---|
| 18S ribosomal RNA | 18S | Hs03003631-g1 | Thermo Scientific |
| Tafazzin | TAZ | Hs00794094_m1 | Thermo Scientific |
| YME1 like 1 ATPase | YME1L1 | Hs00907919-m1 | Thermo Scientific |
| OMA1 zinc metallopeptidase | OMA1 | Hs00377028-m1 | Thermo Scientific |
| Gene | Catalog | Company | |
|---|---|---|---|
| AB mitochondrial gene 7S, encoding D-loop, which is a replication start site of the mtDNA | MT-7S | HS02596861_s1 | Thermo Scientific |
| Hemoglobin subunit beta | HBB | HS00758889_s1 | Thermo Scientific |
| Protein | Dilution (I) | Company | |
|---|---|---|---|
| Translocase of Outer Mitochondrial Membrane 20 kDa protein | TOMM20 | 1:200 | Sigma Aldrich #HPA011562 |
| Phalloidin | Alexa Fluor 488 | 1:200 | Life Tech # A12379 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki-Hatano, S.; Sriramvenugopal, M.; Ramanathan, M.; Soustek, M.; Byrne, B.J.; Cade, W.T.; Kang, P.B.; Pacak, C.A. Increased mtDNA Abundance and Improved Function in Human Barth Syndrome Patient Fibroblasts Following AAV-TAZ Gene Delivery. Int. J. Mol. Sci. 2019, 20, 3416. https://doi.org/10.3390/ijms20143416
Suzuki-Hatano S, Sriramvenugopal M, Ramanathan M, Soustek M, Byrne BJ, Cade WT, Kang PB, Pacak CA. Increased mtDNA Abundance and Improved Function in Human Barth Syndrome Patient Fibroblasts Following AAV-TAZ Gene Delivery. International Journal of Molecular Sciences. 2019; 20(14):3416. https://doi.org/10.3390/ijms20143416
Chicago/Turabian StyleSuzuki-Hatano, Silveli, Mughil Sriramvenugopal, Manash Ramanathan, Meghan Soustek, Barry J. Byrne, W. Todd Cade, Peter B. Kang, and Christina A. Pacak. 2019. "Increased mtDNA Abundance and Improved Function in Human Barth Syndrome Patient Fibroblasts Following AAV-TAZ Gene Delivery" International Journal of Molecular Sciences 20, no. 14: 3416. https://doi.org/10.3390/ijms20143416