Molecular and Clinical Opposite Findings in 11p15.5 Associated Imprinting Disorders: Characterization of Basic Mechanisms to Improve Clinical Management


Abstract
:1. Introduction
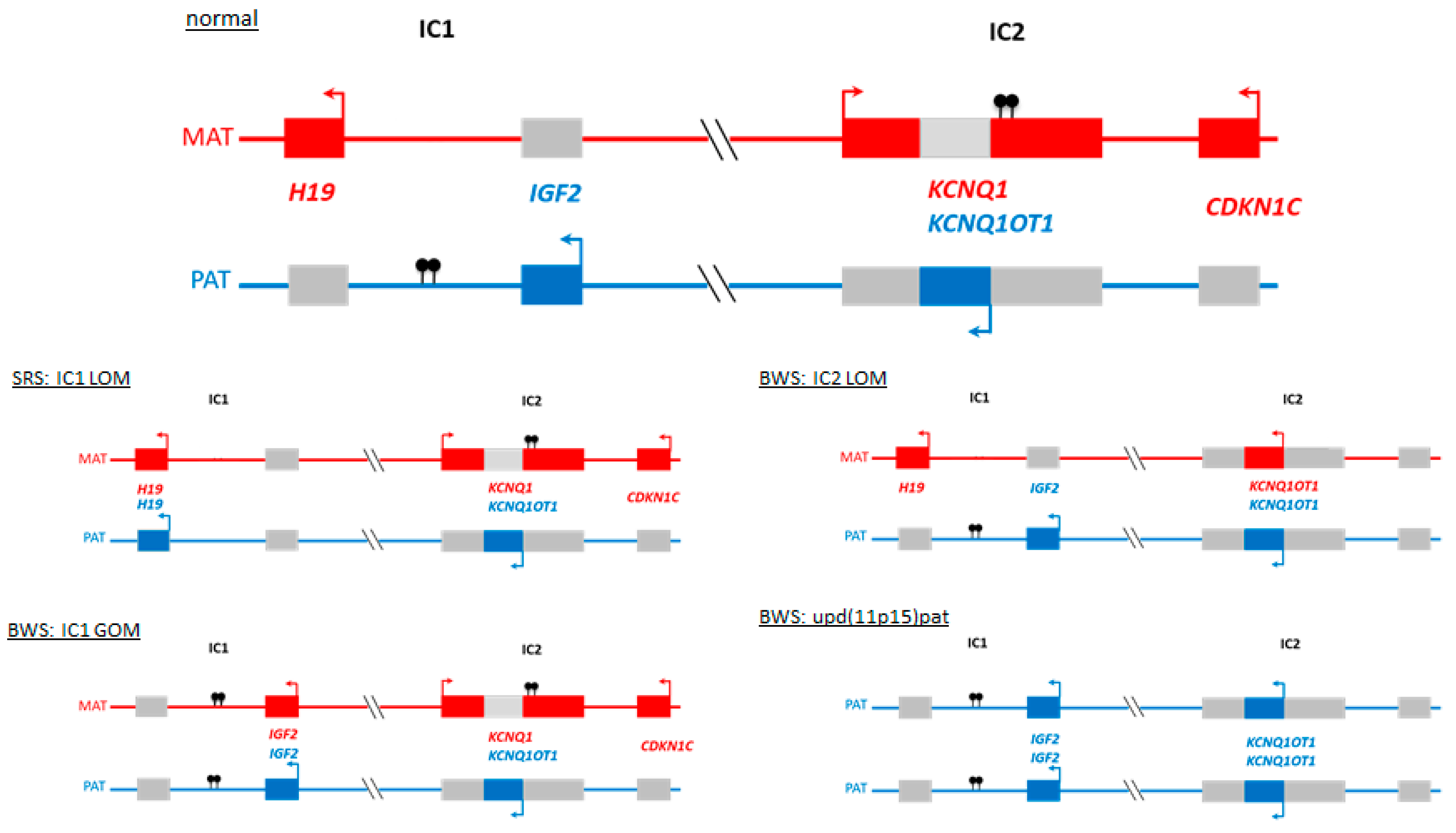
2. Disturbed Imprinting is The Major Molecular Change in BWS and SRS Phenotypes
3. Genomic Alterations Resulting in SRS and BWS Phenotypes
3.1. Genomic Variants in 11p15.5 and Other Imprinted Regions Affecting the Function or the Expression of the Imprinted Genes in This Region
3.2. Variants in the Mothers of MLID Patients Causing Disturbed Imprinting Marks in Their Offspring
3.3. Variants Affecting Non-Imprinted Regions or Genes Resulting in Phenotypes Similar to SRS or BWS, i.e., Differential Diagnoses
4. Imprinting Alterations Contribute to the Understanding of Its Underlying Mechanisms and Its Functional Relevance
4.1. The Imprinted Genes Network
4.2. Multilocus Imprinting Disturbances
4.3. Growth Disturbances As a Functional Result of Altered Imprinting
5. (Epi)Genotype–Phenotype Correlations in SRS and BWS
6. The Molecular Diagnosis Is the Prerequisite for Precise Clinical Management and Counseling
7. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BWS | Beckwith–Wiedemann syndrome |
| CNV | copy number variation |
| DMR | differentially methylated region |
| GOM | gain of methylation |
| IC | imprinting center |
| IGN | imprinted genes network |
| LOM | loss of methylation |
| MLID | multilocus imprinting disturbances |
| MLPA | multiplex ligation-dependent probe amplification |
| MS | methylation-specific |
| ncRNA | non-coding RNA |
| NGS | next generation sequencing |
| PHP1b | Pseudohypoparathyreoidism 1 b |
| SNV | single nucleotide variant |
| SRS | Silver–Russell syndrome |
| TF | transcription factor |
| TGS | third generation sequencing |
| TNDM | transient neonatal diabetes |
| UPD | Uniparental Disomy |
| WES | whole exome sequencing |
| WGS | whole genome sequencing |
References
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.M.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Yakoreva, M.; Kahre, T.; Zordania, R.; Reinson, K.; Teek, R.; Tillmann, V.; Peet, A.; Oiglane-Shlik, E.; Pajusalu, S.; Murumets, U.; et al. A retrospective analysis of the prevalence of imprinting disorders in Estonia from 1998 to 2016. Eur. J. Hum. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019, 20, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Soellner, L.; Begemann, M.; Mackay, D.J.G.; Gronskov, K.; Tumer, Z.; Maher, E.R.; Temple, I.K.; Monk, D.; Riccio, A.; Linglart, A.; et al. Recent Advances in Imprinting Disorders. Clin. Genet. 2017, 91, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Horsthemke, B. Mechanisms of imprint dysregulation. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154C, 321–328. [Google Scholar] [CrossRef]
- Kagami, M.; Mizuno, S.; Matsubara, K.; Nakabayashi, K.; Sano, S.; Fuke, T.; Fukami, M.; Ogata, T. Epimutations of the IG-DMR and the MEG3-DMR at the 14q32.2 imprinted region in two patients with Silver—Russell Syndrome-compatible phenotype. Eur. J. Hum. Genet. 2014, 23, 1062–1067. [Google Scholar] [CrossRef]
