Skin Doctor: Machine Learning Models for Skin Sensitization Prediction that Provide Estimates and Indicators of Prediction Reliability

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Characterization of the LLNA Data Sets
2.1.1. Data Set Composition
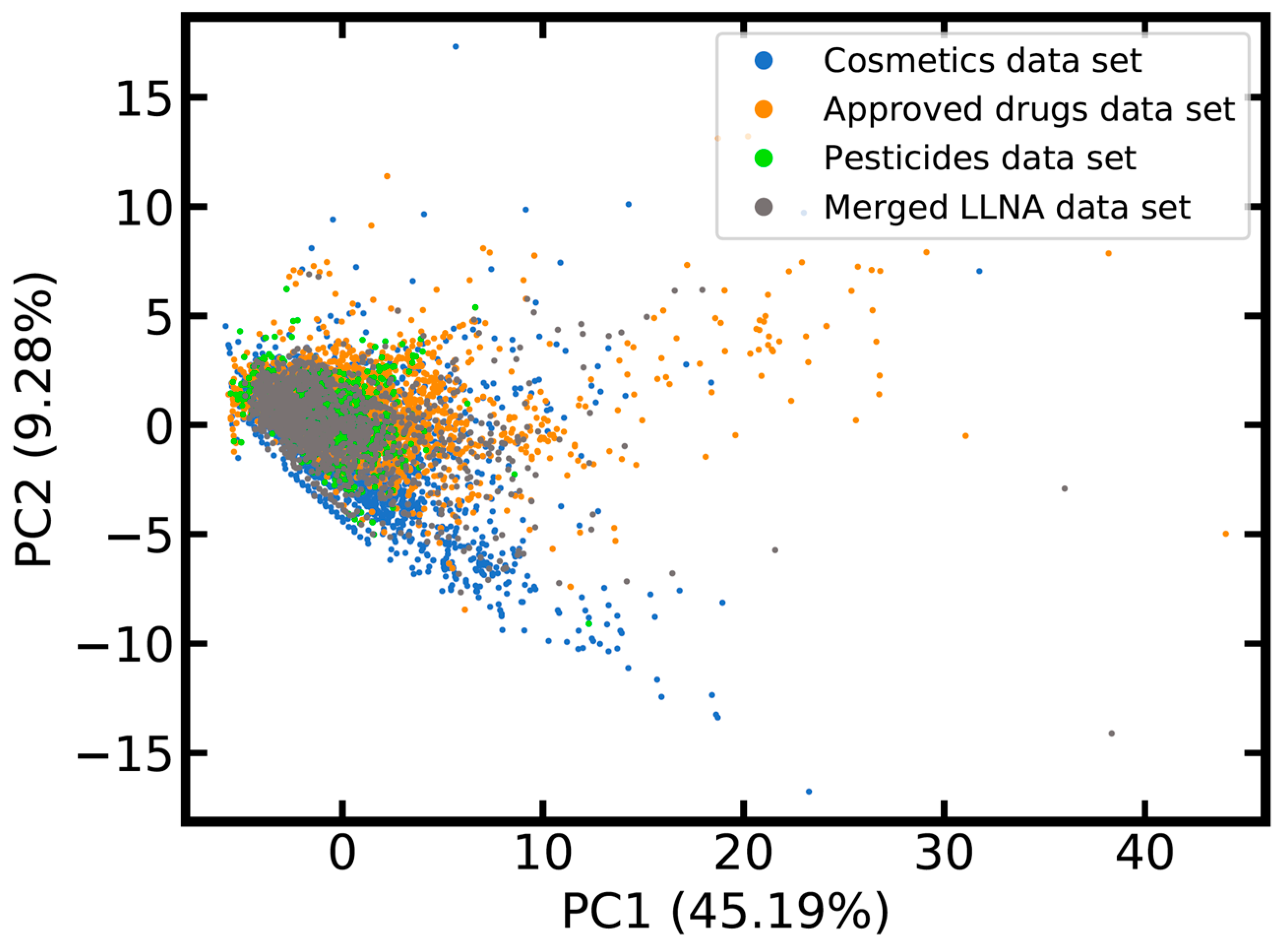
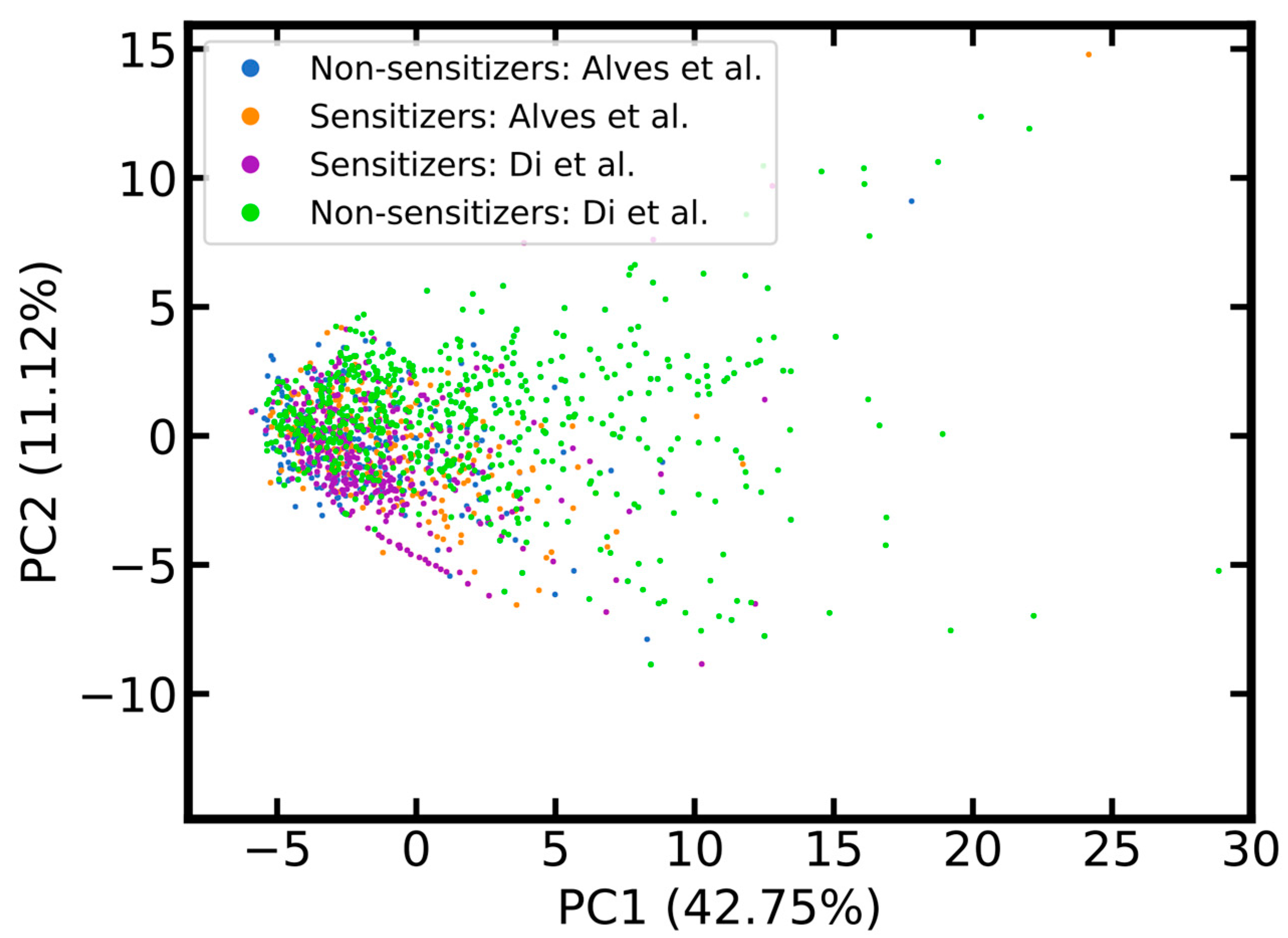
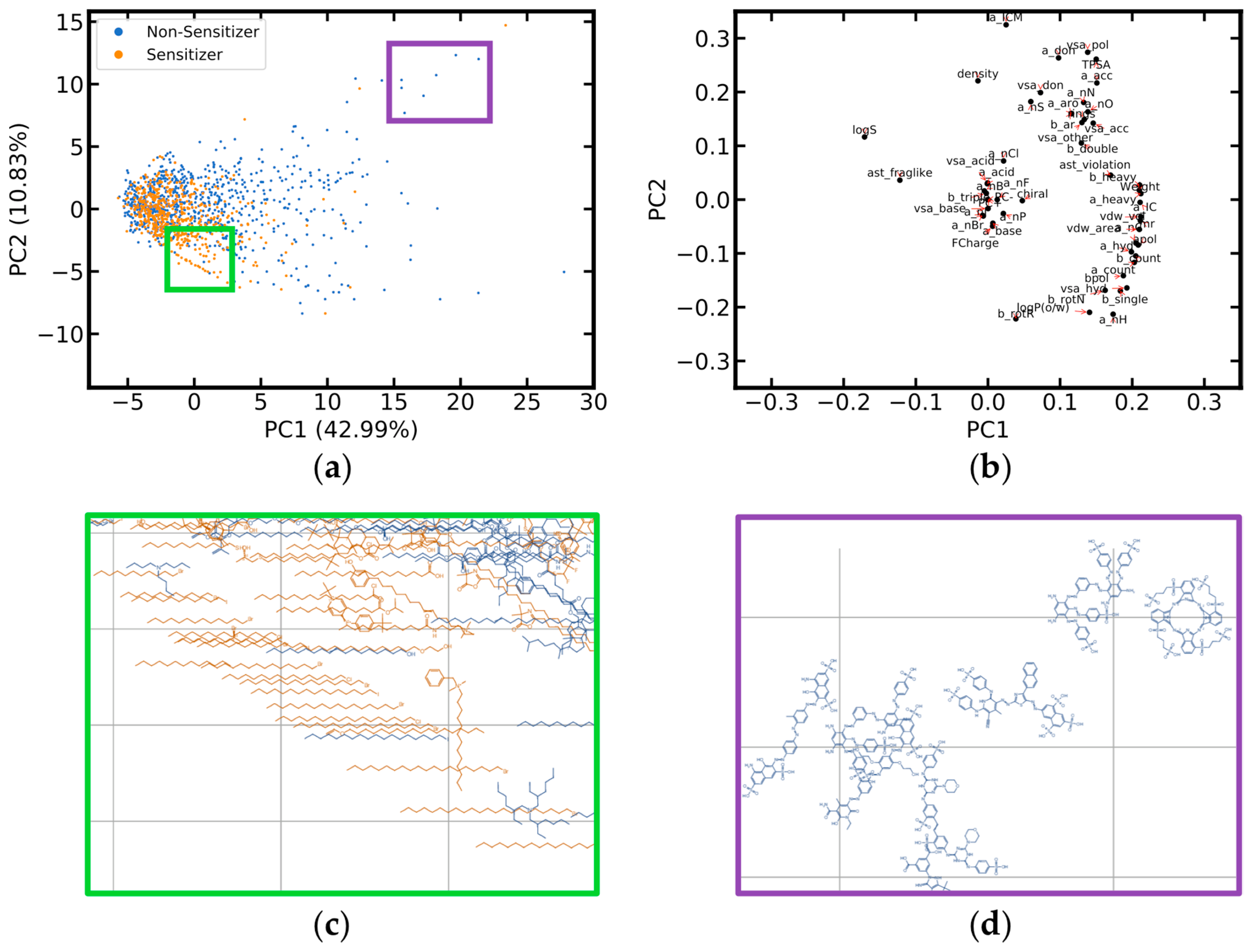
2.1.2. Coverage of Chemical Space
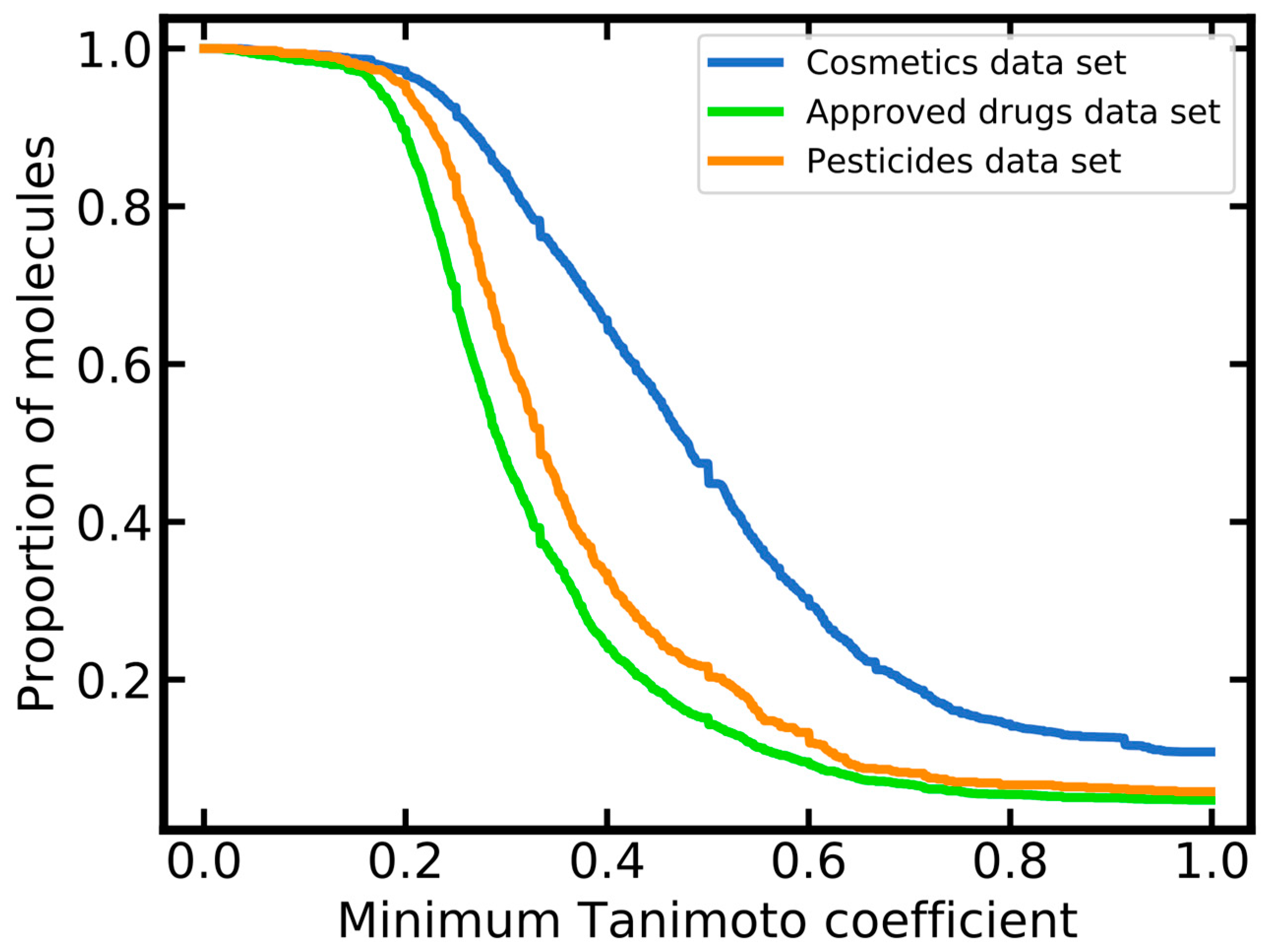
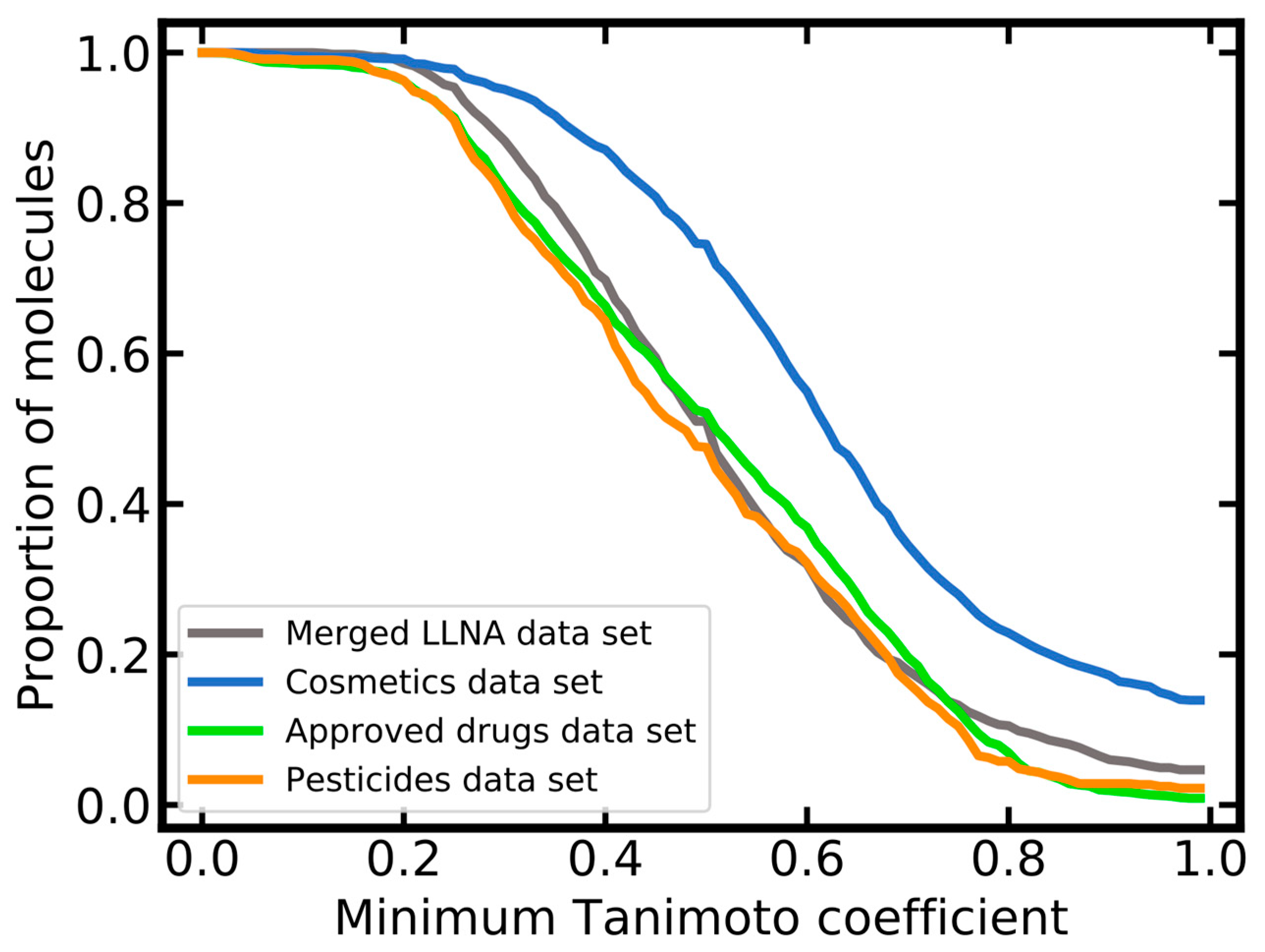
2.1.3. Molecular Diversity
2.2. Molecular Properties of Skin Sensitizers and Non-Sensitizers
2.3. Model Development
2.4. Model Performance
2.4.1. Measures for the Evaluation of Model Performance
- Matthews correlation coefficient (MCC), which is regarded to be one of the best measures of binary classification performance. It is robust against data imbalance and considers the proportion of all four cases of predictions (i.e., true positive, false positive, true negative and false negative predictions). Note that MCC values range from −1 to + 1. A value of + 1 indicates perfect prediction, whereas a value of −1 indicates a prediction that is in total disagreement. A value of 0 indicates a performance which is equal to random.
