The 1,3,5-Triazine Derivatives as Innovative Chemical Family of 5-HT6 Serotonin Receptor Agents with Therapeutic Perspectives for Cognitive Impairment

 ,
,  , ,
, ,
Abstract
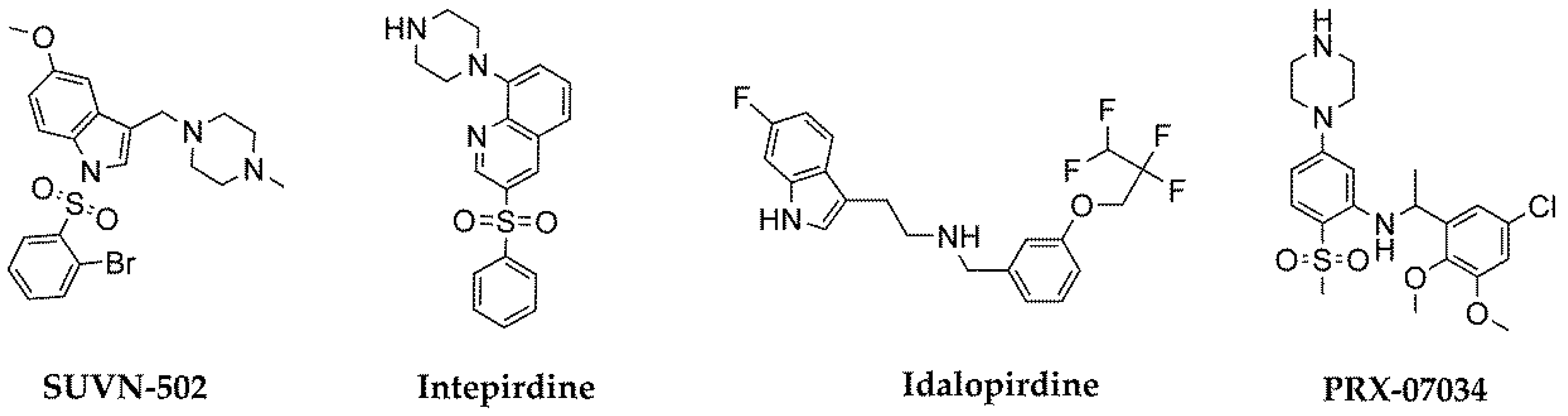
:1. Introduction
2. Results
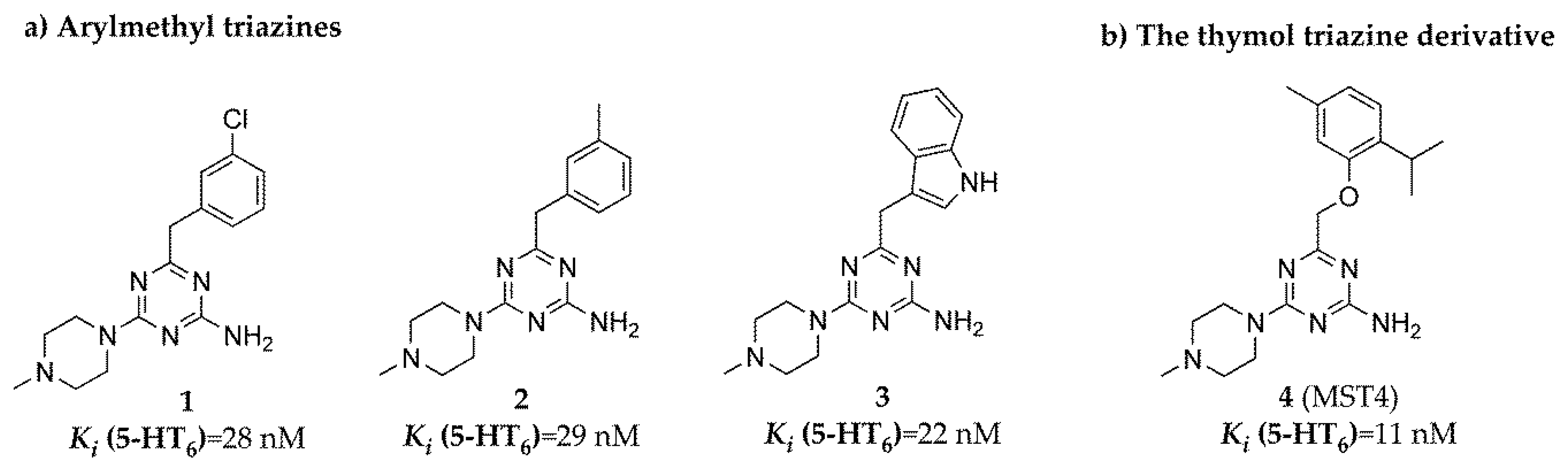
2.1. Radioligand Binding Assay
2.2. ADMET Assays Results
2.2.1. Bioavailability
2.2.2. Pharmacokinetics
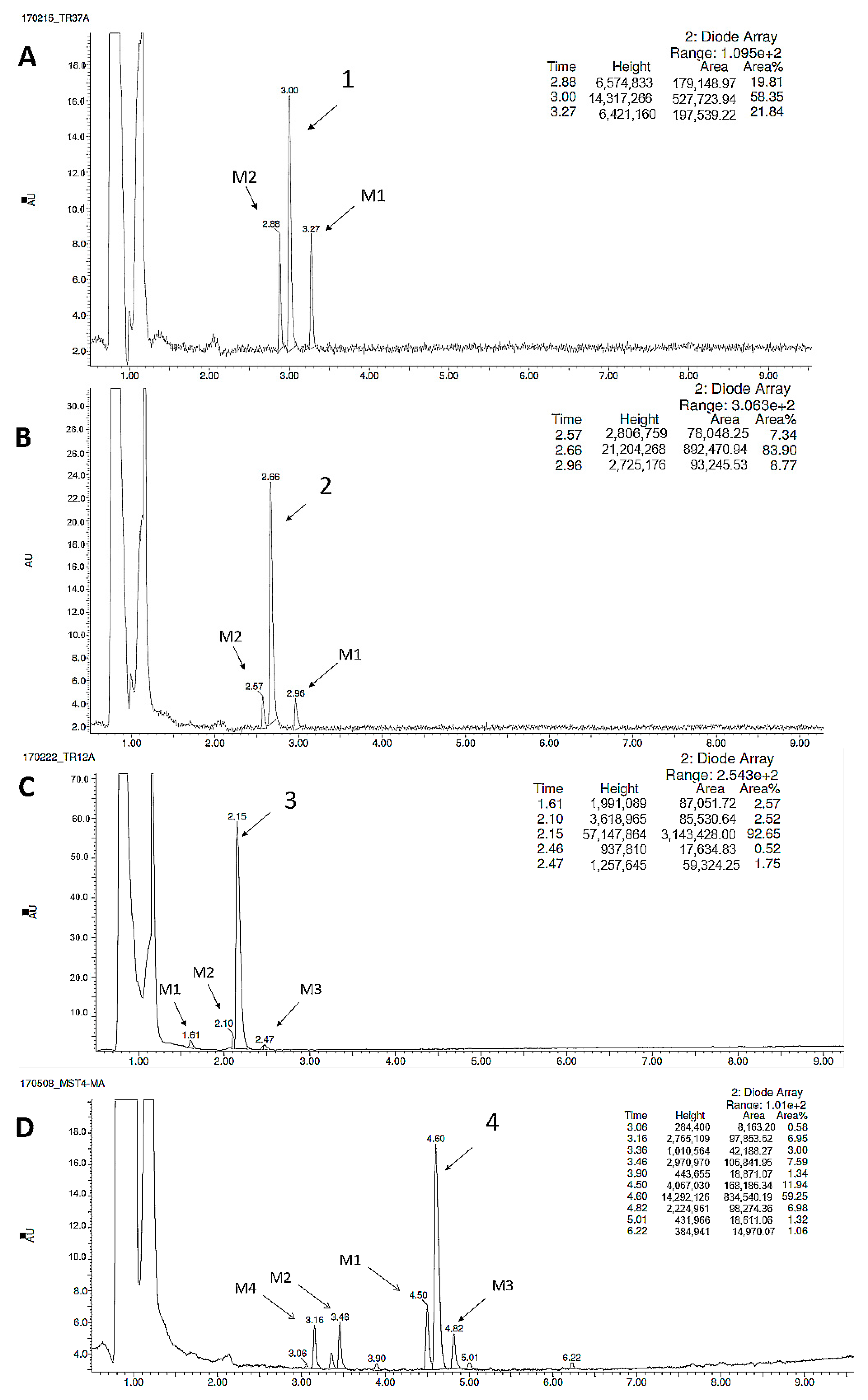
2.2.3. Metabolic Pathways
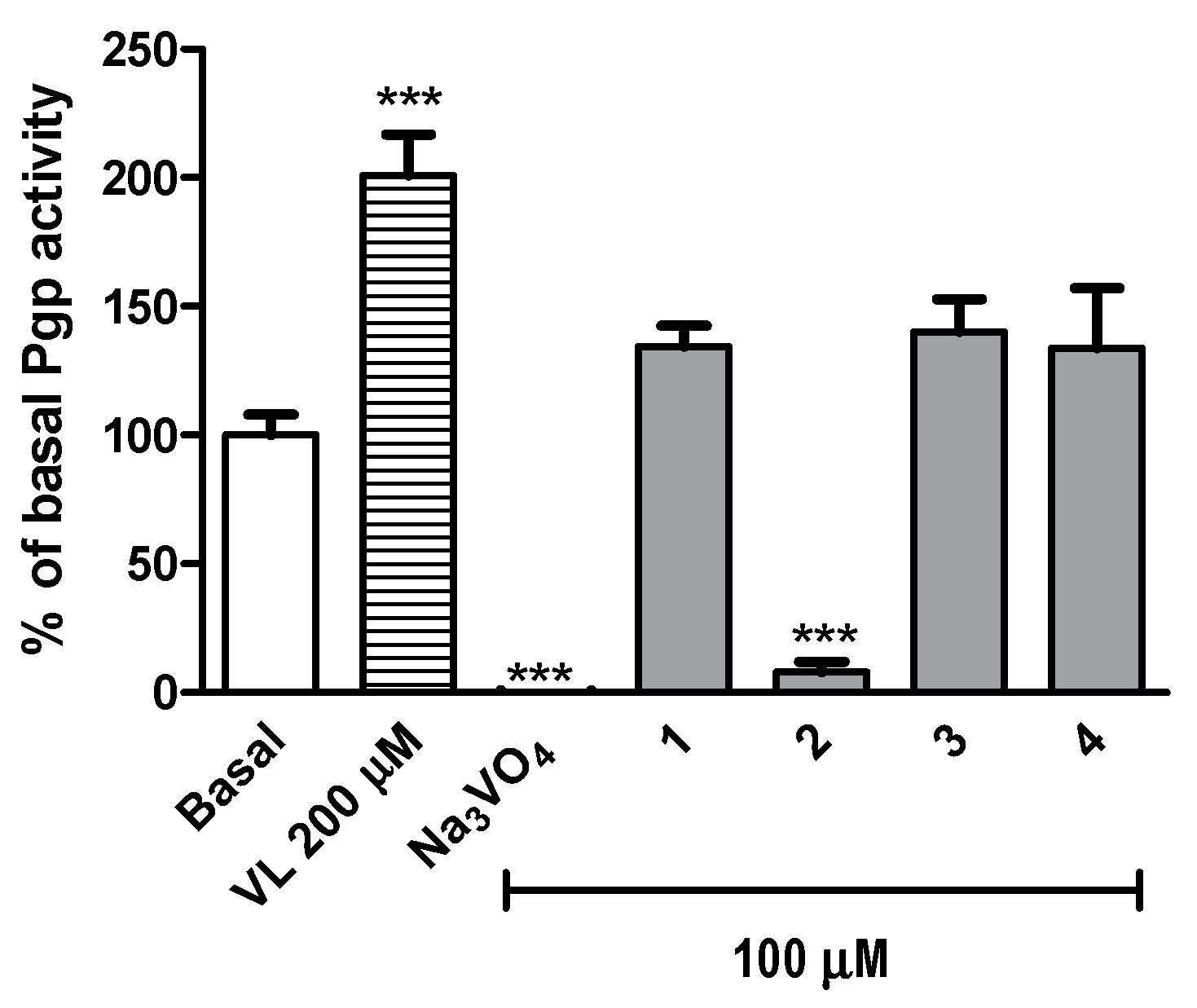
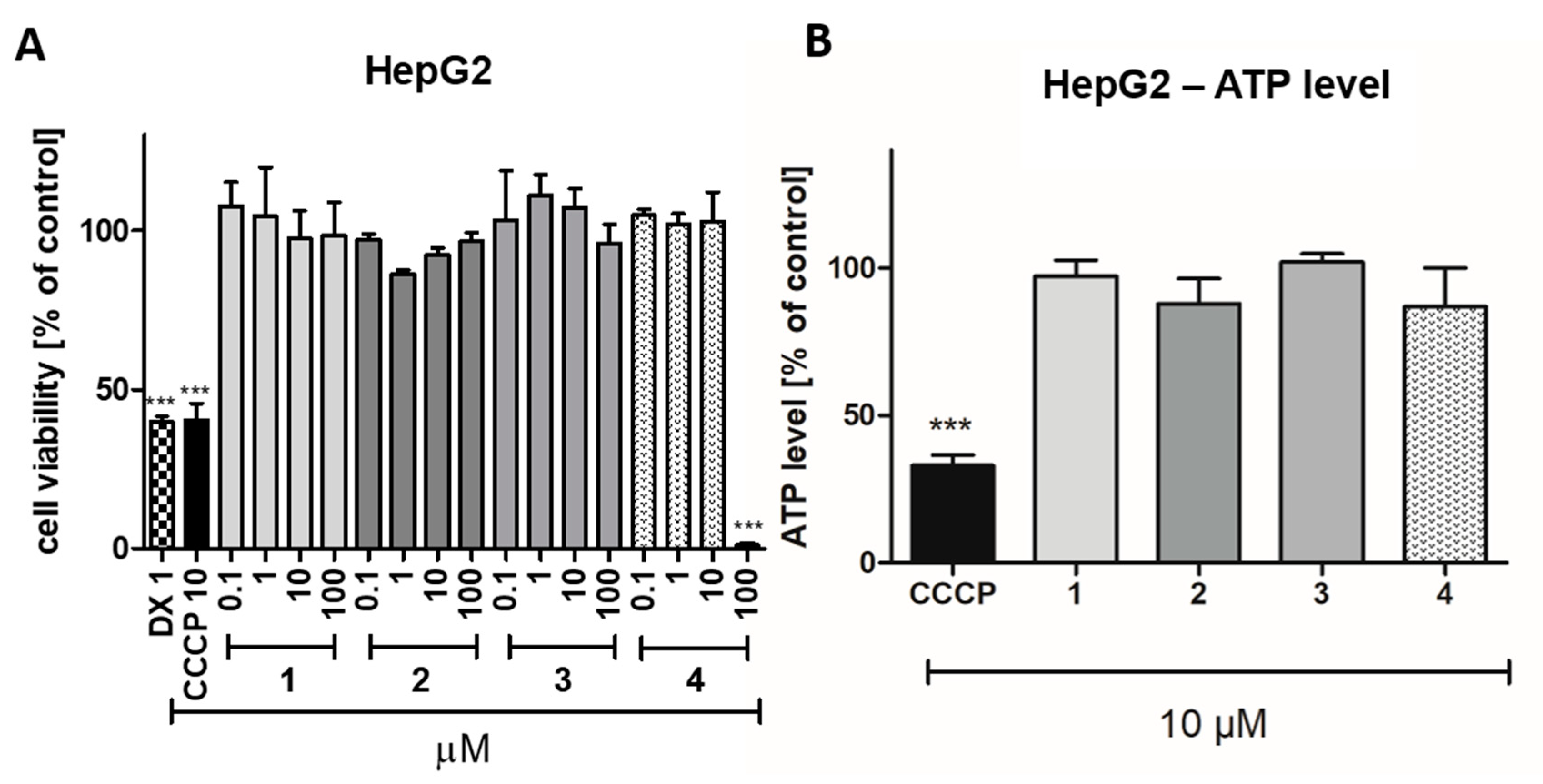
2.2.4. Safety
2.3. Behavioral Tests In Vivo
2.3.1. Anxiolytic-Like Activity of Investigated Compounds
2.3.2. Ability of Investigated Compounds to Reverse Memory Impairment
2.4. Influence on Body Mass In Vivo
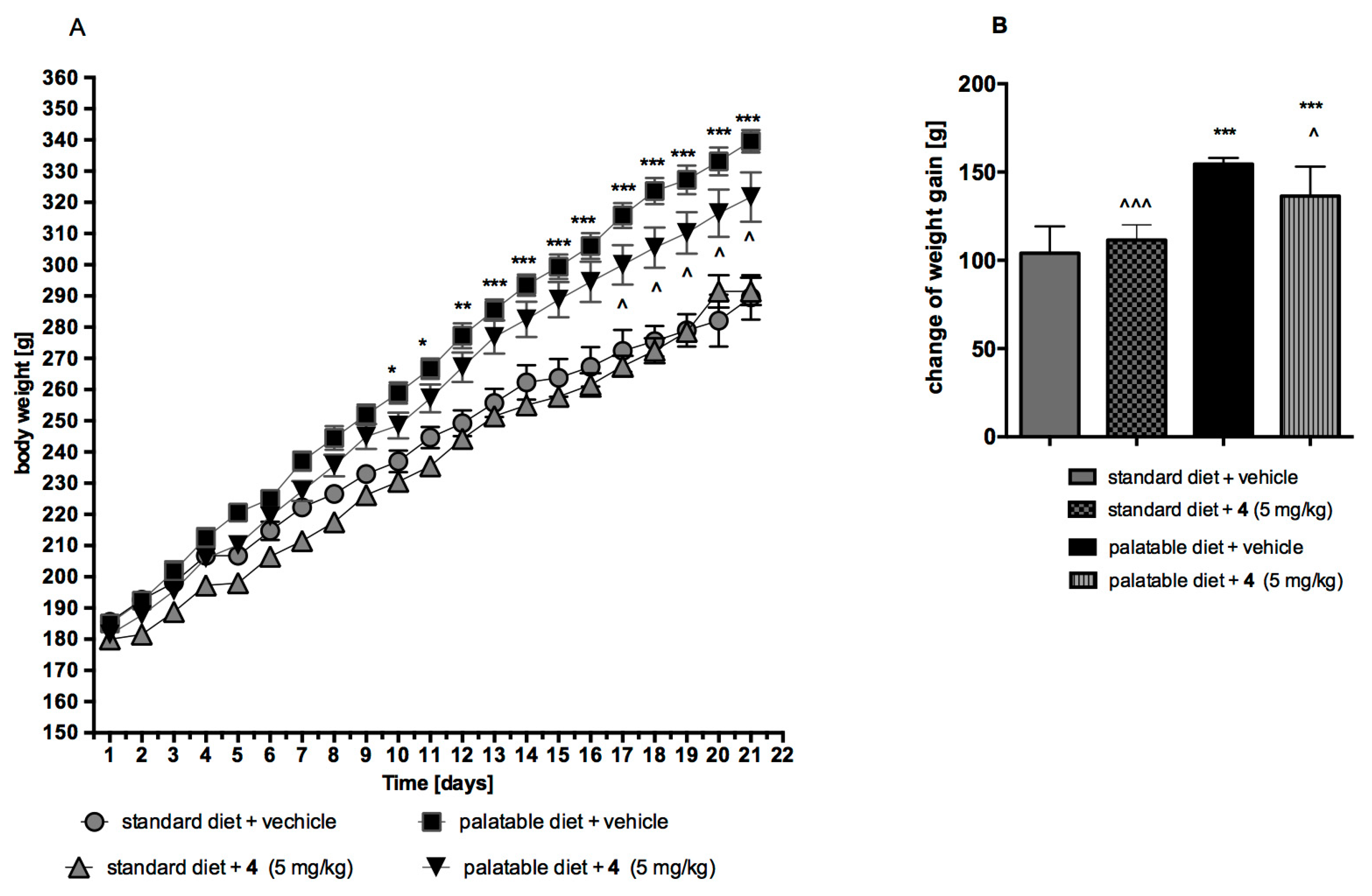
2.4.1. Obesity Induced with Palatable Diet
2.4.2. Effect on Body Weight in Model of Excessive Eating and in Rats Fed Standard Diet
3. Discussion
4. Materials and Methods
4.1. Radioligand Binding Assay
4.2. ADMET
4.2.1. References
4.2.2. Bioavailability
4.2.3. Pharmacokinetics
4.2.4. Safety
4.3. Behavioral Tests In Vivo
4.3.1. Animals
4.3.2. Drugs
4.3.3. Vogel Conflict Drinking Test
4.3.4. Hot Plate and Free-Drinking Tests
4.3.5. Novel Object Recognition Test (NORT)
4.4. Assays of Influence on Body Mass In Vivo
4.4.1. Animals
4.4.2. The Effect of 4 on Body Weight of Non-Obese Rats Fed Palatable Diet (Model of Excessive Eating)
4.4.3. The Effect of 4 on Body Weight of Non-Obese Rats Fed Only with Standard Diet
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Monsma, F.J.; Shen, Y.O.N.G.; Ward, R.P.; Hamblin, M.W.; Sibley, D.R. Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol. Pharmacol. 1993, 43, 320–327. [Google Scholar] [PubMed]
- Ruat, M.; Traiffort, E.; Arrang, J.M.; Tardivellacombe, J.; Diaz, J.; Leurs, R.; Schwartz, J.C. A novel rat serotonin (5-HT6) receptor: molecular cloning, localization and stimulation of cAMP accumulation. Biochem. Biophys. Res. Comm. 1993, 193, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Karila, D.; Freret, T.; Bouet, V.; Boulouard, M.; Dallemagne, P.; Rochais, C. Therapeutic Potential of 5-HT6 Receptor Agonists: Miniperspective. J. Med. Chem. 2015, 5, 7901–7912. [Google Scholar] [CrossRef] [PubMed]
