Inhibition of Mitochondrial Complex I Impairs Release of α-Galactosidase by Jurkat Cells


Abstract
:1. Introduction
2. Results
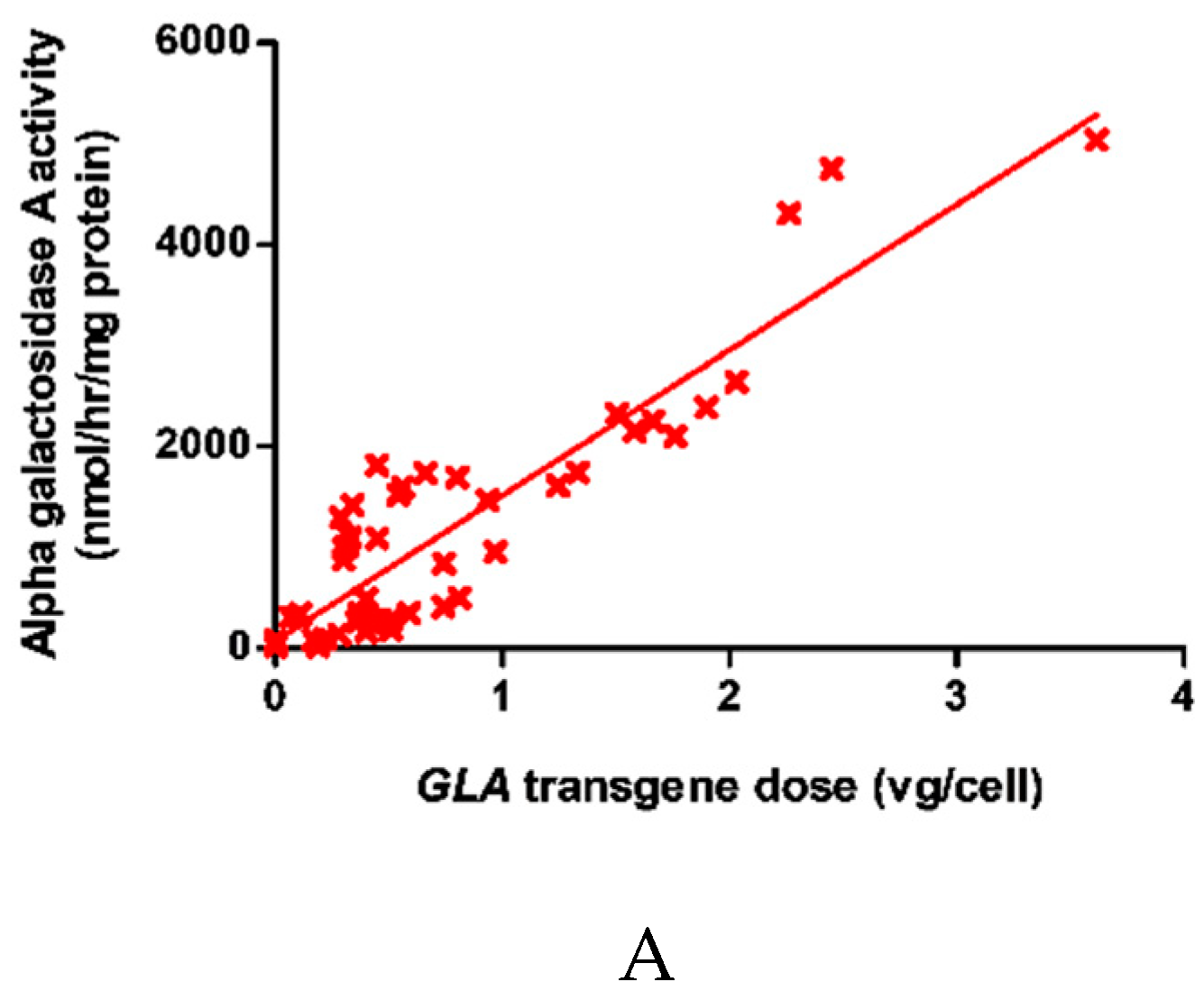
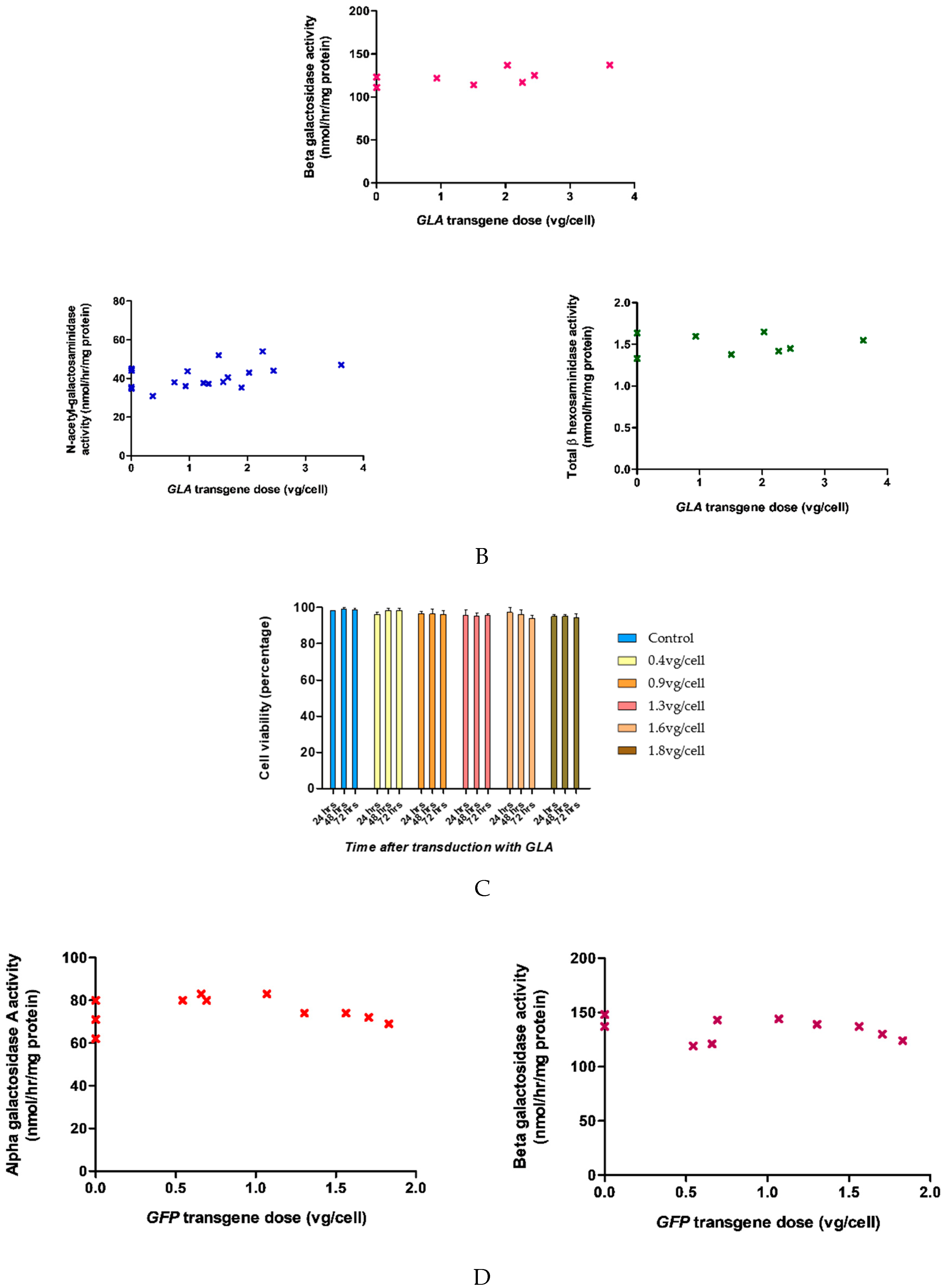
2.1. Dose-Dependent Increase in Intracellular α-galA Activity with Transgene Overexpression
2.2. α-Gal-A Activity and Michaelis Constant Increased in Cells Following Transduction with The GLA Transgene at between 0.4 vg/cell and 1.5 vg/cell
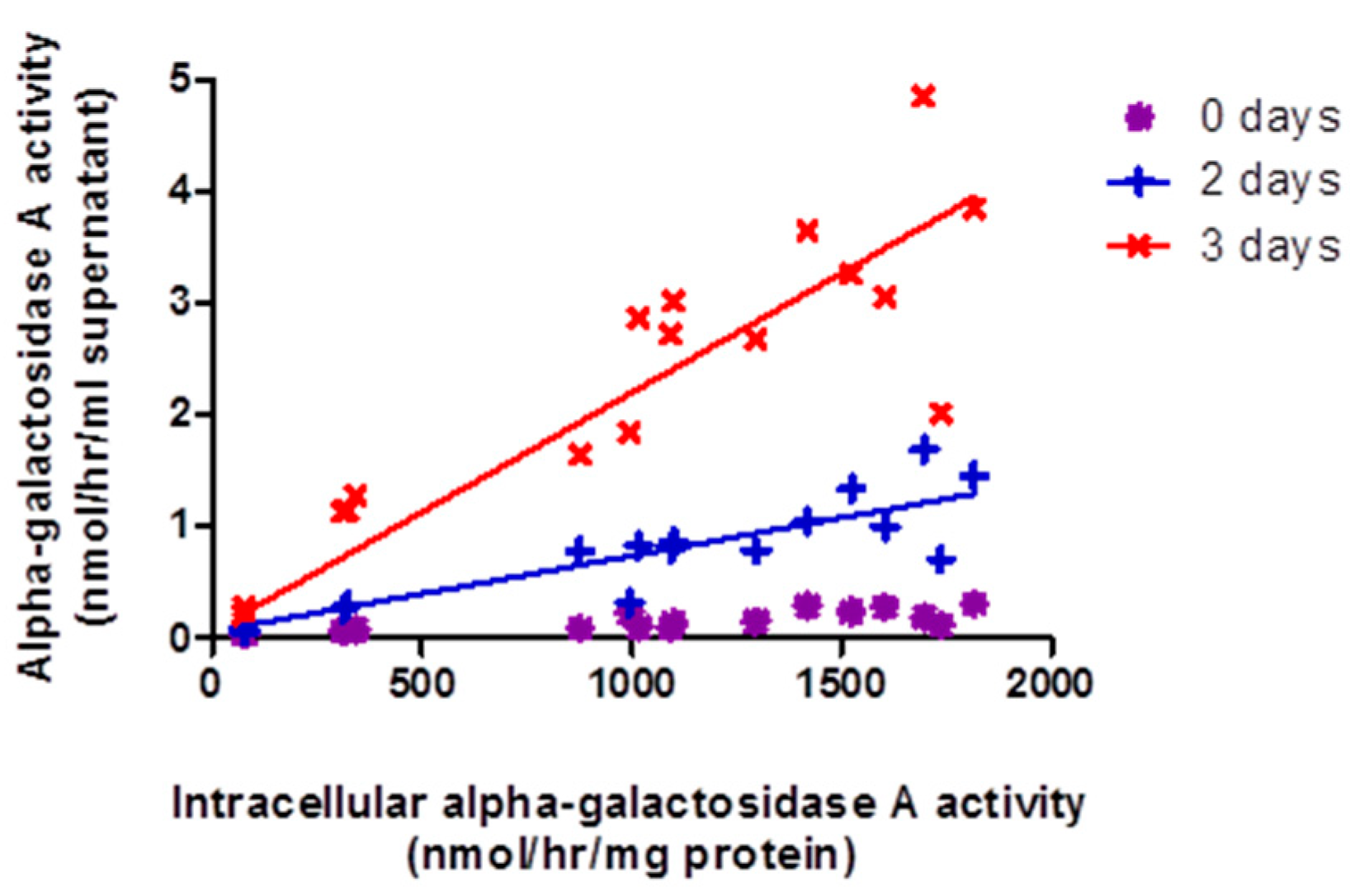
2.3. Release of α-Gal-A into Surrounding Growth Media by GLA-Jurkats Was Both Dose- and Time-Dependent, without Evidence of Cellular Damage
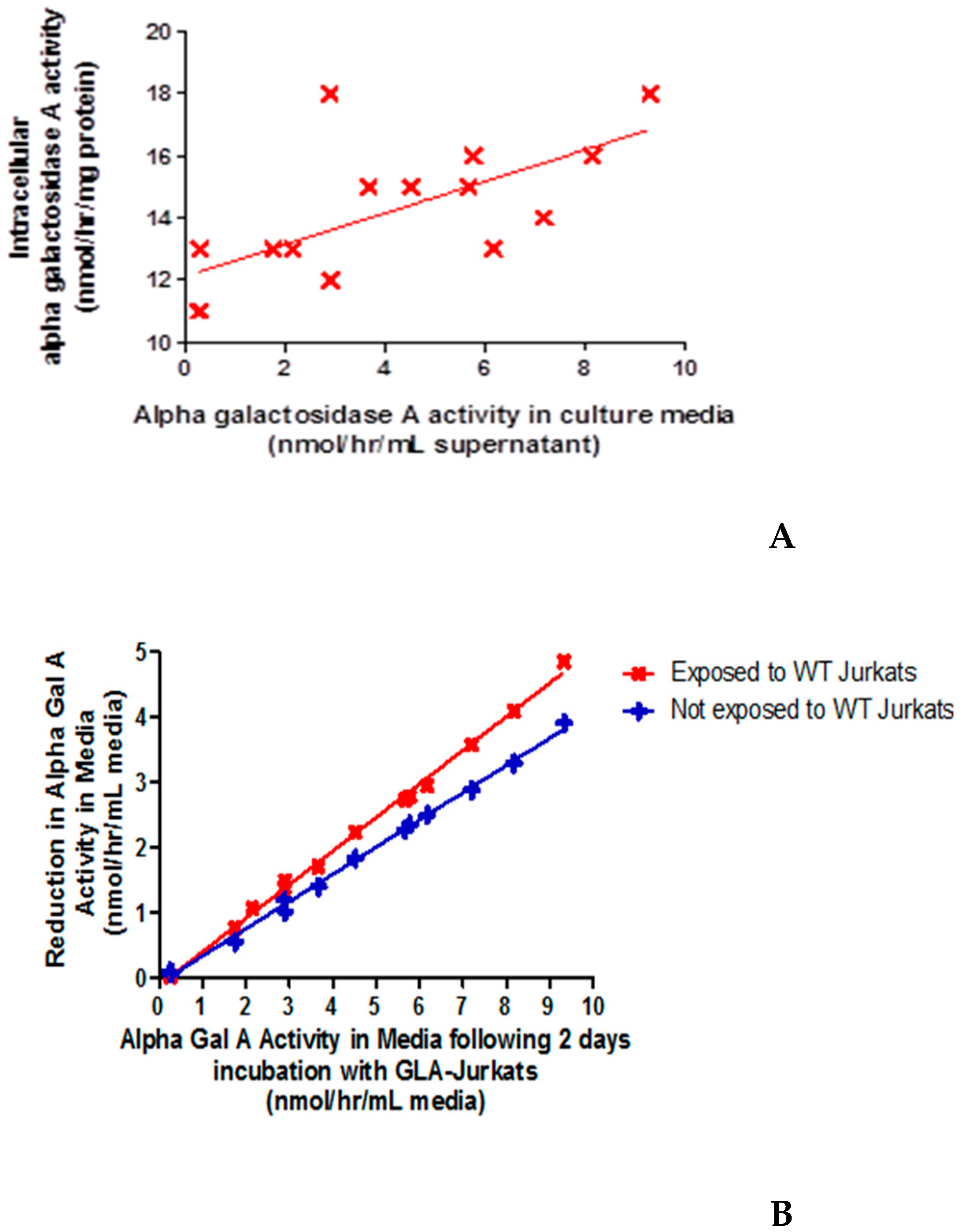
2.4. Wild-Type Jurkats Grown in Media Overexpressing α-Gal-A Showed Increased Intracellular α-galA Activity, Consistent with Enzyme Uptake from the Media
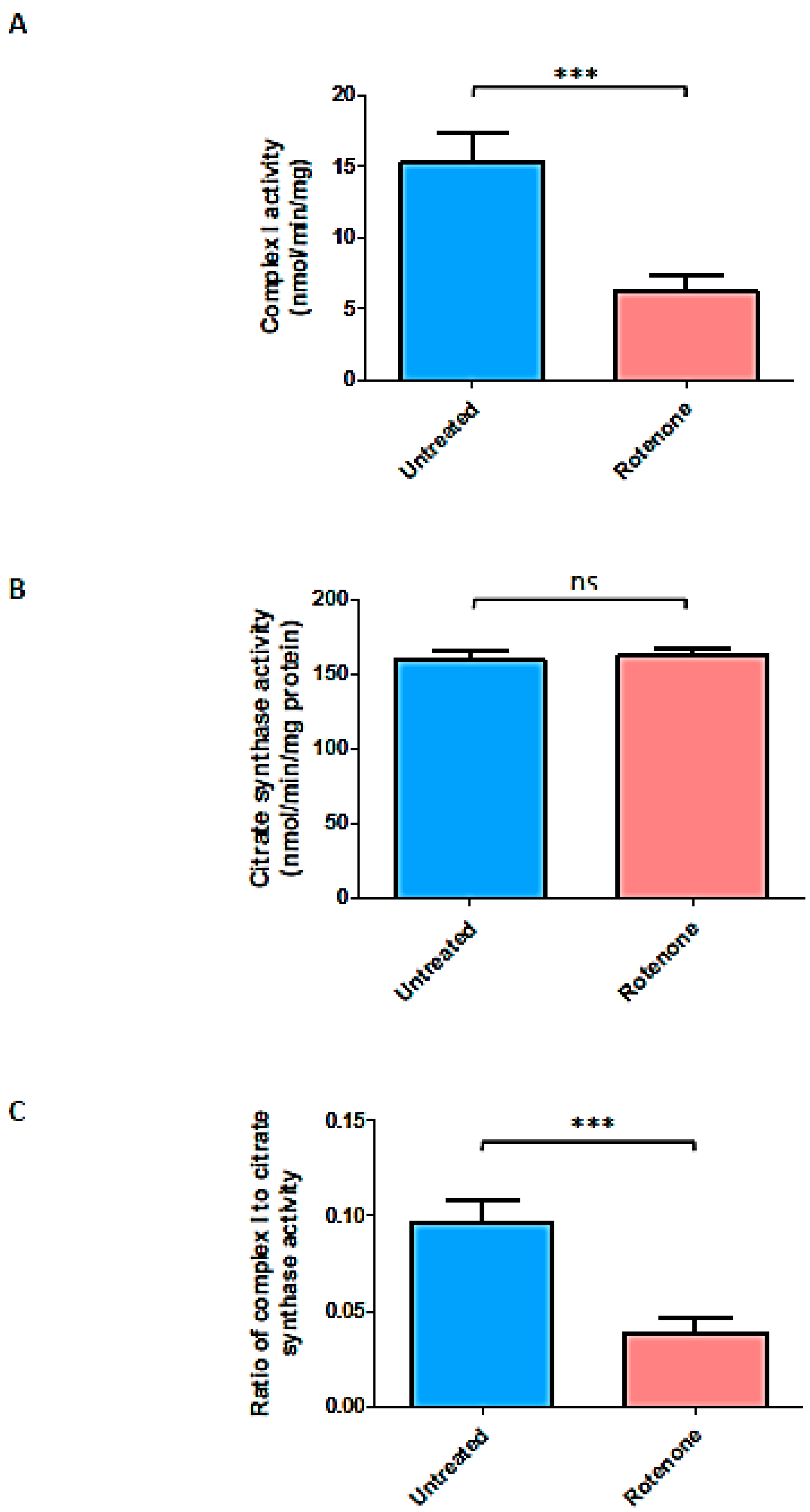
2.5. Rotenone Treatment Resulted in Marked Loss of Complex I Activity in Jurkat Treated Cells
2.6. Rotenone Treatment Did Not Significantly Affect Transduction Efficiency of The Lentivirus Containing GLA Transgene
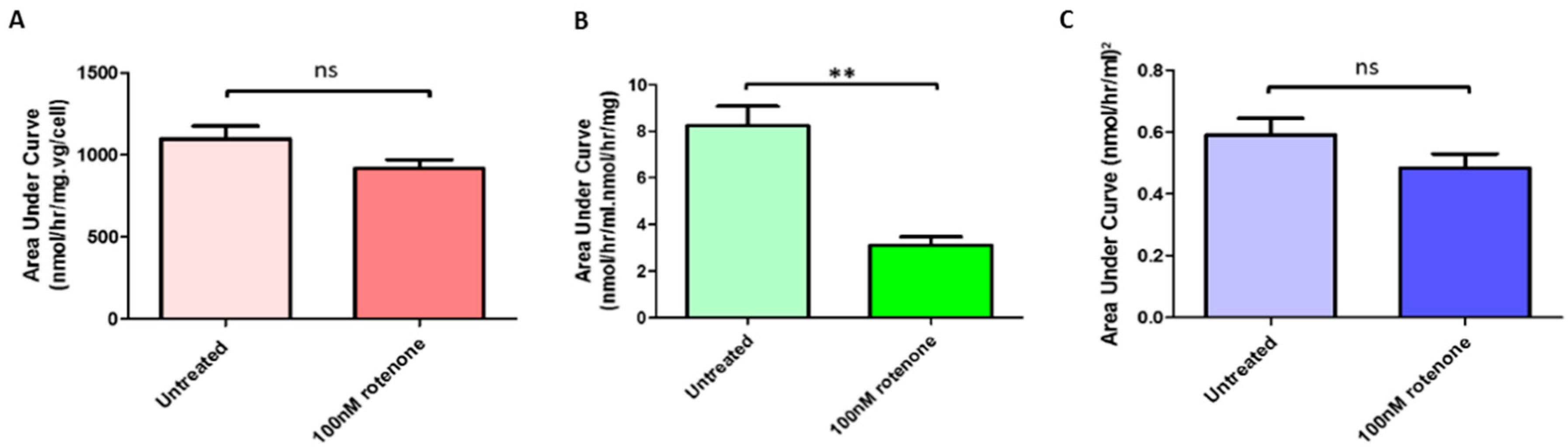
2.7. Rotenone Treatment Significantly Reduced Release by GLA-Jurkats into the Surrounding Media
2.8. Rotenone Treatment Did Not Significantly Alter Uptake of α-Gal-A by Wild-Type Recipient Jurkats from Donor Media Taken from Transduced Jurkats
3. Discussion
4. Materials and Methods
4.1. Jurkat Cells
4.2. Lentiviral Vector Production
4.3. Transduction of Jurkat Cells
4.4. Quantitative PCR
4.5. Cell Toxicity
4.6. Cell Lysis
4.7. Lysosomal Enzyme Activity Assays
4.8. Kinetic Studies
4.9. Cross Correction Studies
4.10. Mitochondrial Complex I Inhibition
4.11. Mitochondrial Complex I and Citrate Synthase Activity
4.12. Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FD | Fabry Disease |
| α-gal-A | α-galactosidase-A |
| Gb3 | globotriaoslyceramide |
| ERT | Enzyme Replacement Therapy |
| GFP | Glial Fibrillary Acid Protein |