- ACC, which has been most commonly used by others to measure the performance of models for the prediction of the skin sensitization potential. It is defined as the proportion of correct predictions within all predictions made.
- Area under the receiver operating characteristic curve (AUC), which in this case quantifies the ability to correctly rank compounds according to their skin sensitization potential. The AUC does not rely on a decision threshold.
- Sensitivity (Se), which in this case quantifies the proportion of correctly identified skin sensitizers.
- Specificity (Sp), which in this case quantifies the proportion of correctly predicted non-sensitizers.
- Positive predictive value (PPV), which reports the proportion of true positive predictions among all positive predictions.
- Negative predictive value (NPV), which reports the proportion of true negative predictions among all negative predictions.
- CCR, which is the mean of Se and Sp.
2.4.2. Model Performance During Cross-Validation
2.4.3. In-Depth Analysis of Selected Models within the Cross-Validation Framework
- SVM_MOE2D+OASIS: the model with highest MCC.
- SVM_PaDEL+OASIS: a model performing comparable to the SVM_MOE2D+OASIS and based on freely available software only.
- SVM_PaDEL: the best model based on a single set of molecular descriptors.
- RF_MACCS: the best model based on a single set of molecular fingerprints.
- SVM_PaDEL+MACCS: a model with good performance, combining the descriptor sets used by the above two models.
2.4.4. Performance of Selected Models on the Test Set
2.4.5. Comparison of Model Performance to that of Existing Models
2.5. Skin Doctor Web Service
3. Materials and Methods
3.1. Data Preparation
3.2. Descriptor Calculation
3.3. Data Analysis
3.4. Compilation of Data Sets for Model Development
3.5. Model Generation
3.6. Hardware and Software
4. Conclusions
- The compound of interest is within the applicability domain of the model.
- The distance between the predicted class probability and the decision threshold is at least 0.15 for RF models and 0.5 for SVM models.
- The predicted activity class for a compound of interest is in agreement with the class assigned to the nearest neighbor in the training data.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC | accuracy |
| ACD | allergic contact dermatitis |
| AUC | area under the receiver operating characteristic curve |
| CCR | correct classification rate |
| LLNA | local lymph node assay |
| MCC | Matthews correlation coefficient |
| MOE | Molecular Operating Environment |
| NERDD | New E-Resource for Drug Discovery |
| NPV | negative predictive value |
| PCA | principal component analysis |
| RBF | radial basis function |
| RF | random forest |
| PPV | positive predictive value |
| Se | sensitivity |
| Sp | specificity |
| SVM | support vector machine |
References
- Kimber, I.; Basketter, D.A.; Gerberick, G.F.; Ryan, C.A.; Dearman, R.J. Chemical allergy: Translating biology into hazard characterization. Toxicol. Sci. 2011, 120 (Suppl. 1), S238–S268. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Linneberg, A.; Menné, T.; Johansen, J.D. The epidemiology of contact allergy in the general population—prevalence and main findings. Contact Dermat. 2007, 57, 287–299. [Google Scholar] [CrossRef]
- Lushniak, B.D. Occupational contact dermatitis. Dermatol. Ther. 2004, 17, 272–277. [Google Scholar] [CrossRef][Green Version]
- Anderson, S.E.; Siegel, P.D.; Meade, B.J. The LLNA: A brief review of recent advances and limitations. J. Allergy 2011, 2011. [Google Scholar] [CrossRef]
- Dent, M.; Amaral, R.T.; Da Silva, P.A.; Ansell, J.; Boisleve, F.; Hatao, M.; Hirose, A.; Kasai, Y.; Kern, P.; Kreiling, R.; et al. Principles underpinning the use of new methodologies in the risk assessment of cosmetic ingredients. Comput. Toxicol. 2018, 7, 20–26. [Google Scholar] [CrossRef]
- Mehling, A.; Eriksson, T.; Eltze, T.; Kolle, S.; Ramirez, T.; Teubner, W.; van Ravenzwaay, B.; Landsiedel, R. Non-animal test methods for predicting skin sensitization potentials. Arch. Toxicol. 2012, 86, 1273–1295. [Google Scholar] [CrossRef]
- Reisinger, K.; Hoffmann, S.; Alépée, N.; Ashikaga, T.; Barroso, J.; Elcombe, C.; Gellatly, N.; Galbiati, V.; Gibbs, S.; Groux, H.; et al. Systematic evaluation of non-animal test methods for skin sensitisation safety assessment. Toxicol. In Vitro 2015, 29, 259–270. [Google Scholar] [CrossRef]
- Ezendam, J.; Braakhuis, H.M.; Vandebriel, R.J. State of the art in non-animal approaches for skin sensitization testing: From individual test methods towards testing strategies. Arch. Toxicol. 2016, 90, 2861–2883. [Google Scholar] [CrossRef]
- Thyssen, J.P.; Giménez-Arnau, E.; Lepoittevin, J.-P.; Menné, T.; Boman, A.; Schnuch, A. The critical review of methodologies and approaches to assess the inherent skin sensitization potential (skin allergies) of chemicals. Part I. Contact Dermat. 2012, 66 (Suppl. 1), 11–24. [Google Scholar] [CrossRef]
- Wilm, A.; Kühnl, J.; Kirchmair, J. Computational approaches for skin sensitization prediction. Crit. Rev. Toxicol. 2018, 48, 738–760. [Google Scholar] [CrossRef]
- ECHA (European Chemicals Agency). The Use of Alternatives to Testing on Animals for the REACH Regulation, Third Report under Article 117(3) of the REACH Regulation. Available online: https://echa.europa.eu/documents/10162/13639/alternatives_test_animals_2017_en.pdf (accessed on 10 July 2019).