- Stefulj, J.; Jernej, B.; Cicin-Sain, L.; Rinner, I.; Schauenstein, K. mRNA expression of serotonin receptors in cells of the immune tissues of the rat. Brain Behav. Immun. 2000, 14, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.B.; Qiu, C.L.; Zhao, H.; Liu, Q.; Shao, Y. Expression of mRNA for multiple serotonin (5-HT) receptor types/subtypes by the peripheral blood mononuclear cells of rhesus macaques. J. Neuroimmunol. 2006, 178, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Leiser, S.C.; Li, Y.; Pehrson, A.L.; Dale, E.; Smagin, G.; Sanchez, C. Serotonergic regulation of prefrontal cortical circuitries involved in cognitive processing: a review of individual 5-HT receptor mechanisms and concerted effects of 5-HT receptors exemplified by the multimodal antidepressant vortioxetine. ACS Neurosci. 2015, 6, 970–986. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A. The procognitive effects of 5-HT6 receptor ligands in animal models of schizophrenia. Rev. Neurosci. 2014, 25, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Sebben, M.; Ansanay, H.; Bockaert, J.; Dumuis, A. 5-HT6 receptors positively coupled to adenylyl cyclase in striatal neurones in culture. Neuroreport 1994, 5, 2553–2557. [Google Scholar] [CrossRef]
- Yun, H.M.; Kim, S.; Kim, H.J.; Kostenis, E.; Kim, J.I.; Seong, J.Y.; Baik, J.H.; Rhim, H. The novel cellular mechanism of human 5-HT6 receptor through an interaction with Fyn. J. Biol. Chem. 2007, 282, 5496–5505. [Google Scholar] [CrossRef]
- Bonsi, P.; Cuomo, D.; Ding, J.; Sciamanna, G.; Ulrich, S.; Tscherter, A.; Bernardi, G.; Surmeier, D.J.; Pisani, A. Endogenous serotonin excites striatal cholinergic interneurons via the activation of 5-HT 2C, 5-HT6, and 5-HT7 serotonin receptors: implications for extrapyramidal side effects of serotonin reuptake inhibitors. Neuropsychopharmacology 2007, 32, 1840–1854. [Google Scholar] [CrossRef]