- Younis, S.; Schonke, M.; Massart, J.; Hjortebjerg, R.; Sundstrom, E.; Gustafson, U.; Bjornholm, M.; Krook, A.; Frystyk, J.; Zierath, J.R.; et al. The ZBED6-IGF2 axis has a major effect on growth of skeletal muscle and internal organs in placental mammals. Proc. Natl. Acad. Sci. USA 2018, 115, E2048–E2057. [Google Scholar] [CrossRef]
- Zhang, P.; Liegeois, N.J.; Wong, C.; Finegold, M.; Hou, H.; Thompson, J.C.; Silverman, A.; Harper, J.W.; DePinho, R.A.; Elledge, S.J. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997, 387, 151–158. [Google Scholar] [CrossRef]
- Neuheuser, L.; Meyer, R.; Begemann, M.; Elbracht, M.; Eggermann, T. Next generation sequencing and imprinting disorders: Current applications and future perspectives: Lessons from Silver-Russell syndrome. Mol. Cell. Probes 2019, 44, 1–7. [Google Scholar] [CrossRef]
- Mackay, D.J.G.; Bliek, J.; Lombardi, M.P.; Russo, S.; Calzari, L.; Guzzetti, S.; Izzi, C.; Selicorni, A.; Melis, D.; Temple, K.; et al. Discrepant molecular and clinical diagnoses in Beckwith-Wiedemann and Silver-Russell syndromes. Genet. Res. (Camb.) 2019, 101, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abi Habib, W.; Azzi, S.; Brioude, F.; Steunou, V.; Thibaud, N.; Das Neves, C.; Le Jule, M.; Chantot-Bastaraud, S.; Keren, B.; Rossi, S.; et al. Extensive investigation of the IGF2/H19 imprinting control region reveals novel OCT4/SOX2 binding site defects associated with specific methylation patterns in Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 2014, 23, 5763–5773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, F.M.; Sparago, A.; Freschi, A.; Hill-Harfe, K.; Maas, S.M.; Frints, S.G.M.; Alders, M.; Pignata, L.; Franzese, M.; Angelini, C.; et al. Transcription alterations of KCNQ1 associated with imprinted methylation defects in the Beckwith-Wiedemann locus. Genet. Med. 2019, 21, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Heide, S.; Chantot-Bastaraud, S.; Keren, B.; Harbison, M.D.; Azzi, S.; Rossignol, S.; Michot, C.; Lys, M.L.; Demeer, B.; Heinrichs, C.; et al. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J. Med. Genet. 2018, 55, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Kraft, F.; Wesseler, K.; Begemann, M.; Kurth, I.; Elbracht, M.; Eggermann, T. Novel familial distal imprinting centre 1 (11p15.5) deletion provides further insights in imprinting regulation. Clin. Epigenetics 2019, 11, 30. [Google Scholar] [CrossRef]
- Cytrynbaum, C.; Chong, K.; Hannig, V.; Choufani, S.; Shuman, C.; Steele, L.; Morgan, T.; Scherer, S.W.; Stavropoulos, D.J.; Basran, R.K.; et al. Genomic imbalance in the centromeric 11p15 imprinting center in three families: Further evidence of a role for IC2 as a cause of Russell-Silver syndrome. Am. J. Med. Genet. Part A 2016, 170, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Oliver-Petit, I.; Blaise, A.; Praz, F.; Rossignol, S.; le Jule, M.; Thibaud, N.; Faussat, A.-M.; Tauber, M.; le Bouc, Y.; et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J. Med. Genet. 2013, 50, 823–830. [Google Scholar] [CrossRef]
- Habib, W.A.; Brioude, F.; Edouard, T.; Bennett, J.T.; Lienhardt-Roussie, A.; Tixier, F.; Salem, J.; Yuen, T.; Azzi, S.; le Bouc, Y.; et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 2018, 20, 250–258. [Google Scholar] [CrossRef]