- Kleinstreuer, N.C.; Hoffmann, S.; Alépée, N.; Allen, D.; Ashikaga, T.; Casey, W.; Clouet, E.; Cluzel, M.; Desprez, B.; Gellatly, N.; et al. Non-animal methods to predict skin sensitization (II): An assessment of defined approaches. Crit. Rev. Toxicol. 2018, 48, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Luechtefeld, T.; Marsh, D.; Rowlands, C.; Hartung, T. Machine learning of toxicological big data enables read-across structure activity relationships (RASAR) outperforming animal test reproducibility. Toxicol. Sci. 2018, 165, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Luechtefeld, T.; Rowlands, C.; Hartung, T. Big-data and machine learning to revamp computational toxicology and its use in risk assessment. Toxicol. Res. 2018, 7, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.M.; Borba, J.; Capuzzi, S.J.; Muratov, E.; Andrade, C.H.; Rusyn, I.; Tropsha, A. Oy vey! A comment on “Machine learning of toxicological big data enables read-across structure activity relationships outperforming animal test reproducibility”. Toxicol. Sci. 2019, 167, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Luechtefeld, T.; Marsh, D.; Hartung, T. Missing the difference between big data and artificial intelligence in RASAR versus traditional QSAR. Toxicol. Sci. 2019, 167, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.-W.; Lin, Y.-H.; Wang, S.-S. Transfer learning for predicting human skin sensitizers. Arch. Toxicol. 2019, 93, 931–940. [Google Scholar] [CrossRef]
- Chilton, M.L.; Macmillan, D.S.; Steger-Hartmann, T.; Hillegass, J.; Bellion, P.; Vuorinen, A.; Etter, S.; Smith, B.P.C.; White, A.; Sterchele, P.; et al. Making reliable negative predictions of human skin sensitisation using an in silico fragmentation approach. Regul. Toxicol. Pharm. 2018, 95, 227–235. [Google Scholar] [CrossRef]
- Braga, R.C.; Alves, V.M.; Muratov, E.N.; Strickland, J.; Kleinstreuer, N.; Trospsha, A.; Andrade, C.H. Pred-Skin: A fast and reliable web application to assess skin sensitization effect of chemicals. J. Chem. Inf. Model. 2017, 57, 1013–1017. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, M.K.; Kim, K.-B.; Kim, H.S.; Lee, B.-M. Quantitative structure–activity and quantitative structure–property relationship approaches as alternative skin sensitization risk assessment methods. J. Toxicol. Environ. Health 2019, 82, 447–472. [Google Scholar] [CrossRef]
- Toropov, A.A.; Toropova, A.P.; Selvestrel, G.; Benfenati, E. Idealization of correlations between optimal simplified molecular input-line entry system-based descriptors and skin sensitization. SAR QSAR Environ. Res. 2019, 30, 447–455. [Google Scholar] [CrossRef]
- Di, P.; Yin, Y.; Jiang, C.; Cai, Y.; Li, W.; Tang, Y.; Liu, G. Prediction of the skin sensitising potential and potency of compounds via mechanism-based binary and ternary classification models. Toxicol. In Vitro 2019, 59, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.M.; Muratov, E.; Fourches, D.; Strickland, J.; Kleinstreuer, N.; Andrade, C.H.; Tropsha, A. Predicting chemically-induced skin reactions. Part I: QSAR models of skin sensitization and their application to identify potentially hazardous compounds. Toxicol. Appl. Pharmacol. 2015, 284, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zheng, M.; Wang, Y.; Shen, Q.; Luo, X.; Jiang, H.; Chen, K. Fragment-based prediction of skin sensitization using recursive partitioning. J. Comput. Aided Mol. Des. 2011, 25, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, Q.; Piclin, N.; Cotterill, J.; Pintore, M.; Price, N.R.; Chrétien, J.R.; Roncaglioni, A. Global QSAR models of skin sensitisers for regulatory purposes. Chem. Cent. J. 2010, 4, S5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Enoch, S.J.; Roberts, D.W. Predicting skin sensitization potency for Michael acceptors in the LLNA using quantum mechanics calculations. Chem. Res. Toxicol. 2013, 26, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S. LLNA variability: An essential ingredient for a comprehensive assessment of non-animal skin sensitization test methods and strategies. ALTEX 2015, 32, 379–383. [Google Scholar] [PubMed]
- Alves, V.M.; Capuzzi, S.J.; Braga, R.C.; Borba, J.V.B.; Silva, A.C.; Luechtefeld, T.; Hartung, T.; Andrade, C.H.; Muratov, E.N.; Tropsha, A. A perspective and a new integrated computational strategy for skin sensitization assessment. ACS Sustain. Chem. Eng. 2018, 6, 2845–2859. [Google Scholar] [CrossRef]