- Meffre, J.; Chaumont-Dubel, S.; la Cour, C.M.; Loiseau, F.; Watson, D.J.; Dekeyne, A.; Séveno, M.; Rivet, J.M.; Gaven, F.; Déléris, P.; et al. 5-HT6 receptor recruitment of mTOR as a mechanism for perturbed cognition in schizophrenia. EMBO Mol. Med. 2012, 4, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Majouga, A.G.; Veselov, M.S.; Chufarova, N.V.; Baranovsky, S.S.; Filkov, G.I. Computational approaches to the design of novel 5-HT6 R ligands. Rev. Neurosci. 2014, 25, 451–467. [Google Scholar] [CrossRef] [PubMed]
- Grychowska, K.; Satala, G.; Kos, T.; Partyka, A.; Colacino, E.; Chaumont-Dubel, S.; Bantreil, X.; Wesołowska, A.; Pawłowski, M.; Martinez, J.; et al. Novel 1H-Pyrrolo[3,2-c]quinoline Based 5-HT6 Receptor Antagonists with Potential Application for the Treatment of Cognitive Disorders Associated with Alzheimer’s Disease. ACS Chem. Neurosci. 2016, 7, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, Á.; Satala, G.; Bojarski, A.; Keserű, G. Spiro [pyrrolidine-3, 3′-oxindoles] and their indoline analogues as new 5-HT6 receptor chemotypes. Molecules 2017, 22, 2221. [Google Scholar] [CrossRef] [PubMed]
- Staroń, J.; Mordalski, S.; Warszycki, D.; Satała, G.; Hogendorf, A.; Bojarski, A.J. Pyrano [2, 3, 4-cd] indole as a Scaffold for Selective Nonbasic 5-HT6R Ligands. ACS Med. Chem. Lett. 2017, 8, 390–394. [Google Scholar] [CrossRef]
- Vanda, D.; Soural, M.; Canale, V.; Chaumont-Dubel, S.; Satała, G.; Kos, T.; Funk, P.; Fülöpová, V.; Lemrová, B.; Koczurkiewicz, P.; et al. Novel non-sulfonamide 5-HT6 receptor partial inverse agonist in a group of imidazo [4, 5-b] pyridines with cognition enhancing properties. Eur. J. Med. Chem. 2018, 144, 716–729. [Google Scholar] [CrossRef]
- Hung, S.Y.; Fu, W.M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement. (N Y) 2017, 3, 367–384. [Google Scholar] [CrossRef]
