The Janus Face of Tumor Microenvironment Targeted by Immunotherapy

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Dynamic Niche of TME
2.1. The Role of ECM
2.2. The Contribution of Stromal Cells
2.3. Development of the Pre-Metastatic Niche (PMN)
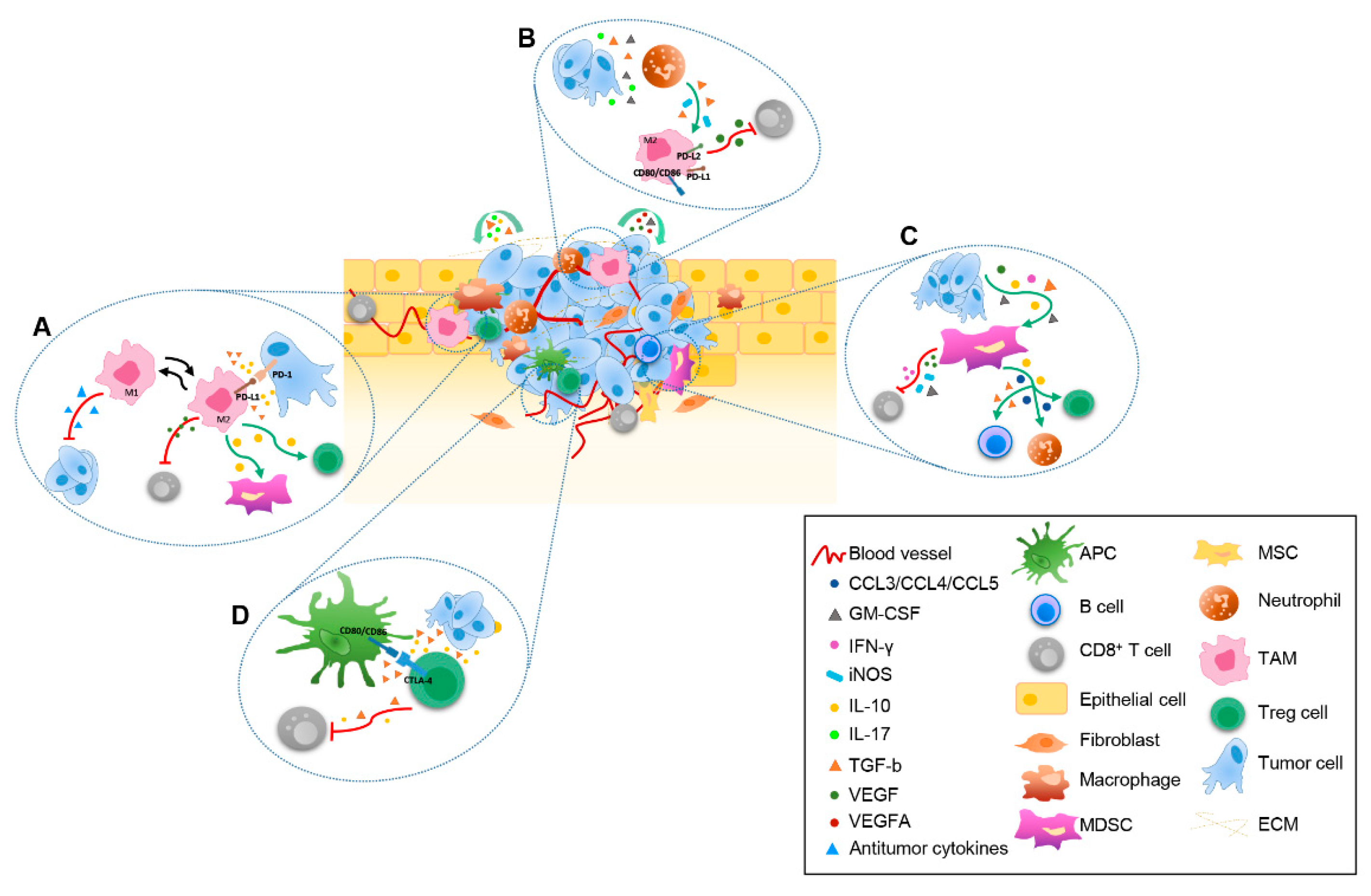
3. The Immunosuppressive Landscape of TME
3.1. TAMs as Major Drivers of Immunosuppressive TME
3.2. The Role of MDSCs
3.3. Tregs-Mediated Immunosuppression of TME
3.4. The Immunosuppressive Plasticity of DCs
3.5. The Role of Neutrophils
3.6. CD8+ T Cells as the Main Players of Antitumor Response
4. The Strength of Targeting TME with Immunotherapy
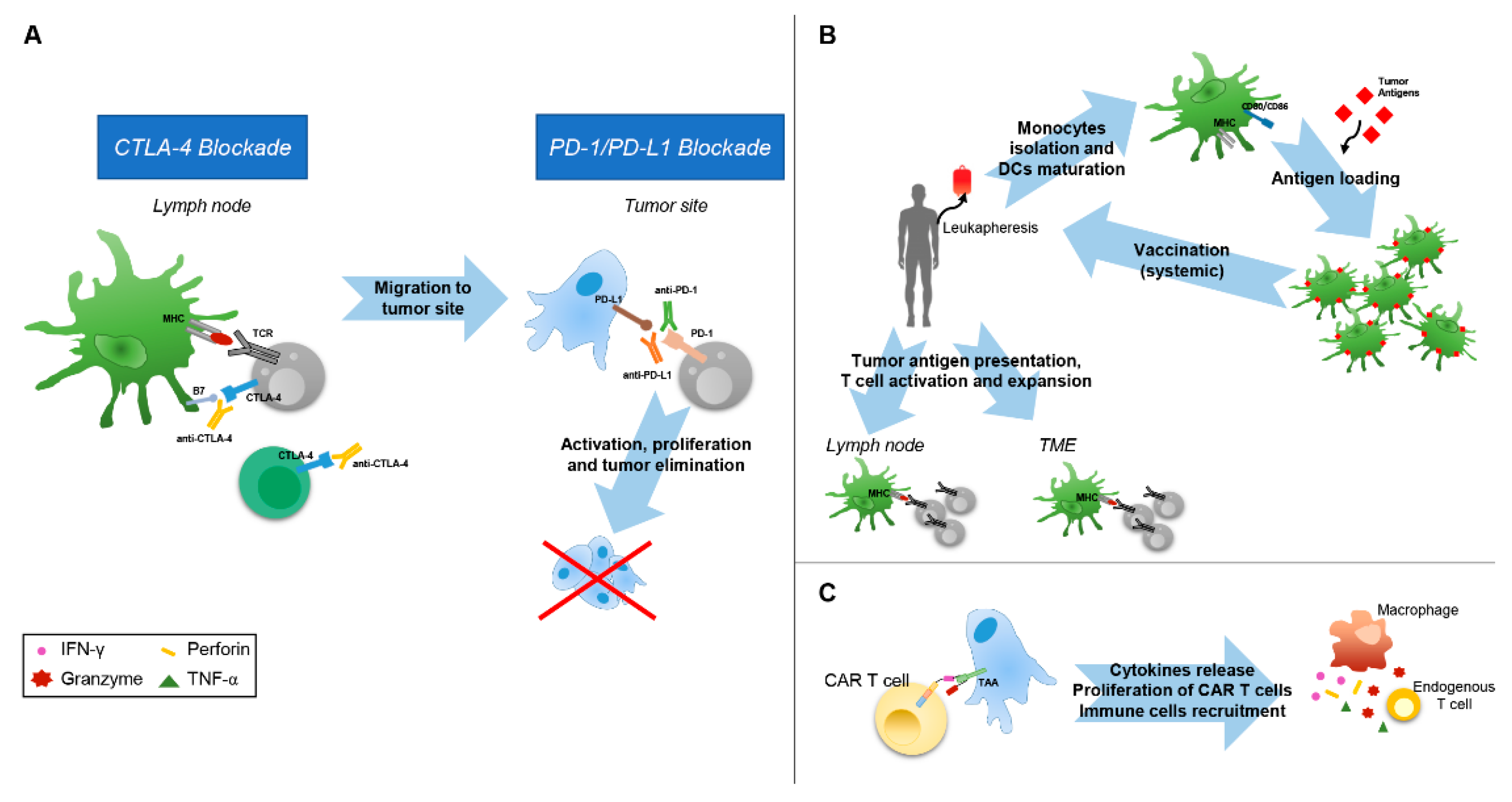
4.1. Immune-Checkpoint Inhibitors Therapies
4.2. Cancer Vaccines in Combination with ICIs and Other Antitumor Therapies
4.3. CAR T cells Therapies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | antigen presenting cell |
| Arg-1 | arginine-1 |
| CAA | cancer-associated adipocytes |
| CAF | cancer-associated fibroblast |
| CAR | chimeric antigen receptor |
| cDC | conventional DC |
| CSC | cancer stem cell |
| CTLA-4 | cytotoxic T lymphocyte antigen 4 |
| DC | dendritic cell |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EMT | epitelial-mesenchymal transition |
| eTreg | effector/activated T reg |
| EV | extracellular vesicle |
| FGF | fibroblast growth factor |
| GITR | glucocorticoid-induced TNFR-related protein |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| G-MDSC | granulocitic myeloid-derived suppressor cell |
| HER2 | human epidermal growth factor receptor 2 |
| ICI | immune checkpoint inhibitor |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| IGF | insulin-like growth factor |
| IL | interleukin |
| IFN | interferon |
| MDSC | myeloid-derived suppressor cell |
| M-MDSC | monocytic-myeloid-derived suppressor cell |
| MMP | matrix metalloproteinase |
| mo-DC | monocytes |
| mRCC | metastatic renal cell carcinoma |
| MSC | mesenchymal stem cells |
| NK | natural killer |
| NO | nitric oxide |
| NSCLC | non-small-cell lung cancer |
| ORR | overall response rate |
| OS | overall survival |
| PD-1 | programmed cell death 1 |
| pDC | plasmacytoid DC |
| PDGF-β | platelet-derived growth factor-beta |
| PD-L1 | programmed cell death ligand 1 |
| PGE-2 | prostaglandin E-2 |
| PMN | pre-metastatic niche |
| PMN-MDSC | polymorphonuclear MDSC |
| ROS | reactive oxygen species |
| RR | response rate |
| SR | survival rate |
| TAA | tumor associated antigens |
| TAM | tumor-associated macrophage |
| TCR | T cell receptor |
| TGF-β | transforming growth factor-beta |
| TIL | tumor-infiltrating lymphocytes |
| TME | tumor microenvironment |
| TNF-α | tumor necrosis factor-alfa |
| Treg | regulatory T cell |
| VEGFA | vascular endothelial growth factor A |
References