References
- Sweeley, C.C.; Klionsky, B. Fabrys Disease—Classification as a Sphingolipidosis and Partial Characterization of a Novel Glycolipid. J. Biol. Chem. 1963, 238, 3148–3150. [Google Scholar]
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar]
- Garman, S.C.; Garboczi, D.N. The molecular defect leading to Fabry disease: Structure of human alpha-galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef]
- Kolter, T.; Sandhoff, K. Sphingolipids—Their metabolic pathways and the pathobiochemistry of neurodegenerative diseases. Angew. Chem. Int. Ed. 1999, 38, 1532–1568. [Google Scholar] [CrossRef]
- Fox, M.F.; DuToit, D.L.; Warnich, L.; Retief, A.E. Regional localization of alpha-galactosidase (GLA) to Xpter→q22, hexosaminidase B (HEXB) to 5q13→qter, and arylsulfatase B (ARSB) to 5pter→q13. Cytogenet. Cell Genet. 1984, 38, 45–49. [Google Scholar]
- Bishop, D.F.; Calhoun, D.H.; Bernstein, H.S.; Hantzopoulos, P.; Quinn, M.; Desnick, R.J. Human alpha-galactosidase A: Nucleotide sequence of a cDNA clone encoding the mature enzyme. Proc. Natl. Acad. Sci. USA 1986, 83, 4859–4863. [Google Scholar]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A., 3rd; Sabnis, S.; Moore, D.F.; Weibel, T.; Balow, J.E.; Brady, R.O. Enzyme replacement therapy in Fabry disease: A randomized controlled trial. JAMA 2001, 285, 2743–2749. [Google Scholar]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and efficacy of recombinant human alpha-galactosidase A-replacement therapy in Fabry’s disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef]
- Markham, A. Migalastat: First Global Approval. Drugs 2016, 76, 1147–1152. [Google Scholar] [CrossRef]
- El Dib, R.; Gomaa, H.; Carvalho, R.P.; Camargo, S.E.; Bazan, R.; Barretti, P.; Barreto, F.C. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst. Rev. 2016, 7, CD006663. [Google Scholar]