- Apt Systemst Ltd. Aptsys.net OASIS. QSAR Toolbox 4.3. Available online: http://oasis-lmc.org/products/software/toolbox.aspx (accessed on 10 July 2019).
- Chembench|Home. Available online: https://chembench.mml.unc.edu (accessed on 26 April 2019).
- CosIng—Cosmetics—GROWTH—European Commission. Available online: http://ec.europa.eu/growth/tools-databases/cosing/index.cfm?fuseaction=search.simple (accessed on 26 April 2019).
- DrugBank Version 5.1.2. Available online: https://www.drugbank.ca (accessed on 7 May 2019).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- EU Pesticides Database—European Commission. Available online: http://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/public/?event=activesubstance.selection&language=EN (accessed on 25 February 2019).
- Chemical Identifier Resolver. Available online: https://cactus.nci.nih.gov/chemical/structure (accessed on 25 February 2019).
- Chemical Computing Group Molecular Operating Environment (MOE)|MOEsaic|PSILO. Available online: https://www.chemcomp.com/Products.htm (accessed on 12 June 2019).
- PaDEL-Descriptor. Available online: http://www.yapcwsoft.com/dd/padeldescriptor/ (accessed on 10 May 2019).
- Yap, C.W. PaDEL-Descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Landrum, G. RDKit. Available online: http://www.rdkit.org (accessed on 26 April 2019).
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Stork, C.; Embruch, G.; Šícho, M.; de Bruyn Kops, C.; Chen, Y.; Svozil, D.; Kirchmair, J. NERDD: A web portal providing access to in silico tools for drug discovery. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Stork, C.; Wagner, J.; Friedrich, N.-O.; de Bruyn Kops, C.; Šícho, M.; Kirchmair, J. Hit Dexter: A machine-learning model for the prediction of frequent hitters. Chem. Med. Chem. 2018, 13, 564–571. [Google Scholar] [CrossRef] [PubMed]
- MolVs. MolVs Version 0.1.1. Available online: https://github.com/mcs07/MolVS (accessed on 26 April 2019).
- Scikit-Learn: Machine Learning in Python—Scikit-Learn 0.21.0 Documentation. Available online: https://scikit-learn.org/stable/ (accessed on 10 May 2019).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LLNA Data Set Compiled by Alves et al. | LLNA Data Set Compiled by Di et al. | Merged LLNA Data Set | Cosmetic Substances and Ingredients Data Set | Approved Drugs Data Set | Pesticides Data Set | |
|---|---|---|---|---|---|---|
| Data source | Chembench [30]1 | Supporting information of Di et al. [22] | LLNA data sets of Alves et al. and Di et al. | CosIng Database [31] | “Approved Drugs” subset of DrugBank [32,33]2 | EU Pesticides Database [34] |
| Number of compounds prior to data preprocessing | 1000 | 1007 | 1993 | 5937 | 2352 | 1383 |
| Number of compounds after data preprocessing | 1000 | 9933 | 14164 (1132/284)5 | 46436 | 21557 | 8128 |
| Number of sensitizers | 481 | 364 | 572 (457/115)5 | n/a | n/a | n/a |
| Number of non-sensitizers | 519 | 629 | 844 (675/169)5 | n/a | n/a | n/a |
| Number of Murcko scaffolds | 312 | 354 | 453 | 856 | 1158 | 329 |
| Proportion of compounds without a Murcko scaffold | 0.32 | 0.29 | 0.31 | 0.42 | 0.13 | 0.24 |
| Proportion of singleton scaffolds | 0.77 | 0.79 | 0.78 | 0.72 | 0.82 | 0.81 |
| Number of Compounds | Data Set Compiled by Alves et al. | Data Set Compiled by Di et al. | Merged LLNA Data Set | |
|---|---|---|---|---|
| Cosmetics | 4643 | 324 | 252 | 387 |
| Approved Drugs | 2155 | 88 | 68 | 97 |
| Pesticides | 812 | 43 | 34 | 44 |
| Descriptor Set | Short Name | Number of Descriptors/Length of the Fingerprint | Calculated with | Number of Successfully Processed Molecules1 | |
|---|---|---|---|---|---|
| Training set | Test set | ||||
| 0D, 1D and 2D descriptors | MOE2D | 206 | MOE [36]; this set corresponds to all descriptors listed as “2D descriptors” in MOE | 1132 | 284 |
| Selection of 0D, 1D and 2D descriptors | MOE2D_53 | 532 | MOE [36] | 1132 | 284 |
| 0D, 1D and 2D descriptors | PaDEL | 1444 | PaDEL [37,38]; this is the complete set of 0D, 1D and 2D descriptors implemented in PaDEL | 1109 | 279 |
| MACCS keys | MACCS | 166 | RDKit [39] | 1132 | 284 |
| Morgan2 fingerprints | Morgan2 | 2048 | RDKit [39] | 1132 | 284 |
| OASIS skin sensitization protein binding fingerprint | OASIS | 5 bit fingerprint | OECD Toolbox [29] | 1128 | 283 |
| PaDEL estate fingerprint | PaDEL_Est | 79 | PaDEL [37,38] | 1132 | 284 |