- Schneider, G.; Schmidt-Supprian, M.; Rad, R.; Saur, D. Tissue-specific tumorigenesis: Context matters. Nat. Rev. Cancer 2017, 17, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.; Fernandes, A. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Hiroshima, Y.; Matsuyama, R.; Homma, Y.; Hoffman, R.M.; Endo, I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Gkretsi, V.; Stylianou, A.; Papageorgis, P.; Polydorou, C.; Stylianopoulos, T. Remodeling Components of the Tumor Microenvironment to Enhance Cancer Therapy. Front. Oncol. 2015, 5, 214. [Google Scholar] [CrossRef]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Mason, B.N.; Starchenko, A.; Williams, R.M.; Bonassar, L.J.; Reinhart-King, C.A. Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothelial cell behavior. Acta Biomater. 2013, 9, 4635–4644. [Google Scholar] [CrossRef] [PubMed]
- Maritzen, T.; Schachtner, H.; Legler, D.F. On the move: Endocytic trafficking in cell migration. Cell. Mol. Life Sci. 2015, 72, 2119–2134. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.; Han, H.; Posner, R.G.; Von Hoff, D.D. Tumor-stromal interactions in pancreatic cancer. Crit. Rev. Oncog. 2013, 18, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.J.; Gallagher, R.; Seaton, A.; Wilson, C.; Scullin, P.; Pettigrew, J.; Stratford, I.J.; Williams, K.J.; Johnston, P.G.; Waugh, D.J.J. HIF-1 and NF-kappaB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. Oncogene 2007, 26, 7333–7345. [Google Scholar] [CrossRef]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [PubMed]
- Alsibai, K.D.; Meseure, D. Significance of Tumor Microenvironment Scoring and Immune Biomarkers in Patient Stratification and Cancer Outcomes. Histopathol. Update 2018, 11. [Google Scholar]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H.; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Angiogenesis and Vascular Functions in Modulation of Obesity, Adipose Metabolism, and Insulin Sensitivity. Cell Metab. 2013, 18, 478–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Hosaka, K.; Nakamura, M.; Cao, Y. Co-option of pre-existing vascular beds in adipose tissue controls tumor growth rates and angiogenesis. Oncotarget 2016, 7, 38282–38291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells - current trends and future prospective. Biosci. Rep. 2015, 35, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2005, 113, 752–760. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Marquez-Curtis, L.A.; Wysoczynski, M.; Ratajczak, M.Z. Enhancing effect of platelet-derived microvesicles on the invasive potential of breast cancer cells. Transfusion 2006, 46, 1199–1209. [Google Scholar] [CrossRef]
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef]