- Nirogi, R.; Abraham, R.; Benade, V.; Medapati, R.B.; Jayarajan, P.; Bhyrapuneni, G.; Muddana, N.; Mekala, V.R.; Subramanian, R.; Shinde, A.; et al. SUVN-502, a novel, potent, pure, and orally active 5-HT6 receptor antagonist: pharmacological, behavioral, and neurochemical characterization. Behav. Pharmacol. 2019, 30, 16–35. [Google Scholar] [CrossRef]
- Liu, K.G.; Robichaud, A.J. 5-HT6 antagonists as potential treatment for cognitive dysfunction. Drug Dev. Res. 2009, 70, 145–168. [Google Scholar] [CrossRef]
- Benhamú, B.; Martin-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; Lopez-Rodriguez, M.L. Serotonin 5-HT6 receptor antagonists for the treatment of cognitive deficiency in Alzheimer’s disease. J. Med. Chem. 2014, 57, 7160–7181. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Kurczab, R.; Więcek, M.; Kamińska, K.; Satała, G.; Jastrzębska-Więsek, M.; Partyka, A.; Bojarski, A.J.; Wesołowska, A.; Kieć-Kononowicz, K.; et al. The computer-aided discovery of novel family of the 5-HT6 serotonin receptor ligands among derivatives of 4-benzyl-1, 3, 5-triazine. Eur. J. Med. Chem. 2017, 135, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Kurczab, R.; Więcek, M.; Satała, G.; Kieć-Kononowicz, K.; Handzlik, J. Synthesis and computer-aided analysis of the role of linker for novel ligands of the 5-HT6 serotonin receptor among substituted 1, 3, 5-triazinylpiperazines. Bioorg. Chem. 2019, 84, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Ali, W.; Więcek, M.; Łażewska, D.; Kurczab, R.; Jastrzębska-Więsek, M.; Satała, G.; Kucwaj-Brysz, K.; Lubelska, A.; Głuch-Lutwin, M.; Mordyl, B.; et al. Synthesis and computer-aided SAR studies for derivatives of phenoxyalkyl-1,3,5-triazine as the new potent ligands for serotonin receptors 5-HT6. Eur. J. Med. Chem. 2019, 178, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Guioca, V. New substituted triazines and their diuretic activity. Ann. Pharm. Fr. 1973, 31, 283–292. [Google Scholar] [PubMed]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Sobiło, A.; Olejarz, A.; Kucwaj-Brysz, K.; Satała, G.; Bojarski, A.J.; Wesołowska, A.; et