| PaDEL extended fingerprint | PaDEL_Ext | 1024 | PaDEL [37,38] | 1132 | 284 |
| Name | Number of Descriptors | Number of Compounds in Training Data | ACC | ACC STDEV | MCC | MCCSTDEV | AUC | CCR | Se | SP | PPV | NPV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SVM_MOE2D+OASIS | 211 | 1128 | 0.78 | 0.054 | 0.55 | 0.109 | 0.83 | 0.78 | 0.77 | 0.78 | 0.71 | 0.83 |
| SVM_PaDEL+MACCS | 1610 | 1108 | 0.76 | 0.035 | 0.51 | 0.069 | 0.83 | 0.76 | 0.75 | 0.76 | 0.69 | 0.82 |
| SVM_PaDEL+Morgan2 | 3492 | 1108 | 0.76 | 0.036 | 0.51 | 0.078 | 0.82 | 0.75 | 0.66 | 0.83 | 0.73 | 0.78 |
| SVM_PaDEL+PaDEL-Ext | 2468 | 1109 | 0.76 | 0.039 | 0.51 | 0.075 | 0.84 | 0.76 | 0.74 | 0.78 | 0.7 | 0.81 |
| SVM_MOE2D+MACCS | 372 | 1132 | 0.76 | 0.047 | 0.5 | 0.096 | 0.81 | 0.74 | 0.68 | 0.81 | 0.71 | 0.79 |
| SVM_MOE2D+Morgan2 | 2254 | 1132 | 0.75 | 0.041 | 0.5 | 0.081 | 0.83 | 0.75 | 0.77 | 0.73 | 0.66 | 0.83 |
| SVM_MOE2D+PaDEL | 1680 | 1109 | 0.76 | 0.039 | 0.5 | 0.079 | 0.83 | 0.75 | 0.74 | 0.77 | 0.69 | 0.81 |
| SVM_MOE2D+PaDEL-Est | 285 | 1132 | 0.76 | 0.039 | 0.5 | 0.081 | 0.81 | 0.75 | 0.68 | 0.81 | 0.71 | 0.79 |
| SVM_MOE2D+PaDEL-Ext | 1230 | 1132 | 0.75 | 0.054 | 0.5 | 0.105 | 0.83 | 0.75 | 0.75 | 0.76 | 0.68 | 0.81 |
| SVM_PaDEL | 1444 | 1109 | 0.75 | 0.038 | 0.5 | 0.075 | 0.83 | 0.75 | 0.75 | 0.75 | 0.68 | 0.81 |
| SVM_PaDEL+OASIS | 1449 | 1109 | 0.75 | 0.038 | 0.5 | 0.075 | 0.83 | 0.75 | 0.75 | 0.75 | 0.68 | 0.81 |
| SVM_PaDEL+PaDEL-Est | 1523 | 1109 | 0.75 | 0.038 | 0.5 | 0.075 | 0.83 | 0.75 | 0.75 | 0.75 | 0.68 | 0.81 |
| RF_PaDEL+MACCS | 1610 | 1108 | 0.76 | 0.018 | 0.49 | 0.037 | 0.82 | 0.73 | 0.62 | 0.85 | 0.74 | 0.77 |
| RF_PaDEL+Morgan2 | 3492 | 1108 | 0.76 | 0.02 | 0.49 | 0.042 | 0.82 | 0.74 | 0.64 | 0.84 | 0.73 | 0.77 |
| RF_PaDEL+OASIS | 1449 | 1109 | 0.76 | 0.02 | 0.49 | 0.043 | 0.82 | 0.74 | 0.62 | 0.85 | 0.74 | 0.77 |
| RF_PaDEL+PaDEL-Ext | 2468 | 1109 | 0.76 | 0.022 | 0.49 | 0.048 | 0.82 | 0.73 | 0.61 | 0.86 | 0.75 | 0.76 |
| SVM_PaDEL-Est+MACCS | 245 | 1132 | 0.75 | 0.051 | 0.49 | 0.106 | 0.81 | 0.74 | 0.69 | 0.8 | 0.7 | 0.79 |
| RF_MOE2D+PaDEL | 1680 | 1109 | 0.75 | 0.034 | 0.48 | 0.072 | 0.83 | 0.73 | 0.62 | 0.84 | 0.73 | 0.77 |
| RF_Morgan2+PaDEL-Est | 2127 | 1132 | 0.76 | 0.033 | 0.48 | 0.071 | 0.82 | 0.73 | 0.63 | 0.84 | 0.73 | 0.77 |
| RF_PaDEL | 1444 | 1109 | 0.75 | 0.015 | 0.48 | 0.033 | 0.82 | 0.73 | 0.62 | 0.84 | 0.73 | 0.76 |
| RF_PaDEL-Est+OASIS | 84 | 1128 | 0.75 | 0.043 | 0.48 | 0.091 | 0.8 | 0.74 | 0.65 | 0.82 | 0.72 | 0.78 |
| SVM_MACCS+OASIS | 171 | 1128 | 0.75 | 0.047 | 0.48 | 0.102 | 0.82 | 0.74 | 0.69 | 0.79 | 0.69 | 0.79 |
| SVM_MOE2D | 206 | 1132 | 0.74 | 0.037 | 0.48 | 0.067 | 0.82 | 0.74 | 0.75 | 0.74 | 0.66 | 0.82 |
| SVM_Morgan2+PaDEL-Ext | 3072 | 1132 | 0.75 | 0.044 | 0.48 | 0.09 | 0.82 | 0.74 | 0.68 | 0.8 | 0.7 | 0.79 |
| RF_MACCS | 166 | 1132 | 0.75 | 0.039 | 0.47 | 0.088 | 0.81 | 0.73 | 0.61 | 0.84 | 0.73 | 0.76 |
| RF_MACCS+OASIS | 171 | 1128 | 0.75 | 0.034 | 0.47 | 0.074 | 0.8 | 0.73 | 0.6 | 0.85 | 0.74 | 0.76 |
| RF_PaDEL+PaDEL-Est | 1523 | 1109 | 0.75 | 0.028 | 0.47 | 0.06 | 0.83 | 0.73 | 0.61 | 0.85 | 0.73 | 0.76 |
| SVM_MACCS | 166 | 1132 | 0.74 | 0.057 | 0.47 | 0.12 | 0.81 | 0.73 | 0.69 | 0.78 | 0.68 | 0.79 |
| SVM_PaDEL-Est+OASIS | 84 | 1128 | 0.74 | 0.048 | 0.47 | 0.099 | 0.8 | 0.74 | 0.71 | 0.76 | 0.67 | 0.8 |
| SVM_PaDEL-Est+PaDEL-Ext | 1103 | 1132 | 0.74 | 0.039 | 0.47 | 0.08 | 0.81 | 0.74 | 0.7 | 0.78 | 0.68 | 0.79 |
| SVM_PaDEL-Ext | 1024 | 1132 | 0.74 | 0.046 | 0.47 | 0.093 | 0.81 | 0.73 | 0.7 | 0.77 | 0.68 | 0.79 |
| SVM_PaDEL-Ext+OASIS | 1029 | 1128 | 0.74 | 0.036 | 0.47 | 0.072 | 0.82 | 0.74 | 0.7 | 0.77 | 0.68 | 0.79 |
| RF_MOE2D+Morgan2 | 2254 | 1132 | 0.74 | 0.033 | 0.46 | 0.071 | 0.81 | 0.72 | 0.62 | 0.82 | 0.71 | 0.76 |
| RF_PaDEL-Est+MACCS | 245 | 1132 | 0.75 | 0.045 | 0.46 | 0.1 | 0.81 | 0.72 | 0.59 | 0.85 | 0.73 | 0.76 |
| RF_Morgan2 | 2048 | 1132 | 0.74 | 0.039 | 0.46 | 0.081 | 0.81 | 0.73 | 0.64 | 0.81 | 0.7 | 0.77 |
| SVM_Morgan2+MACCS | 2214 | 1132 | 0.74 | 0.058 | 0.46 | 0.117 | 0.8 | 0.73 | 0.68 | 0.78 | 0.68 | 0.78 |
| SVM_PaDEL-Ext+MACCS | 1190 | 1132 | 0.74 | 0.047 | 0.46 | 0.097 | 0.81 | 0.73 | 0.68 | 0.77 | 0.68 | 0.78 |
| RF_MOE2D+OASIS | 211 | 1128 | 0.74 | 0.041 | 0.45 | 0.09 | 0.81 | 0.71 | 0.6 | 0.83 | 0.71 | 0.75 |
| RF_MOE2D+PaDEL-Est | 285 | 1132 | 0.74 | 0.032 | 0.45 | 0.07 | 0.81 | 0.72 | 0.6 | 0.84 | 0.72 | 0.75 |