- Shimizu, K.; Iyoda, T.; Okada, M.; Yamasaki, S.; Fujii, S. Immune suppression and reversal of the suppressive tumor microenvironment. Int. Immunol. 2018, 30, 445–455. [Google Scholar] [CrossRef]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.-F.; Liu, H.; Gao, R.; Zhou, M.; Ma, J.; Zhang, Y.; Zhao, J.; Chen, Y.; Zhang, T.; Huang, F.; et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J. Immunother. Cancer 2018, 6, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, Y.; Zhang, M.; Zhao, J.; Ma, X.; Huang, T.; Pang, H.; Li, J.; Song, J. Inhibition of TLR4 Signalling-Induced Inflammation Attenuates Secondary Injury after Diffuse Axonal Injury in Rats. Mediators Inflamm. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh, E.; Orangi, M.; Mohammadi, H.; Babaie, F.; Baradaran, B. Myeloid-derived suppressor cells: Important contributors to tumor progression and metastasis. J. Cell. Physiol. 2018, 233, 3024–3036. [Google Scholar] [CrossRef]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Douville, C.; Cohen, J.D.; Yen, T.-T.; Kinde, I.; Sundfelt, K.; Kjær, S.K.; Hruban, R.H.; Shih, I.-M.; et al. Evaluation of liquid from the Papanicolaou test and other liquid biopsies for the detection of endometrial and ovarian cancers. Sci. Transl. Med. 2018, 10, eaap8793. [Google Scholar] [CrossRef]
- Otvos, B.; Finke, J.; Vogelbaum, M.; Lathia, J.D. Interrogating the interactions between myeloid derived suppressor cells and cancer stem cells in glioblastoma. J. Immunother. Cancer 2013, 1, P268. [Google Scholar] [CrossRef]
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K.; et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells 2016, 34, 2026–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.Y.K.; Yuen, V.W.H.; Wong, C.C.L. Hypoxia and the metastatic niche. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE 2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Larmonier, N.; Marron, M.; Zeng, Y.; Cantrell, J.; Romanoski, A.; Sepassi, M.; Thompson, S.; Chen, X.; Andreansky, S.; Katsanis, E. Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol. Immunother. 2007, 56, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Frick, J.-S.; Grünebach, F.; Autenrieth, I.B. Immunomodulation by semi-mature dendritic cells: A novel role of Toll-like receptors and interleukin-6. Int. J. Med. Microbiol. 2010, 300, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through education: How tolerogenic dendritic cells shape immunity. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M. Tumor-altered dendritic cell function: Implications for anti-tumor immunity. Front. Immunol. 2013, 4, 192. [Google Scholar] [CrossRef]
- Mellor, A.L.; Lemos, H.; Huang, L. Indoleamine 2,3-Dioxygenase and tolerance: Where Are We Now? Front. Immunol. 2017, 8, 1360. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, T.S.; Chew, V.; Sieow, J.L.; Goh, S.; Yeong, J.P.S.; Soon, A.L.; Ricciardi-Castagnoli, P. PD-1 expression on dendritic cells suppresses CD8+T cell function and antitumor immunity. Oncoimmunology 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- De Mingo Pulido, Á.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103+ Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, S.; Herfs, M.; Delvenne, P.; Hubert, P. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: Insight into the molecular mechanisms. J. Leukoc. Biol. 2013, 93, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; De Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-associated neutrophils in cancer: Going pro. Cancers 2019, 11, 564. [Google Scholar] [CrossRef]