- Begemann, M.; Zirn, B.; Santen, G.; Wirthgen, E.; Soellner, L.; Buttel, H.-M.; Schweizer, R.; van Workum, W.; Binder, G.; Eggermann, T. Paternally Inherited IGF2 Mutation and Growth Restriction. N. Engl. J. Med. 2015, 373, 349–356. [Google Scholar] [CrossRef]
- Nakamura, A.; Muroya, K.; Ogata-Kawata, H.; Nakabayashi, K.; Matsubara, K.; Ogata, T.; Kurosawa, K.; Fukami, M.; Kagami, M. A case of paternal uniparental isodisomy for chromosome 7 associated with overgrowth. J. Med. Genet. 2018, 55, 567–570. [Google Scholar] [CrossRef]
- Hannula-Jouppi, K.; Muurinen, M.; Lipsanen-Nyman, M.; Reinius, L.E.; Ezer, S.; Greco, D.; Kere, J. Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics 2014, 9, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Tumer, Z.; Lopez-Hernandez, J.A.; Netchine, I.; Elbracht, M.; Gronskov, K.; Gede, L.B.; Sachwitz, J.; den Dunnen, J.T.; Eggermann, T. Structural and sequence variants in patients with Silver-Russell syndrome or similar features-Curation of a disease database. Hum. Mutat. 2018, 39, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, T.; Heilsberg, A.-K.; Bens, S.; Siebert, R.; Beygo, J.; Buiting, K.; Begemann, M.; Soellner, L. Additional molecular findings in 11p15-associated imprinting disorders: An urgent need for multi-locus testing. J. Mol. Med. (Berl.) 2014, 92, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Mackay, D.J.G.; Hahnemann, J.M.D.; Boonen, S.E.; Poerksen, S.; Bunyan, D.J.; White, H.E.; Durston, V.J.; Thomas, N.S.; Robinson, D.O.; Shield, J.P.H.; et al. Epimutation of the TNDM locus and the Beckwith-Wiedemann syndrome centromeric locus in individuals with transient neonatal diabetes mellitus. Hum. Genet. 2006, 119, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Kagami, M. Kagami-Ogata syndrome: A clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J. Hum. Genet. 2016, 61, 87–94. [Google Scholar] [CrossRef]
- Kagami, M.; Nagasaki, K.; Kosaki, R.; Horikawa, R.; Naiki, Y.; Saitoh, S.; Tajima, T.; Yorifuji, T.; Numakura, C.; Mizuno, S.; et al. Temple syndrome: Comprehensive molecular and clinical findings in 32 Japanese patients. Genet. Med. 2017, 19, 1356–1366. [Google Scholar] [CrossRef]
- Baujat, G.; Rio, M.; Rossignol, S.; Sanlaville, D.; Lyonnet, S.; le Merrer, M.; Munnich, A.; Gicquel, C.; Colleaux, L.; Cormier-Daire, V. Clinical and molecular overlap in overgrowth syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2005, 137C, 4–11. [Google Scholar] [CrossRef]
- Sachwitz, J.; Meyer, R.; Fekete, G.; Spranger, S.; Matuleviciene, A.; Kucinskas, V.; Bach, A.; Luczay, A.; Bruchle, N.O.; Eggermann, K.; et al. NSD1 duplication in Silver-Russell syndrome (SRS): Molecular karyotyping in patients with SRS features. Clin. Genet. 2017, 91, 73–78. [Google Scholar] [CrossRef]
- Keppler-Noreuil, K.M.; Parker, V.E.R.; Darling, T.N.; Martinez-Agosto, J.A. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2016, 172, 402–421. [Google Scholar] [Green Version]
- Dyment, D.A.; Smith, A.C.; Alcantara, D.; Schwartzentruber, J.A.; Basel-Vanagaite, L.; Curry, C.J.; Temple, I.K.; Reardon, W.; Mansour, S.; Haq, M.R.; et al. Mutations in PIK3R1 cause SHORT syndrome. Am. J. Hum. Genet. 2013, 93, 158–166. [Google Scholar] [CrossRef]