al. In the search for a lead structure among series of potent and selective hydantoin 5-HT7R agents: The drug-likeness in vitro study. Chem. Biol. Drug Design 2017, 90, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Kucwaj-Brysz, K.; Wesołowska, A.; Kieć-Kononowicz, K.; Handzlik, J. MF-8, a novel promising arylpiperazine-hydantoin based 5-HT7 receptor antagonist: In vitro drug-likeness studies and in vivo pharmacological evaluation. Bioorg. Med. Chem. Lett. 2018, 28, 878–883. [Google Scholar] [CrossRef]
- Latacz, G.; Hogendorf, A.S.; Hogendorf, A.; Lubelska, A.; Wierońska, J.M.; Woźniak, M.; Cieślik, P.; Kieć-Kononowicz, K.; Handzlik, J.; Bojarski, A.J. Search for a 5-CT alternative. In vitro and in vivo evaluation of novel pharmacological tools: 3-(1-alkyl-1 H-imidazol-5-yl)-1 H-indole-5-carboxamides, low-basicity 5-HT7 receptor agonists. MedChemComm 2019, 9, 1882–1890. [Google Scholar] [CrossRef]
- Socała, K.; Mogilski, S.; Pieróg, M.; Nieoczym, D.; Abram, M.; Szulczyk, B.; Lubelska, A.; Latacz, G.; Doboszewska, U.; Wlaź, P.; et al. KA-11, a novel pyrrolidine-2, 5-dione derived broad-spectrum anticonvulsant: its antiepileptogenic, antinociceptive properties and in vitro characterization. ACS Chem. Neurosci. 2018, 10, 636–648. [Google Scholar] [CrossRef]
- Obach, R.S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar]
- Nassar, A.F. Drug metabolism handbook: concepts and applications; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 147–158. ISBN 9780470118030. [Google Scholar]
- Kamińska, K.; Ziemba, J.; Ner, J.; Schwed, J.S.; Łażewska, D.; Więcek, M.; Karcz, T.; Olejarz, A.; Latacz, G.; Kuder, K.; et al. (2-Arylethenyl)-1, 3, 5-triazin-2-amines as a novel histamine H4 receptor ligands. Eur. J. Med. Chem. 2015, 103, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Łażewska, D.; Latacz, G.; Olejarz, A.; Makuch, W.; Stark, H.; Kieć-Kononowicz, K.; Mika, J. Antinociceptive effects of novel histamine H3 and H4 receptor antagonists and their influence on morphine analgesia of neuropathic pain in the mouse. Br. J. Pharmacol. 2018, 175, 2897–2910. [Google Scholar] [CrossRef] [PubMed]