- Ortiz, A.; Abiose, A.; Bichet, D.G.; Cabrera, G.; Charrow, J.; Germain, D.P.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase beta: Data from the Fabry Registry. J. Med. Genet. 2016, 53, 495–502. [Google Scholar] [CrossRef]
- Rombach, S.M.; Smid, B.E.; Bouwman, M.G.; Linthorst, G.E.; Dijkgraaf, M.G.; Hollak, C.E. Long term enzyme replacement therapy for Fabry disease: Effectiveness on kidney, heart and brain. Orphanet J. Rare Dis. 2013, 8, 47. [Google Scholar] [CrossRef]
- Hughes, D.; Bichet, D.G.; Germain, D.P.; Giugliani, R.; Schiffmann, R.; Wilcox, W.; Castelli, J.P.; Benjamin, E.; Skuban, N.; Barth, J. Phenotype of Fabry disease in patients with mutations amenable to migalastat. Mol. Genet. Metab. 2016, 117, S58–S59. [Google Scholar] [CrossRef]
- Guérard, N.; Oder, D.; Nordbeck, P.; Zwingelstein, C.; Morand, O.; Welford, R.W.; Dingemanse, J.; Wanner, C. Lucerastat, an Iminosugar for Substrate Reduction Therapy: Tolerability, Pharmacodynamics, and Pharmacokinetics in Patients With Fabry Disease on Enzyme Replacement. Clin. Pharmacol. Ther. 2018, 103, 703–711. [Google Scholar] [CrossRef]
- Friedmann, T.; Roblin, R. Gene therapy for human genetic disease? Science 1972, 175, 949–955. [Google Scholar] [CrossRef]
- Huang, J.; Khan, A.; Au, B.C.; Barber, D.L.; López-Vásquez, L.; Prokopishyn, N.L.; Boutin, M.; Rothe, M.; Rip, J.W.; Abaoui, M.; et al. Lentivector Iterations and Pre-Clinical Scale-Up/Toxicity Testing: Targeting Mobilized CD34(+) Cells for Correction of Fabry Disease. Mol. Ther. Methods Clin. Dev. 2017, 5, 241–258. [Google Scholar] [CrossRef]
- Jung, S.-C.; Han, I.P.; Limaye, A.; Xu, R.; Gelderman, M.P.; Zerfas, P.; Tirumalai, K.; Murray, G.J.; During, M.J.; Brady, R.O.; et al. Adeno-associated viral vector-mediated gene transfer results in long-term enzymatic and functional correction in multiple organs of Fabry mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2676–2681. [Google Scholar] [CrossRef] [Green Version]