- Priolo, M.; Sparago, A.; Mammi, C.; Cerrato, F.; Lagana, C.; Riccio, A. MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur. J. Hum. Genet. 2008, 16, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Delgado, M.; Riccio, A.; Eggermann, T.; Maher, E.R.; Lapunzina, P.; Mackay, D.; Monk, D. Causes and Consequences of Multi-Locus Imprinting Disturbances in Humans. Trends Genet. 2016, 32, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzi, S.; Blaise, A.; Steunou, V.; Harbison, M.D.; Salem, J.; Brioude, F.; Rossignol, S.; Habib, W.A.; Thibaud, N.; Neves, C.D.; et al. Complex tissue-specific epigenotypes in Russell-Silver Syndrome associated with 11p15 ICR1 hypomethylation. Hum. Mutat. 2014, 35, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Beygo, J.; Burger, J.; Strom, T.M.; Kaya, S.; Buiting, K. Disruption of KCNQ1 prevents methylation of the ICR2 and supports the hypothesis that its transcription is necessary for imprint establishment. Eur. J. Hum. Genet. 2019, 27, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Beygo, J.; Citro, V.; Sparago, A.; de Crescenzo, A.; Cerrato, F.; Heitmann, M.; Rademacher, K.; Guala, A.; Enklaar, T.; Anichini, C.; et al. The molecular function and clinical phenotype of partial deletions of the IGF2/H19 imprinting control region depends on the spatial arrangement of the remaining CTCF-binding sites. Hum. Mol. Genet. 2013, 22, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Sparago, A.; Cerrato, F.; Riccio, A. Is ZFP57 binding to H19/IGF2:IG-DMR affected in Silver-Russell syndrome? Clin. Epigenet. 2018, 10, 23. [Google Scholar] [CrossRef]
- Monk, D.; Sanchez-Delgado, M.; Fisher, R. NLRPs, the subcortical maternal complex and genomic imprinting. Reproduction 2017, 154, R161–R170. [Google Scholar] [CrossRef] [Green Version]
- van den Veyver, I.B.; Al-Hussaini, T.K. Biparental hydatidiform moles: A maternal effect mutation affecting imprinting in the offspring. Hum. Reprod. Update 2006, 12, 233–242. [Google Scholar] [CrossRef]
- Begemann, M.; Rezwan, F.I.; Beygo, J.; Docherty, L.E.; Kolarova, J.; Schroeder, C.; Buiting, K.; Chokkalingam, K.; Degenhardt, F.; Wakeling, E.L.; et al. Maternal variants in NLRP and other maternal effect proteins are associated with multilocus imprinting disturbance in offspring. J. Med. Genet. 2018, 55, 497–504. [Google Scholar] [CrossRef]
- Soellner, L.; Kraft, F.; Sauer, S.; Begemann, M.; Kurth, I.; Elbracht, M.; Eggermann, T. Search for cis-acting factors and maternal effect variants in Silver-Russell patients with ICR1 hypomethylation and their mothers. Eur. J. Hum. Genet. 2019, 27, 42–48. [Google Scholar] [CrossRef]
- Patten, M.M.; Cowley, M.; Oakey, R.J.; Feil, R. Regulatory links between imprinted genes: Evolutionary predictions and consequences. Proc. R. Soc. B Biol. Sci. 2016, 283. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, Y.; Sagi, I.; Yanuka, O.; Eiges, R.; Benvenisty, N. The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome. Nat. Genet. 2014, 46, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Varrault, A.; Gueydan, C.; Delalbre, A.; Bellmann, A.; Houssami, S.; Aknin, C.; Severac, D.; Chotard, L.; Kahli, M.; le Digarcher, A.; et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 2006, 11, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Platas, I.; Martin-Trujillo, A.; Petazzi, P.; Guillaumet-Adkins, A.; Esteller, M.; Monk, D. Altered expression of the imprinted transcription factor PLAGL1 deregulates a network of genes in the human IUGR placenta. Hum. Mol. Genet. 