- Wesołowska, A.; Nikiforuk, A. Effects of the brain-penetrant and selective 5-HT6 receptor antagonist SB-399885 in animal models of anxiety and depression. Neuropharmacology 2007, 52, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Frölich, L.; Ballard, C.; Tariot, P.N.; Molinuevo, J.L.; Boneva, N.; Windfeld, K.; Raket, L.L.; Cummings, J.L. Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: three randomized clinical trials. JAMA 2018, 319, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Ennaceur, A.; Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef]
- Klinkenberg, I.; Blokland, A. The validity of scopolamine as a pharmacological model for cognitive impairment: a review of animal behavioral studies. Neurosci. Biobehav. Rev. 2010, 34, 1307–1350. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, R.K.; Jaggi, A.S.; Singh, N. Animal models of dementia and cognitive dysfunction. Life Sci. 2014, 109, 73–86. [Google Scholar] [CrossRef]
- Kurczab, R.; Ali, W.; Łażewska, D.; Kotańska, M.; Jastrzębska-Więsek, M.; Satała, G.; Więcek, M.; Lubelska, A.; Latacz, G.; Partyka, A.; et al. Computer-aided studies for novel arylhydantoin 1,3,5-triazine derivatives as 5-HT6 serotonin receptor ligands with antidepressive-like, anxiolytic and antiobesity action in vivo. Molecules 2018, 2529, 2529. [Google Scholar] [CrossRef]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A novel design of artificial membrane for improving the PAMPA model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef]
- Cruciani, G.; Carosati, E.; De Boeck, B.; Ethirajulu, K.; Mackie, C.; Howe, T.; Vianello, R. MetaSite: understanding metabolism in human cytochromes from the perspective of the chemist. J. Med. Chem. 2005, 48, 6970–6979. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.R.; Beer, B.; Clody, D.E. A simple and reliable conflict procedure for testing anti-anxiety agents. Psychopharmacologia 1971, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Eddy, N.B.; Leimbach, D. Synthetic analgesics. II. Dithienylbutenyl-and dithienylbutylamines. J. Pharmacol. Exp. Ther. 1953, 107, 385–393. [Google Scholar] [PubMed]