- Medin, J.A.; Tudor, M.; Simovitch, R.; Quirk, J.M.; Jacobson, S.; Murray, G.J.; Brady, R.O. Correction in trans for Fabry disease: Expression, secretion and uptake of alpha-galactosidase A in patient-derived cells driven by a high-titer recombinant retroviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 7917–7922. [Google Scholar] [CrossRef]
- Yoshimitsu, M.; Sato, T.; Tao, K.; Walia, J.S.; Rasaiah, V.I.; Sleep, G.T.; Murray, G.J.; Poeppl, A.G.; Underwood, J.; West, L.; et al. Bioluminescent imaging of a marking transgene and correction of Fabry mice by neonatal injection of recombinant lentiviral vectors. Proc. Natl. Acad. Sci. USA 2004, 101, 16909–16914. [Google Scholar] [CrossRef] [Green Version]
- Yoshimitsu, M.; Higuchi, K.; Ramsubir, S.; Nonaka, T.; Rasaiah, V.I.; Siatskas, C.; Liang, S.B.; Murray, G.J.; Brady, R.O.; Medin, J.A. Efficient correction of Fabry mice and patient cells mediated by lentiviral transduction of hematopoietic stem/progenitor cells. Gene Ther. 2007, 14, 256–265. [Google Scholar] [CrossRef]
- Takenaka, T.; Qin, G.; Brady, R.O.; Medin, J.A. Circulating alpha-galactosidase A derived from transduced bone marrow cells: Relevance for corrective gene transfer for Fabry disease. Hum. Gene 1999, 10, 1931–1939. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Machann, W.; Breunig, F.; Weidemann, F.; Sandstede, J.; Hahn, D.; Köstler, H.; Neubauer, S.; Wanner, C.; Beer, M. Cardiac energy metabolism is disturbed in Fabry disease and improves with enzyme replacement therapy using recombinant human galactosidase A. Eur. J. Heart Fail. 2011, 13, 278–283. [Google Scholar] [CrossRef] [Green Version]
- Palecek, T.; Bultas, J.; Hájek, M.; Karetová, D.; Kuchynka, P.; Kautzner, J.; Elleder, M.; Linhart, A. Association between cardiac energy metabolism and gain of left ventricular mass in Fabry disease. Int. J. Cardiol. 2010, 144, 337–339. [Google Scholar] [CrossRef]
- Liebau, M.C.; Braun, F.; Höpker, K.; Weitbrecht, C.; Bartels, V.; Müller, R.-U.; Brodesser, S.; Saleem, M.A.; Benzing, T.; Schermer, B.; et al. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS ONE 2013, 8, e63506. [Google Scholar] [CrossRef]