2014, 23, 6275–6285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Cullen, B.R. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 2007, 13, 313–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Charalambous, M.; Zarse, K.; Mori, M.A.; Kleinridders, A.; Ristow, M.; Ferguson-Smith, A.C.; Kahn, C.R. Insulin and insulin-like growth factor 1 receptors are required for normal expression of imprinted genes. Proc. Natl. Acad. Sci. USA 2014, 111, 14512–14517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macneil, L.T.; Walhout, A.J.M. Gene regulatory networks and the role of robustness and stochasticity in the control of gene expression. Genome Res. 2011, 21, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Mackay, D.J.G.; Callaway, J.L.A.; Marks, S.M.; White, H.E.; Acerini, C.L.; Boonen, S.E.; Dayanikli, P.; Firth, H.V.; Goodship, J.A.; Haemers, A.P.; et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 2008, 40, 949–951. [Google Scholar] [CrossRef]
- Li, X.; Ito, M.; Zhou, F.; Youngson, N.; Zuo, X.; Leder, P.; Ferguson-Smith, A.C. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev. Cell 2008, 15, 547–557. [Google Scholar] [CrossRef]
- Quenneville, S.; Verde, G.; Corsinotti, A.; Kapopoulou, A.; Jakobsson, J.; Offner, S.; Baglivo, I.; Pedone, P.V.; Grimaldi, G.; Riccio, A.; et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell 2011, 44, 361–372. [Google Scholar] [CrossRef]
- Strogantsev, R.; Krueger, F.; Yamazawa, K.; Shi, H.; Gould, P.; Goldman-Roberts, M.; McEwen, K.; Sun, B.; Pedersen, R.; Ferguson-Smith, A.C. Allele-specific binding of ZFP57 in the epigenetic regulation of imprinted and non-imprinted monoallelic expression. Genome Biol. 2015, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, S.; Djuric, U.; Mazhar, B.; Seoud, M.; Khan, R.; Kuick, R.; Bagga, R.; Kircheisen, R.; Ao, A.; Ratti, B.; et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat. Genet. 2006, 38, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.M.P.; Slim, R. Genetics Epigenetics of Recurrent Hydatidiform Moles: Basic Science Genetic Counselling. Curr. Obstet. Gynecol. Rep. 2014, 3, 55–64. [Google Scholar] [CrossRef]
- Meyer, E.; Lim, D.; Pasha, S.; Tee, L.J.; Rahman, F.; Yates, J.R.W.; Woods, C.G.; Reik, W.; Maher, E.R. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PLoS Genet. 2009, 5, e1000423. [Google Scholar] [CrossRef]
- Docherty, L.E.; Rezwan, F.I.; Poole, R.L.; Turner, C.L.S.; Kivuva, E.; Maher, E.R.; Smithson, S.F.; Hamilton-Shield, J.P.; Patalan, M.; Gizewska, M.; et al. Mutations in NLRP5 are associated with reproductive wastage multilocus imprinting disorders in humans. Nat. Commun. 2015, 6, 8086. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Wang, L.; Yu, F.; Gao, H.; Lei, T.; Li, P.; Liu, P.; Zheng, X.; Hu, X.; Chen, Y.; et al. Imp2 regulates GBM progression by activating IGF2/PI3K/Akt pathway. Cancer Biol. Ther. 2015, 16, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Avila, M.; Dyment, D.A.; Sagen, J.V.; St-Onge, J.; Moog, U.; Chung, B.H.Y.; Mo, S.; Mansour, S.; Albanese, A.; Garcia, S.; et al. Clinical reappraisal of SHORT syndrome with PIK3R1 mutations: Toward recommendation for molecular testing management. Clin. Genet. 2016, 89, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, J.; Yulug, I.G.; Egan, S.E.; Fisher, E.M. The gene that encodes the phosphatidylinositol-3 kinase regulatory subunit (p85 alpha) maps to chromosome 13 in the mouse. Genomics 1994, 24, 400–402. [Google Scholar] [CrossRef]