- Zajdel, P.; Kos, T.; Marciniec, K.; Satała, G.; Canale, V.; Kamiński, K.; Hołuj, M.; Lenda, T.; Koralewski, R.; Bednarski, M.; et al. Novel multi-target azinesulfonamides of cyclic amine derivatives as potential antipsychotics with pro-social and pro-cognitive effects. Eur. J. Med. Chem. 2018, 145, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Kotańska, M.; Śniecikowska, J.; Jastrzębska-Więsek, M.; Kołaczkowski, M.; Pytka, K. Metabolic and Cardiovascular Benefits and Risks of EMD386088-A 5-HT6 Receptor Partial Agonist and Dopamine Transporter Inhibitor. Front. Neurosci. 2017, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Kotańska, M.; Lustyk, K.; Bucki, A.; Marcinkowska, M.; Śniecikowska, J.; Kołaczkowski, M. Idalopirdine, a selective 5-HT 6 receptor antagonist, reduces food intake and body weight in a model of excessive eating. Metab. Brain Dis. 2018, 33, 733–740. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | Ki (nM) | ||||
|---|---|---|---|---|---|
| 5-HT6 [3H]-LSD | D2L [3H]- Raclopride | 5-HT1A [3H]-8-OH-DPAT | 5-HT2A [3H]- Ketanserin | 5-HT7 [3H]-5-CT | |
| 1 | 28 a | 838 a | 4954 a | 272 | 8710 a |
| 2 | 29 a | 1192 a | 10,220 a | 329 | 16,160 a |
| 3 | 22 a | 1946 a | 9453 a | 1757 | 16,790 a |
| 4 | 11 a | 1094 | 12,530 | 430 | 11,950 |
| Comp. | 1Pe (10−6 cm/s) ± SD |
|---|---|
| CFN | 15.1 ± 0.4 |
| NFX | 0.56 ± 0.1 |
| 1 | 23.6 ± 2.14 |
| 2 | 19.9 ± 2.68 |
| 3 | 17.9 ± 5.76 |
| 4 | 12.3 ± 1.98 |
| Comp. | t1/2 (min) | 1Clint (mL·min−1·kg−1) |
|---|---|---|
| 1 | 83 | 7.56 |
| 2 | 158 | 3.96 |
| 3 | 267 | 2.34 |
| 4 | 99 | 6.30 |
| Substrate | Molecular Mass (m/z) | Amount of Metabolites | Molecular Mass of the Metabolite (m/z) | Metabolic Pathway |
|---|---|---|---|---|