- Reading, J.L.; Gálvez-Cancino, F.; Swanton, C.; Lladser, A.; Peggs, K.S.; Quezada, S.A. The function and dysfunction of memory CD8 + T cells in tumor immunity. Immunol. Rev. 2018, 283, 194–212. [Google Scholar] [CrossRef]
- Schouppe, E.; Mommer, C.; Movahedi, K.; Laoui, D.; Morias, Y.; Gysemans, C.; Luyckx, A.; De Baetselier, P.; Van Ginderachter, J.A. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur. J. Immunol. 2013, 43, 2930–2942. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 34, 690. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; Mihm, M.C.; Soiffer, R.J.; Haluska, F.G.; Butler, M.; Seiden, M.V.; Davis, T.; Henry-Spires, R.; MacRae, S.; Willman, A.; et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003, 100, 4712–4717. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Camacho, L.H.; Lopez-Berestein, G.; Pavlov, D.; Bulanhagui, C.A.; Millham, R.; Comin-Anduix, B.; Reuben, J.M.; Seja, E.; Parker, C.A.; et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J. Clin. Oncol. 2005, 23, 8968–8977. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency - Supplementary Appendix. N. Engl. J. Med. 2015, 373, 1979. [Google Scholar]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus Ipilimumab in Patients with Metastatic Melanoma. Cancer Immunol. Res. 2014, 2, 923. [Google Scholar] [CrossRef]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Lawrence, D.; McDermott, D.; Zhou, J.; Rodig, S.; Hodi, F.S. VEGF Neutralization Plus CTLA-4 Blockade Alters Soluble and Cellular Factors Associated with Enhancing Lymphocyte Infiltration and Humoral Recognition in Melanoma. Cancer Immunol. Res. 2016, 4, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic cancer vaccine and combinations with antiangiogenic therapies and immune checkpoint blockade. Front. Immunol. 2019, 10, 467. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Castiello, L.; Sabatino, M.; Jin, P.; Clayberger, C.; Marincola, F.M.; Krensky, A.M.; Stroncek, D.F. Monocyte-derived DC maturation strategies and related pathways: A transcriptional view. Cancer Immunol. Immunother. 