- Vanaja, K.G.; Timp, W.; Feinberg, A.P.; Levchenko, A. A Loss of Epigenetic Control Can Promote Cell Death through Reversing the Balance of Pathways in a Signaling Network. Mol. Cell 2018, 72, 60–70. [Google Scholar] [CrossRef]
- Loh, A.H.P.; Brennan, R.C.; Lang, W.H.; Hickey, R.J.; Malkas, L.H.; Sandoval, J.A. Dissecting the PI3K Signaling Axis in Pediatric Solid Tumors: Novel Targets for Clinical Integration. Front. Oncol. 2013, 3, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, R.R.; Vanhaesebroeck, B.; Semple, R.K. Cancer-Associated PIK3CA Mutations in Overgrowth Disorders. Trends Mol. Med. 2018, 24, 856–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, G.; Liebl, M.; Woelfle, J.; Eggermann, T.; Blumenstock, G.; Schweizer, R. Adult height epigenotype in children with Silver-Russell syndrome treated with GH. Horm. Res. Paediatr. 2013, 80, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, E.L.; Amero, S.A.; Alders, M.; Bliek, J.; Forsythe, E.; Kumar, S.; Lim, D.H.; MacDonald, F.; Mackay, D.J.; Maher, E.R.; et al. Epigenotype-phenotype correlations in Silver-Russell syndrome. J. Med. Genet. 2010, 47, 760–768. [Google Scholar] [CrossRef] [Green Version]
- Geoffron, S.; Habib, W.A.; Chantot-Bastaraud, S.; Dubern, B.; Steunou, V.; Azzi, S.; Afenjar, A.; Busa, T.; Canton, A.P.; Chalouhi, C.; et al. Chromosome 14q32.2 Imprinted Region Disruption as an Alternative Molecular Diagnosis of Silver-Russell Syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 2436–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeets, C.C.J.; Zandwijken, G.R.J.; Renes, J.S.; Hokken-Koelega, A.C.S. Long-Term Results of GH Treatment in Silver-Russell Syndrome (SRS): Do They Benefit the Same as Non-SRS Short-SGA? J. Clin. Endocrinol. Metab. 2016, 101, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Smeets, C.C.J.; Renes, J.S.; van der Steen, M.; Hokken-Koelega, A.C.S. Metabolic Health Long-Term Safety of Growth Hormone Treatment in Silver-Russell Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Ballard, L.M.; Jenkinson, E.; Byrne, C.D.; Child, J.C.; Davies, J.H.; Inskip, H.; Lokulo-Sodipe, O.; Mackay, D.J.G.; Wakeling, E.L.; Temple, I.K.; et al. Lived experience of Silver-Russell syndrome: Implications for management during childhood into adulthood. Arch. Dis. Child. 2019, 104, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.; Giaccardi, M.; Capra, V.; Napoli, F.; Cangemi, G.; Notarnicola, S.; Guzzetti, S.; Russo, S.; Maghnie, M.; di Iorgi, N. Clinical Manifestations Metabolic Outcomes of Seven Adults With Silver-Russell Syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 2225–2233. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Ward, A.J.; Chun, S.; Bennett, C.F.; Beaudet, A.L.; Rigo, F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 2015, 518, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Pulst, S.M. Oligonucleotide therapeutics in neurodegenerative diseases. RNA Biol. 2018, 15, 707–714. [Google Scholar] [CrossRef]