| RF_MOE2D+PaDEL-Ext | 1230 | 1132 | 0.74 | 0.017 | 0.45 | 0.037 | 0.82 | 0.72 | 0.58 | 0.85 | 0.73 | 0.75 |
| RF_MOE2D | 206 | 1132 | 0.73 | 0.036 | 0.44 | 0.078 | 0.81 | 0.71 | 0.59 | 0.83 | 0.71 | 0.75 |
| RF_MOE2D+MACCS | 372 | 1132 | 0.73 | 0.033 | 0.44 | 0.072 | 0.81 | 0.71 | 0.58 | 0.84 | 0.71 | 0.75 |
| RF_Morgan2+MACCS | 2214 | 1132 | 0.73 | 0.039 | 0.44 | 0.086 | 0.8 | 0.72 | 0.63 | 0.8 | 0.68 | 0.76 |
| RF_Morgan2+OASIS | 2053 | 1128 | 0.74 | 0.029 | 0.44 | 0.063 | 0.82 | 0.71 | 0.59 | 0.83 | 0.71 | 0.75 |
| RF_Morgan2+PaDEL-Ext | 3072 | 1132 | 0.73 | 0.036 | 0.44 | 0.081 | 0.81 | 0.71 | 0.56 | 0.85 | 0.72 | 0.74 |
| SVM_MOE2D53 | 53 | 1132 | 0.71 | 0.037 | 0.44 | 0.069 | 0.78 | 0.72 | 0.76 | 0.68 | 0.62 | 0.81 |
| SVM_PaDEL-Est | 79 | 1132 | 0.72 | 0.037 | 0.44 | 0.073 | 0.77 | 0.72 | 0.71 | 0.73 | 0.64 | 0.79 |
| RF_PaDEL-Est | 79 | 1132 | 0.73 | 0.022 | 0.43 | 0.042 | 0.77 | 0.71 | 0.64 | 0.79 | 0.67 | 0.76 |
| RF_PaDEL-Ext+MACCS | 1190 | 1132 | 0.73 | 0.037 | 0.43 | 0.081 | 0.81 | 0.7 | 0.55 | 0.85 | 0.72 | 0.74 |
| RF_PaDEL-Ext+OASIS | 1029 | 1128 | 0.73 | 0.033 | 0.43 | 0.072 | 0.8 | 0.7 | 0.57 | 0.84 | 0.71 | 0.74 |
| RF_PaDEL-Ext+PaDEL-Est | 1103 | 1132 | 0.73 | 0.034 | 0.43 | 0.074 | 0.8 | 0.7 | 0.56 | 0.85 | 0.72 | 0.74 |
| SVM_Morgan2+OASIS | 2053 | 1128 | 0.73 | 0.038 | 0.43 | 0.089 | 0.8 | 0.69 | 0.51 | 0.88 | 0.75 | 0.73 |
| SVM_Morgan2+PaDEL-Est | 2127 | 1132 | 0.72 | 0.035 | 0.43 | 0.064 | 0.79 | 0.72 | 0.69 | 0.75 | 0.65 | 0.78 |
| RF_MOE2D53 | 53 | 1132 | 0.73 | 0.039 | 0.42 | 0.086 | 0.78 | 0.7 | 0.58 | 0.83 | 0.69 | 0.74 |
| RF_PaDEL-Ext | 1024 | 1132 | 0.72 | 0.039 | 0.42 | 0.088 | 0.79 | 0.7 | 0.55 | 0.84 | 0.71 | 0.73 |
| SVM_Morgan2 | 2048 | 1132 | 0.72 | 0.031 | 0.39 | 0.072 | 0.8 | 0.68 | 0.49 | 0.87 | 0.72 | 0.71 |
| SVM_OASIS | 5 | 1128 | 0.67 | 0.064 | 0.29 | 0.151 | 0.63 | 0.62 | 0.37 | 0.87 | 0.68 | 0.67 |
| RF_OASIS | 5 | 1128 | 0.66 | 0.054 | 0.27 | 0.122 | 0.64 | 0.63 | 0.43 | 0.82 | 0.62 | 0.68 |
| Machine Learning Approach | Parameter | Explored Values |
|---|---|---|
| RF | n_estimators 1 | 10, 50, 100, 250, 500, 1000 |
| max_features 2 | ‘sqrt’, 0.2, 0.4, 0.6, 0.8, None | |
| SVM | C 3 | 0.01, 0.1, 1, 10, 100, 1000 |
| gamma 4 | 1, 0.1, 0.01, 0.001, 0.0001, 0.00001 |
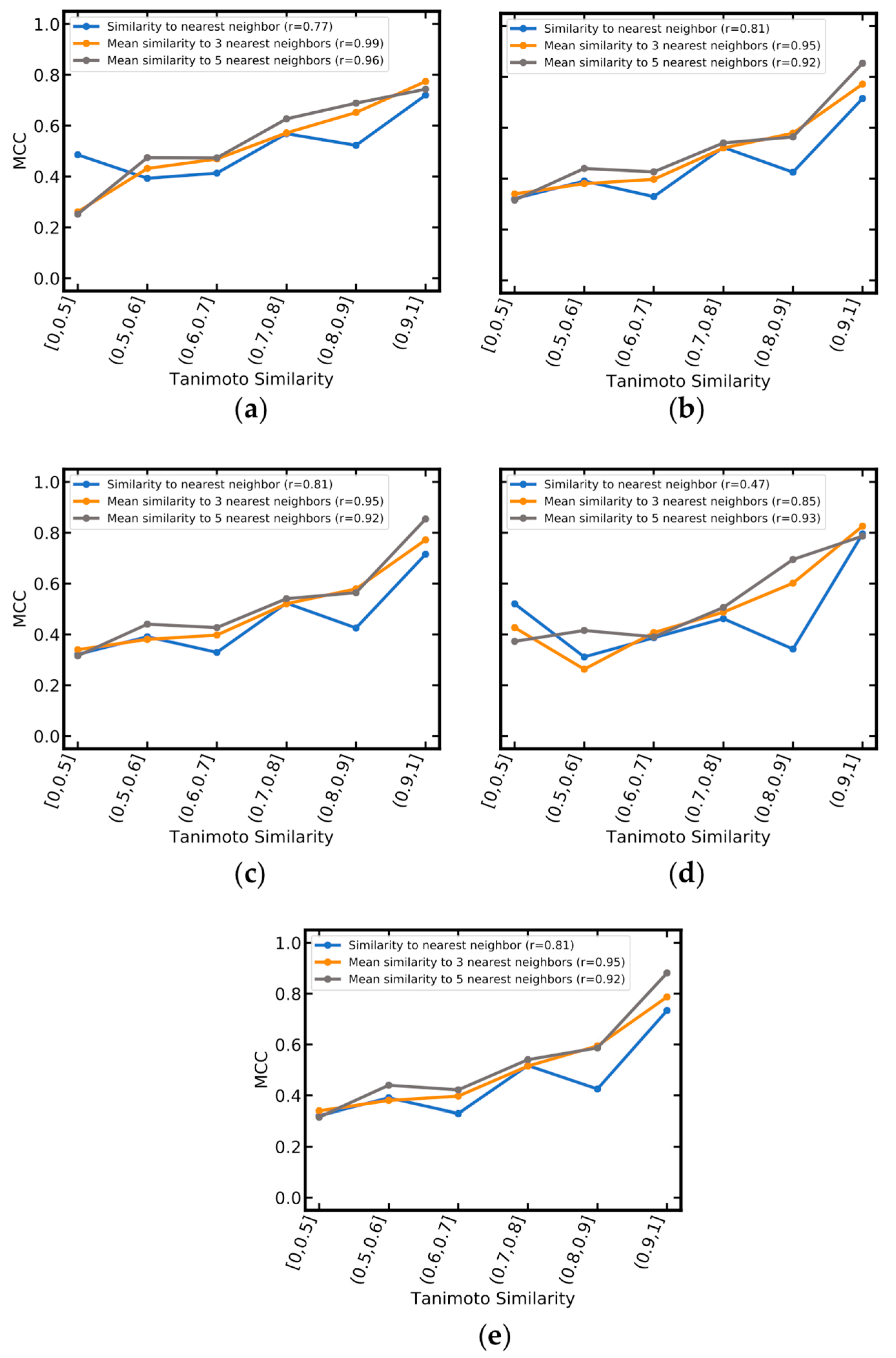
| NAME | Mean Tanimoto Similarity to the Five Nearest Neighbors | Number of Compounds | ACC | MCC | AUC | CCR | Se | Sp | PPV | NPV |
|---|---|---|---|---|---|---|---|---|---|---|
| RF_MACCS | ≥0 | 284 | 0.72 | 0.41 | 0.82 | 0.70 | 0.57 | 0.82 | 0.69 | 0.74 |
| RF_MACCS | ≥0.5 | 273 | 0.73 | 0.43 | 0.82 | 0.71 | 0.6 | 0.82 | 0.69 | 0.75 |
| RF_MACCS | ≥0.75 | 79 | 0.78 | 0.59 | 0.91 | 0.81 | 0.89 | 0.73 | 0.64 | 0.92 |
| RF_MACCS | <0.5 | 11 | 0.45 | −0.29 | 0.60 | 0.42 | 0.00 | 0.83 | 0.00 | 0.50 |
| SVM_MOE_2D+OASIS | ≥0 | 283 | 0.76 | 0.52 | 0.83 | 0.76 | 0.81 | 0.72 | 0.66 | 0.85 |
| SVM_MOE_2D+OASIS | ≥0.5 | 273 | 0.76 | 0.53 | 0.84 | 0.77 | 0.82 | 0.72 | 0.67 | 0.86 |
| SVM_MOE_2D+OASIS | ≥0.75 | 79 | 0.81 | 0.64 | 0.89 | 0.84 | 0.93 | 0.75 | 0.67 | 0.95 |
| SVM_MOE2D+OASIS | <0.5 | 10 | 0.60 | 0.20 | 0.60 | 0.60 | 0.60 | 0.60 | 0.60 | 0.60 |
| SVM_PaDEL | ≥0 | 279 | 0.74 | 0.47 | 0.82 | 0.74 | 0.76 | 0.72 | 0.65 | 0.82 |
| SVM_PaDEL | ≥0.5 | 269 | 0.74 | 0.49 | 0.83 | 0.75 | 0.77 | 0.73 | 0.65 | 0.83 |
| SVM_PaDEL | ≥0.75 | 79 | 0.80 | 0.63 | 0.89 | 0.83 | 0.93 | 0.73 | 0.65 | 0.95 |
| SVM_PaDEL | <0.5 | 10 | 0.60 | 0.20 | 0.56 | 0.60 | 0.60 | 0.60 | 0.60 | 0.60 |
| SVM_PaDEL+MACCS | ≥0 | 279 | 0.75 | 0.50 | 0.82 | 0.75 | 0.78 | 0.73 | 0.66 | 0.83 |
| SVM_PaDEL+MACCS | ≥0.5 | 269 | 0.75 | 0.51 | 0.83 | 0.76 | 0.79 | 0.73 | 0.66 | 0.84 |
| SVM_PaDEL+MACCS | ≥0.75 | 79 | 0.80 | 0.63 | 0.89 | 0.83 | 0.93 | 0.73 | 0.65 | 0.95 |
| SVM_PaDEL+MACCS | <0.5 | 10 | 0.60 | 0.20 | 0.56 | 0.60 | 0.60 | 0.60 | 0.60 | 0.60 |
| SVM_PaDEL+OASIS | ≥0 | 279 | 0.74 | 0.48 | 0.82 | 0.74 | 0.76 | 0.73 | 0.65 | 0.82 |
| SVM_PaDEL+OASIS | ≥0.5 | 271 | 0.75 | 0.49 | 0.83 | 0.75 | 0.77 | 0.73 | 0.65 | 0.83 |
| SVM_PaDEL+OASIS | ≥0.75 | 79 | 0.80 | 0.63 | 0.89 | 0.83 | 0.93 | 0.73 | 0.65 | 0.95 |
| SVM_PaDEL+OASIS | <0.5 | 10 | 0.60 | 0.20 | 0.56 | 0.60 | 0.60 | 0.6 | 0.60 | 0.60 |
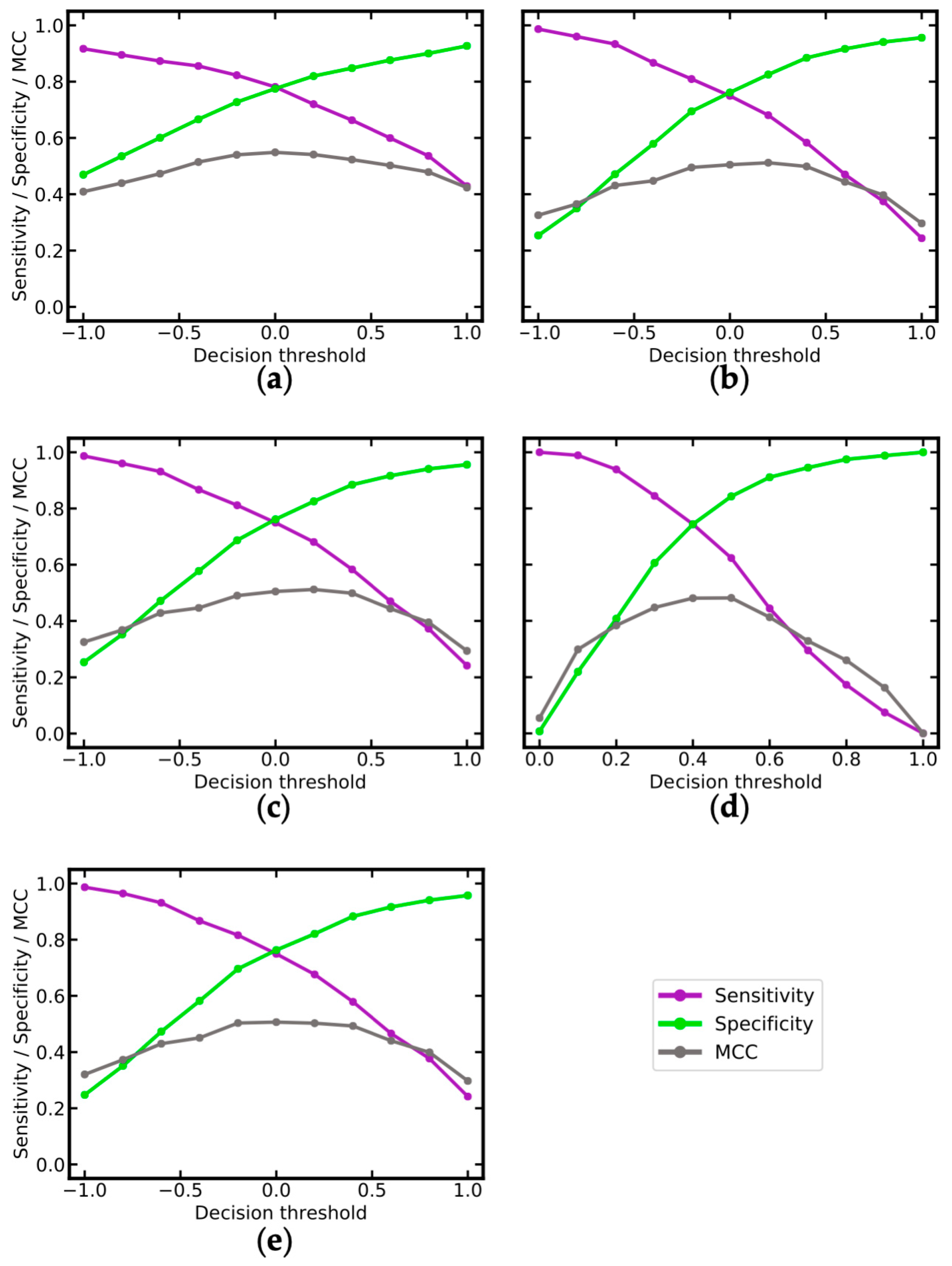
| Name | Distance to Decision Threshold 1 | Number of Compounds | ACC | MCC | AUC | CCR | Se | Sp | PPV | NPV |
|---|---|---|---|---|---|---|---|---|---|---|
| RF-MACCS | ≥0.15 | 175 | 0.85 | 0.67 | 0.46 | 0.84 | 0.81 | 0.87 | 0.76 | 0.90 |