- Nelson, M.P.; Tse, T.E.; O’Quinn, D.B.; Percival, S.M.; Jaimes, E.A.; Warnock, D.G.; Shacka, J.J. Autophagy-lysosome pathway associated neuropathology and axonal degeneration in the brains of alpha-galactosidase A-deficient mice. Acta Neuropathol. Commun. 2014, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Chimenti, C.; Scopelliti, F.; Vulpis, E.; Tafani, M.; Villanova, L.; Verardo, R.; De Paulis, R.; Russo, M.A.; Frustaci, A. Increased oxidative stress contributes to cardiomyocyte dysfunction and death in patients with Fabry disease cardiomyopathy. Hum. Pathol. 2015, 46, 1760–1768. [Google Scholar] [CrossRef]
- Lenaz, G.; Bovina, C.; D’Aurelio, M.; Fato, R.; Formiggini, G.; Genova, M.L.; Giuliano, G.; Pich, M.M.; Paolucci, U.; Castelli, G.P.; et al. Role of mitochondria in oxidative stress and aging. Ann. N. Y. Acad. Sci. 2002, 959, 199–213. [Google Scholar] [CrossRef]
- Das, A.M.; Naim, H.Y. Biochemical basis of Fabry disease with emphasis on mitochondrial function and protein trafficking. Adv. Clin. Chem. 2009, 49, 57–71. [Google Scholar]
- Lucke, T.; Hoppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, E.L.; Henson, P.M. Biochemical characteristics of ATP-induced secretion of lysosomal enzymes from rabbit polymorphonuclear leukocytes. Inflamm 1975, 1, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; Koenig, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Hughes, A.L.; Madeo, F.; Ruckenstuhl, C. The crucial impact of lysosomes in aging and longevity. Ageing Res. Rev. 2016, 32, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Swenson, C.; Sasagawa, S.; Sakatani, T.; Watanabe, M.; Kobayashi, M.; Fujikura, T. Age-related decline in lysosomal enzyme release from polymorphonuclear leukocytes after N-formyl-methionyl-leucyl-phenylalanine stimulation. Exp. Hematol. 1983, 11, 1005–1013. [Google Scholar] [PubMed]