- Tan, W.-H.; Bird, L.M. Angelman syndrome: Current emerging therapies in 2016. Am. J. Med. Genet. Part C Semin. Med. Genet. 2016, 172, 384–401. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wang, S.E.; Jiang, Y.-H. Epigenetic therapy of Prader-Willi syndrome. Transl. Res. 2019, 208, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Min, H.Y.; Lee, S.C.; Woo, J.K.; Jung, H.J.; Park, K.H.; Jeong, H.M.; Hyun, S.Y.; Cho, J.; Lee, W.; Park, J.E.; et al. Essential Role of DNA Methyltransferase 1-mediated Transcription of Insulin-like Growth Factor 2 in Resistance to Histone Deacetylase Inhibitors. Clin. Cancer Res. 2017, 23, 1299–1311. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Genomic Region | Alteration | BWS/Overgrowth | SRS/Growth Retardation | Reference |
|---|---|---|---|---|
| Alterations within imprinted regions | ||||
| 11p15.5: IC1 | GOM | 5%–10% (in up to 20% SNPs/SNVs in transcription factors binding sites in the IC1 can be detected) | NR | [1,12] |
| LOM | single cases * | 40%–50% | [2,11] | |
| 11p15.5: IC2 | LOM | 50% (in some patients disturbance of the KCNQ1 transcript) | Single cases * | [1,11,13] |
| 11p15.5: IC1 and IC2 | Duplication | Maternal: <3% | Paternal: <1% | [14] |
| UPD | upd(11)pat: 20% | upd(11)mat: 1 case | [1] | |
| 11p15.5: IC1 OR IC2 | Small CNVs ** | Single cases | Single cases | [15,16] |
| 11p15.5 | CDKN1C | LoF: 5% of sporadic, 40% of familial cases | GoF: single cases | [1,17] |
| IGF2 | NR | Familial and rare sporadic cases | [18,19] | |
| Chromosome 7 | UPD | upd(7)pat: 1 case | upd(7)mat: 7%–10% | [2,20] |
| Segmental UPD7q | upd(7q)mat: single patients | [21] | ||
| CNVs | Dup 7p13: GRB10 Del 7q32: MEST | [22] | ||
| Chromosome 6 | UPD | upd(6)pat (TNDM) | upd(6)mat | [23,24] |
| Chromosome 14q32 | Epimutation | MEG3 GOM (KOS14) | MEG3 LOM (TS14) | [25,26] |
| CNVs ** | Del14q32 (TS14) [26] | [26] | ||
| UPD | upd(14)mat | [26] | ||
| Several imprinted regions | MLID *** | 30% of IC2 LOM | 15%–38% of IC1 LOM | [1,2] |
| Genomic variants in non-imprinted genes *** | ||||
| NSD1 | Del | Dup | [27,28] | |
| PIK3 function | PIK3CA overgrowth | PIK3R1 SHORT | [29,30] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wesseler, K.; Kraft, F.; Eggermann, T. Molecular and Clinical Opposite Findings in 11p15.5 Associated Imprinting Disorders: Characterization of Basic Mechanisms to Improve Clinical Management. Int. J. Mol. Sci. 2019, 20, 4219. https://doi.org/10.3390/ijms20174219
Wesseler K, Kraft F, Eggermann T. Molecular and Clinical Opposite Findings in 11p15.5 Associated Imprinting Disorders: Characterization of Basic Mechanisms to Improve Clinical Management. International Journal of Molecular Sciences. 2019; 20(17):4219. https://doi.org/10.3390/ijms20174219
Chicago/Turabian StyleWesseler, Katharina, Florian Kraft, and Thomas Eggermann. 2019. "Molecular and Clinical Opposite Findings in 11p15.5 Associated Imprinting Disorders: Characterization of Basic Mechanisms to Improve Clinical Management" International Journal of Molecular Sciences 20, no. 17: 4219. https://doi.org/10.3390/ijms20174219