| RF-MACCS | ≥0.35 | 66 | 0.91 | 0.78 | 0.42 | 0.89 | 0.85 | 0.93 | 0.85 | 0.93 |
| RF-MACCS | <0.15 | 109 | 0.51 | 0.04 | 0.42 | 0.52 | 0.32 | 0.72 | 0.55 | 0.50 |
| SVM_MOE2D+OASIS | ≥0.5 | 203 | 0.82 | 0.64 | 0.42 | 0.83 | 0.88 | 0.78 | 0.73 | 0.90 |
| SVM_MOE2D+OASIS | ≥1.25 | 106 | 0.89 | 0.76 | 0.41 | 0.89 | 0.89 | 0.88 | 0.81 | 0.94 |
| SVM_MOE2D+OASIS | <0.50 | 80 | 0.60 | 0.20 | 0.52 | 0.60 | 0.62 | 0.58 | 0.50 | 0.70 |
| SVM_PaDEL | ≥0.5 | 183 | 0.80 | 0.61 | 0.48 | 0.81 | 0.86 | 0.76 | 0.71 | 0.89 |
| SVM_PaDEL | ≥1.25 | 34 | 0.88 | 0.78 | 0.45 | 0.91 | 1.00 | 0.82 | 0.75 | 1.00 |
| SVM_PaDEL | <0.50 | 96 | 0.61 | 0.21 | 0.36 | 0.60 | 0.55 | 0.66 | 0.51 | 0.69 |
| SVM_PaDEL+MACCS | ≥0.5 | 183 | 0.80 | 0.62 | 0.49 | 0.82 | 0.88 | 0.75 | 0.71 | 0.90 |
| SVM_PaDEL+MACCS | ≥1.25 | 37 | 0.86 | 0.75 | 0.52 | 0.9 | 1.00 | 0.80 | 0.71 | 1.00 |
| SVM_PaDEL+MACCS | <0.50 | 96 | 0.65 | 0.27 | 0.39 | 0.63 | 0.58 | 0.69 | 0.55 | 0.71 |
| SVM_PaDEL+OASIS | ≥0.5 | 183 | 0.80 | 0.61 | 0.49 | 0.81 | 0.86 | 0.76 | 0.71 | 0.89 |
| SVM_PaDEL+OASIS | ≥1.25 | 34 | 0.88 | 0.78 | 0.45 | 0.91 | 1.00 | 0.82 | 0.75 | 1.00 |
| SVM_PaDEL+OASIS | <0.50 | 96 | 0.62 | 0.22 | 0.37 | 0.61 | 0.55 | 0.67 | 0.52 | 0.70 |
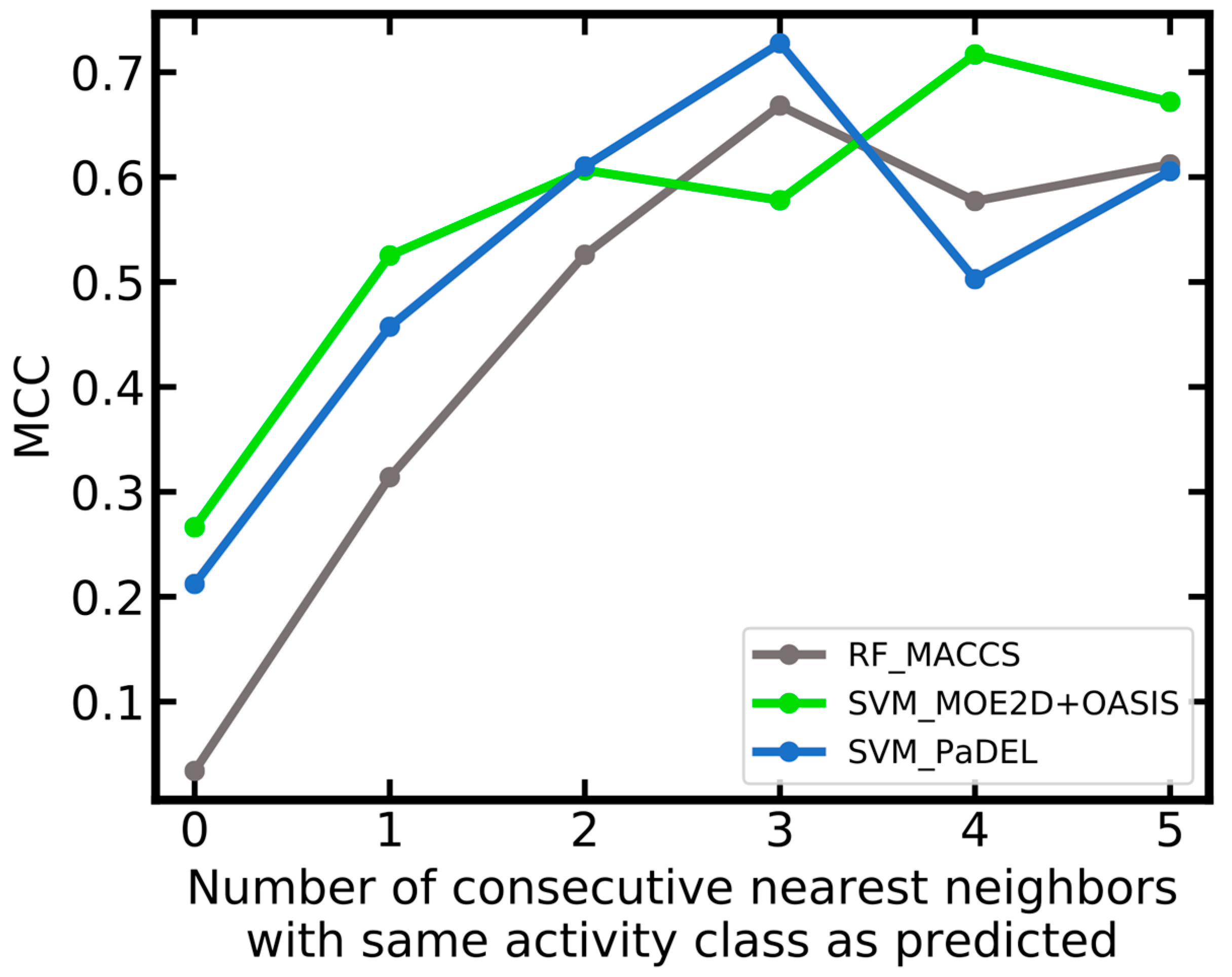
| Name | Number of Concordant Neighbors1 | Number of Compounds | ACC | MCC | AUC | CCR | Se | Sp | PPV | NPV |
|---|---|---|---|---|---|---|---|---|---|---|
| RF_MACCS | 0 | 87 | 0.33 | -0.35 | 0.32 | 0.33 | 0.19 | 0.48 | 0.26 | 0.38 |
| RF_MACCS | ≥1 | 197 | 0.89 | 0.77 | 0.97 | 0.87 | 0.81 | 0.94 | 0.89 | 0.89 |
| RF_MACCS | ≥2 | 147 | 0.96 | 0.90 | 1.00 | 0.94 | 0.89 | 0.99 | 0.98 | 0.95 |
| RF_MACCS | ≥3 | 113 | 0.99 | 0.98 | 1.00 | 0.98 | 0.97 | 1.00 | 1.00 | 0.99 |
| SVM_MOE2D+OASIS | 0 | 85 | 0.56 | 0.13 | 0.56 | 0.57 | 0.62 | 0.51 | 0.55 | 0.58 |
| SVM_MOE2D+OASIS | ≥1 | 198 | 0.84 | 0.69 | 0.94 | 0.85 | 0.92 | 0.79 | 0.72 | 0.94 |
| SVM_MOE2D+OASIS | ≥2 | 146 | 0.91 | 0.81 | 0.99 | 0.92 | 0.95 | 0.89 | 0.79 | 0.98 |
| SVM_MOE2D+OASIS | ≥3 | 115 | 0.91 | 0.80 | 0.99 | 0.92 | 0.94 | 0.90 | 0.79 | 0.97 |
| SVM_PaDEL | 0 | 86 | 0.53 | 0.07 | 0.52 | 0.54 | 0.56 | 0.51 | 0.51 | 0.56 |
| SVM_PaDEL | ≥1 | 193 | 0.83 | 0.66 | 0.92 | 0.84 | 0.87 | 0.8 | 0.72 | 0.92 |
| SVM_PaDEL | ≥2 | 147 | 0.89 | 0.78 | 0.96 | 0.91 | 0.96 | 0.86 | 0.76 | 0.98 |
| SVM_PaDEL | ≥3 | 113 | 0.90 | 0.79 | 0.97 | 0.92 | 0.97 | 0.88 | 0.76 | 0.99 |
| SVM_PaDEL+MACCS | 0 | 86 | 0.55 | 0.10 | 0.53 | 0.55 | 0.59 | 0.51 | 0.52 | 0.57 |
| SVM_PaDEL+MACCS | ≥1 | 193 | 0.84 | 0.68 | 0.91 | 0.85 | 0.89 | 0.81 | 0.73 | 0.93 |
| SVM_PaDEL+MACCS | ≥2 | 147 | 0.90 | 0.80 | 0.96 | 0.92 | 0.96 | 0.88 | 0.79 | 0.98 |
| SVM_PaDEL+MACCS | ≥3 | 113 | 0.91 | 0.81 | 0.97 | 0.93 | 0.97 | 0.89 | 0.78 | 0.99 |
| SVM_PaDEL+OASIS | 0 | 86 | 0.53 | 0.07 | 0.52 | 0.54 | 0.56 | 0.51 | 0.51 | 0.56 |
| SVM_PaDEL+OASIS | ≥1 | 193 | 0.83 | 0.67 | 0.92 | 0.84 | 0.87 | 0.81 | 0.73 | 0.92 |
| SVM_PaDEL+OASIS | ≥2 | 147 | 0.9 | 0.79 | 0.96 | 0.91 | 0.96 | 0.87 | 0.77 | 0.98 |
| SVM_PaDEL+OASIS | ≥3 | 113 | 0.91 | 0.81 | 0.97 | 0.93 | 0.97 | 0.89 | 0.78 | 0.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilm, A.; Stork, C.; Bauer, C.; Schepky, A.; Kühnl, J.; Kirchmair, J. Skin Doctor: Machine Learning Models for Skin Sensitization Prediction that Provide Estimates and Indicators of Prediction Reliability. Int. J. Mol. Sci. 2019, 20, 4833. https://doi.org/10.3390/ijms20194833
Wilm A, Stork C, Bauer C, Schepky A, Kühnl J, Kirchmair J. Skin Doctor: Machine Learning Models for Skin Sensitization Prediction that Provide Estimates and Indicators of Prediction Reliability. International Journal of Molecular Sciences. 2019; 20(19):4833. https://doi.org/10.3390/ijms20194833
Chicago/Turabian StyleWilm, Anke, Conrad Stork, Christoph Bauer, Andreas Schepky, Jochen Kühnl, and Johannes Kirchmair. 2019. "Skin Doctor: Machine Learning Models for Skin Sensitization Prediction that Provide Estimates and Indicators of Prediction Reliability" International Journal of Molecular Sciences 20, no. 19: 4833. https://doi.org/10.3390/ijms20194833
APA StyleWilm, A., Stork, C., Bauer, C., Schepky, A., Kühnl, J., & Kirchmair, J. (2019). Skin Doctor: Machine Learning Models for Skin Sensitization Prediction that Provide Estimates and Indicators of Prediction Reliability. International Journal of Molecular Sciences, 20(19), 4833. https://doi.org/10.3390/ijms20194833