- Lambert, J.R.A. Less Is More: The Efficacy of Gene Therapy to Treat. Fabry Disease; UCL Library Open Access University College: London, UK, 2018. [Google Scholar]
- Desnick, R.J.; Dean, K.J.; Grabowski, G.; Bishop, D.F.; Sweeley, C.C. Enzyme therapy in Fabry disease: Differential in vivo plasma clearance and metabolic effectiveness of plasma and splenic alpha-galactosidase A isozymes. Proc. Natl. Acad. Sci. USA 1979, 76, 5326–5330. [Google Scholar] [CrossRef]
- Nagano, T.; Nakatsuka, S.-I.; Fujita, S.; Kanda, T.; Uematsu, M.; Ikeda, Y.; Ishibashi-Ueda, H.; Yutani, C. Myocardial fibrosis pathology in Anderson-Fabry disease: Evaluation of autopsy cases in the long- and short-term enzyme replacement therapy, and non-therapy case. IJC Metab. Endocr. 2016, 12, 46–51. [Google Scholar] [CrossRef]
- Itoh, Y.; Esaki, T.; Cook, M.; Qasba, P.; Shimoji, K.; Alroy, J.; Brady, R.O.; Sokoloff, L.; Moore, D.F. Local and global cerebral blood flow and glucose utilization in the alpha-galactosidase A knockout mouse model of Fabry disease. J. Neurochem. 2001, 79, 1217–1224. [Google Scholar] [CrossRef]
- Aylett, S.-B.; Neergheen, V.; Hargreaves, I.P.; Eaton, S.; Land, J.M.; Rahman, S.; Heales, S.J. Levels of 5-methyltetrahydrofolate and ascorbic acid in cerebrospinal fluid are correlated: Implications for the accelerated degradation of folate by reactive oxygen species. Neurochem. Int. 2013, 63, 750–755. [Google Scholar] [CrossRef]
- Al Sharani, M. Mitochondiral function, Oxidative Stress and Parkinson’s Disease; UCL Library Open Access University College: London, UK, 2018. [Google Scholar]
- Salamero, J.; Sztul, E.S.; Howell, K.E. Exocytic transport vesicles generated in vitro from the trans-Golgi network carry secretory and plasma membrane proteins. Proc. Natl. Acad. Sci. USA 1990, 87, 7717–7721. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Mellett, N.; Hein, L.K.; Brooks, D.A.; Meikle, P.J. Absence of alpha-galactosidase cross-correction in Fabry heterozygote cultured skin fibroblasts. Mol. Genet. Metab. 