2011, 60, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; De Vries, I.J.M.; Torensma, R.; Figdor, C.G. Dendritic-cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol. 2007, 7, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Pinho, M.P.; Sundarasetty, B.S.; Bergami-Santos, P.C.; Steponavicius-Cruz, K.; Ferreira, A.K.; Stripecke, R.; Barbuto, J.A.M. Dendritic-tumor cell hybrids induce tumor-specific immune responses more effectively than the simple mixture of dendritic and tumor cells. Cytotherapy 2016, 18, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.C.; Castiello, L.; Mattei, M.; Santodonato, L.; D’Agostino, G.; Muraro, E.; Martorelli, D.; Lapenta, C.; Di Napoli, A.; Di Landro, F.; et al. Clinical and antitumor immune responses In Relapsed/Refractory Follicular Lymphoma patients after intranodal injections of IFNα-Dendritic Cells and Rituximab. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; De Vries, I.J.M.; Figdor, C.G. Dendritic cell-based immunotherapy: State of the art and beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef]
- Curran, M.A.; Glisson, B.S. New Hope for Therapeutic Cancer Vaccines in the Era of Immune Checkpoint Modulation. Annu. Rev. Med. 2018, 70, 409–424. [Google Scholar] [CrossRef]
- Karaki, S.; Anson, M.; Tran, T.; Giusti, D.; Blanc, C.; Oudard, S.; Tartour, E. Is There Still Room for Cancer Vaccines at the Era of Checkpoint Inhibitors. Vaccines 2016, 4, 37. [Google Scholar] [CrossRef]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patientswith pretreated advanced melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Feng, L.; Lee, J.J.; Tran, H.; Kim, Y.U.; et al. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients with Incurable Human Papillomavirus 16-Related Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, A.; Maus, M.V. Making CAR T cells a solid option for solid tumors. Front. Immunol. 2018, 9, 2593. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.Z.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2018, 6, 36–46. [Google Scholar] [CrossRef]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-Specific Chimeric Antigen Receptor mRNA-Engineered T Cells Induce Antitumor Activity in Solid Malignancies. Cancer Immunol. Res. 2014, 3, 217. [Google Scholar] [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin - Specific Chimeric Antibody Receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buoncervello, M.; Gabriele, L.; Toschi, E. The Janus Face of Tumor Microenvironment Targeted by Immunotherapy. Int. J. Mol. Sci. 2019, 20, 4320. https://doi.org/10.3390/ijms20174320
Buoncervello M, Gabriele L, Toschi E. The Janus Face of Tumor Microenvironment Targeted by Immunotherapy. International Journal of Molecular Sciences. 2019; 20(17):4320. https://doi.org/10.3390/ijms20174320
Chicago/Turabian StyleBuoncervello, Maria, Lucia Gabriele, and Elena Toschi. 2019. "The Janus Face of Tumor Microenvironment Targeted by Immunotherapy" International Journal of Molecular Sciences 20, no. 17: 4320. https://doi.org/10.3390/ijms20174320
APA StyleBuoncervello, M., Gabriele, L., & Toschi, E. (2019). The Janus Face of Tumor Microenvironment Targeted by Immunotherapy. International Journal of Molecular Sciences, 20(17), 4320. https://doi.org/10.3390/ijms20174320