| 1 | 319.13 | 2 | 335.08 (M1) | hydroxylation |
| 305.11 (M2) | demethylation | |||
| 2 | 299.19 | 2 | 315.14 (M1) | hydroxylation |
| 285.10 (M2) | demethylation | |||
| 3 | 324.18 | 3 | 340.33 (M1) | hydroxylation |
| 310.18 (M2) | demethylation | |||
| 340.13 (M3) | hydroxylation | |||
| 4 | 357.21 | 4 | 342.19 (M1) | demethylation |
| 373.23 (M2) | hydroxylation | |||
| 373.23 (M3) | hydroxylation | |||
| 373.23 (M4) | hydroxylation |
| Treatment | Dose (mg/kg) | Hot Plate Test Time of Reaction (s) | Water Consumption (g/5 min) |
|---|---|---|---|
| Vehicle | 0 | 12.4 ± 1.4 | 4.3 ± 0.4 |
| 3 | 1 | 10.8 ± 1.2 | 4.4 ± 0.1 |
| F(1,12) = 0.6934; ns | F(1,14) = 0.0635; ns | ||
| 4 | 0.3 | 13.8 ± 1.5 | 4.6 ± 0.2 |
| F(1,13) = 0.4406; ns | F(1,14) = 0.2834; ns |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Marć, M.A.; Satała, G.; Wilczyńska, D.; Kotańska, M.; Więcek, M.; Kamińska, K.; et al. The 1,3,5-Triazine Derivatives as Innovative Chemical Family of 5-HT6 Serotonin Receptor Agents with Therapeutic Perspectives for Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 3420. https://doi.org/10.3390/ijms20143420
Latacz G, Lubelska A, Jastrzębska-Więsek M, Partyka A, Marć MA, Satała G, Wilczyńska D, Kotańska M, Więcek M, Kamińska K, et al. The 1,3,5-Triazine Derivatives as Innovative Chemical Family of 5-HT6 Serotonin Receptor Agents with Therapeutic Perspectives for Cognitive Impairment. International Journal of Molecular Sciences. 2019; 20(14):3420. https://doi.org/10.3390/ijms20143420
Chicago/Turabian StyleLatacz, Gniewomir, Annamaria Lubelska, Magdalena Jastrzębska-Więsek, Anna Partyka, Małgorzata Anna Marć, Grzegorz Satała, Daria Wilczyńska, Magdalena Kotańska, Małgorzata Więcek, Katarzyna Kamińska, and et al. 2019. "The 1,3,5-Triazine Derivatives as Innovative Chemical Family of 5-HT6 Serotonin Receptor Agents with Therapeutic Perspectives for Cognitive Impairment" International Journal of Molecular Sciences 20, no. 14: 3420. https://doi.org/10.3390/ijms20143420