2015, 114, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral vectors: Basic to translational. Biochem. J. 2012, 443, 603–618. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Krohn, R.; Hermanson, G.; Mallia, A.; Gartner, F.; Provenzano, M.; Fujimoto, E.; Goeke, N.; Olson, B.; Klenk, D. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Donnai, P.; Donnai, D.; Harris, R.; Stephens, R.; Young, E.; Campbell, S. Antenatal diagnosis of Niemann-Pick disease in a twin pregnancy. J. Med. Genet. 1981, 18, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.H.; Rudge, P.; Smith, S.J.; Bronstein, A.M.; Kendall, B.E.; Holly, E.; Young, E.P.; Crawfurd, M.D.; Bannister, R. The neurological complications of Anderson-Fabry disease (alpha-galactosidase A deficiency)-investigation of symptomatic and presymptomatic patients. Q. J. Med. 1990, 75, 491–507. [Google Scholar] [PubMed]
- Mayes, J.S.; Scheerer, J.B.; Sifers, R.N.; Donaldson, M.L. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. Clin. Chim. Acta Int. J. Clin. Chem. 1981, 112, 247–251. [Google Scholar] [CrossRef]
- Young, I.D.; Young, E.P.; Mossman, J.; Fielder, A.R.; Moore, J.R. Neuraminidase deficiency: Case report and review of the phenotype. J. Med. Genet. 1987, 24, 283–290. [Google Scholar] [CrossRef]
- Chabas, A.; Coll, M.J.; Aparicio, M.; Rodriguez Diaz, E. Mild phenotypic expression of alpha-N-acetylgalactosaminidase deficiency in two adult siblings. J. Inherit. Metab. Dis. 1994, 17, 724–731. [Google Scholar] [CrossRef]
- Brett, E.M.; Ellis, R.B.; Haas, L.; Ikonne, J.U.; Lake, B.D.; Patrick, A.D.; Stephens, R. Late onset GM2-gangliosidosis. Clinical, pathological, and biochemical studies on 8 patients. Arch. Dis. Child. 1973, 48, 775–785. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Okada, S.; Chen, A.; Fillerup, D.L. Tay-sachs disease. Detection of heterozygotes and homozygotes by serum hexosaminidase assay. N. Engl. J. Med. 1970, 283, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Fratantoni, J.C.; Hall, C.W.; Neufeld, E.F. Hurler and Hunter syndromes: Mutual correction of the defect in cultured fibroblasts. Science 1968, 162, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, D.; Garland, P.B. The kinetic properties of citrate synthase from rat liver mitochondria. Biochem. J. 1969, 114, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’ Donnel, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J.R. The ketogenic diet component increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2015, 129, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondiral content in skeletal muscle of healthy young subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Parameter | Wild Type | 0.4 vg/cell | 1.5 vg/cell | 2.0 vg/cell | 1.8 vg/cell (Dilute) |
|---|---|---|---|---|---|
| Km (mM) | 1.57 ± 0.11 | 1.52 ± 0.13 | 2.43 ± 0.12 | 2.30 ± 0.10 | 2.53 ± 0.29 |
| Vmax (nmol/hr) | 1.95 ± 0.04 | 8.31 ± 0.02 | 67.30 ± 1.12 | 82.1 ± 1.20 | 1.17 ± 0.06 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambert, J.R.A.; Howe, S.J.; Rahim, A.A.; Burke, D.G.; Heales, S.J.R. Inhibition of Mitochondrial Complex I Impairs Release of α-Galactosidase by Jurkat Cells. Int. J. Mol. Sci. 2019, 20, 4349. https://doi.org/10.3390/ijms20184349
Lambert JRA, Howe SJ, Rahim AA, Burke DG, Heales SJR. Inhibition of Mitochondrial Complex I Impairs Release of α-Galactosidase by Jurkat Cells. International Journal of Molecular Sciences. 2019; 20(18):4349. https://doi.org/10.3390/ijms20184349
Chicago/Turabian StyleLambert, Jonathan R. A., Steven J. Howe, Ahad A. Rahim, Derek G. Burke, and Simon J. R. Heales. 2019. "Inhibition of Mitochondrial Complex I Impairs Release of α-Galactosidase by Jurkat Cells" International Journal of Molecular Sciences 20, no. 18: 4349. https://doi.org/10.3390/ijms20184349