New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question

 , , and
, , and 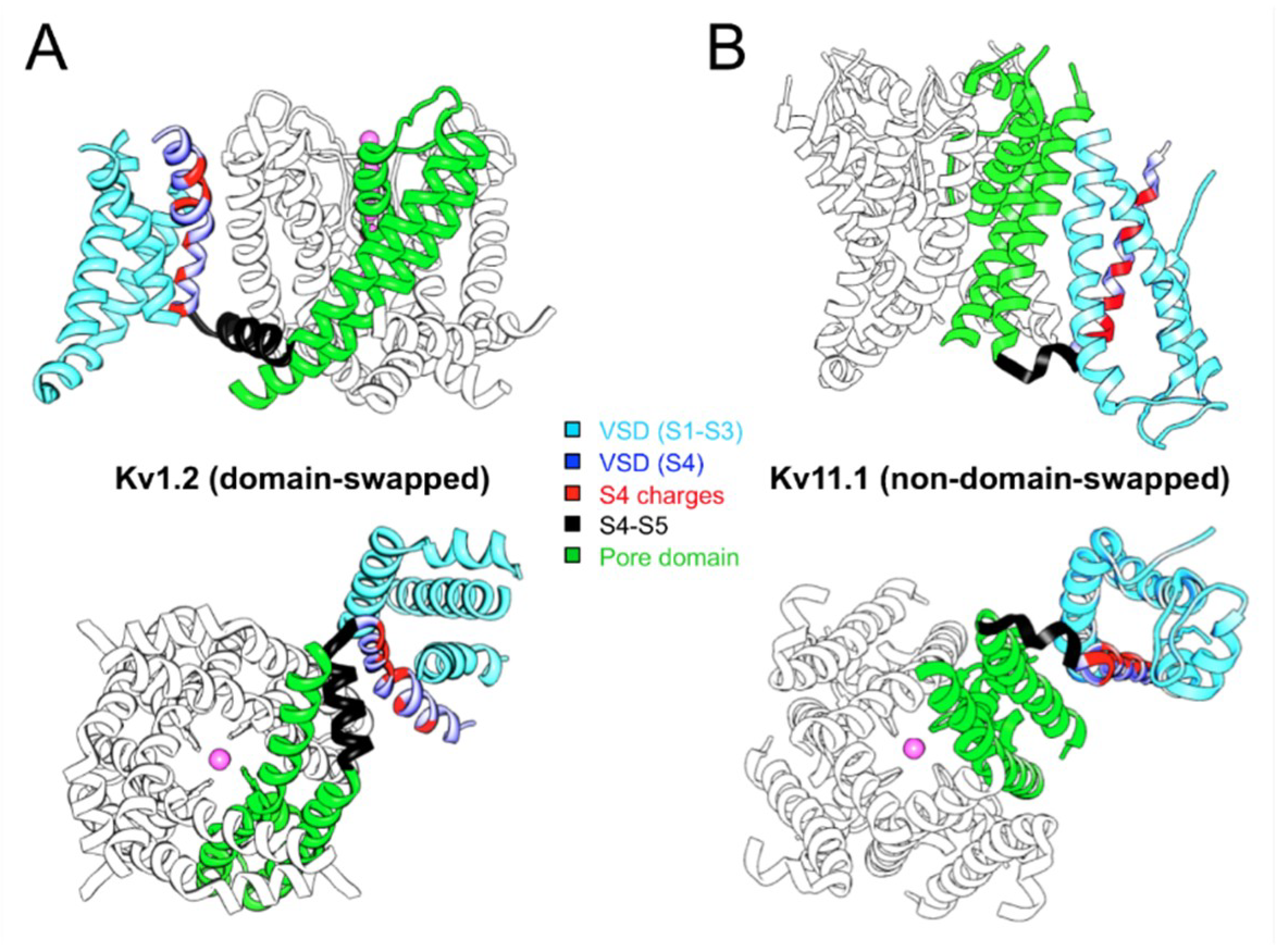{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cryo-EM: A New Catalog of Kv and Other Ion Channel Structures
3. Swapped or Non-Swapped Organization of the Kv Structures?
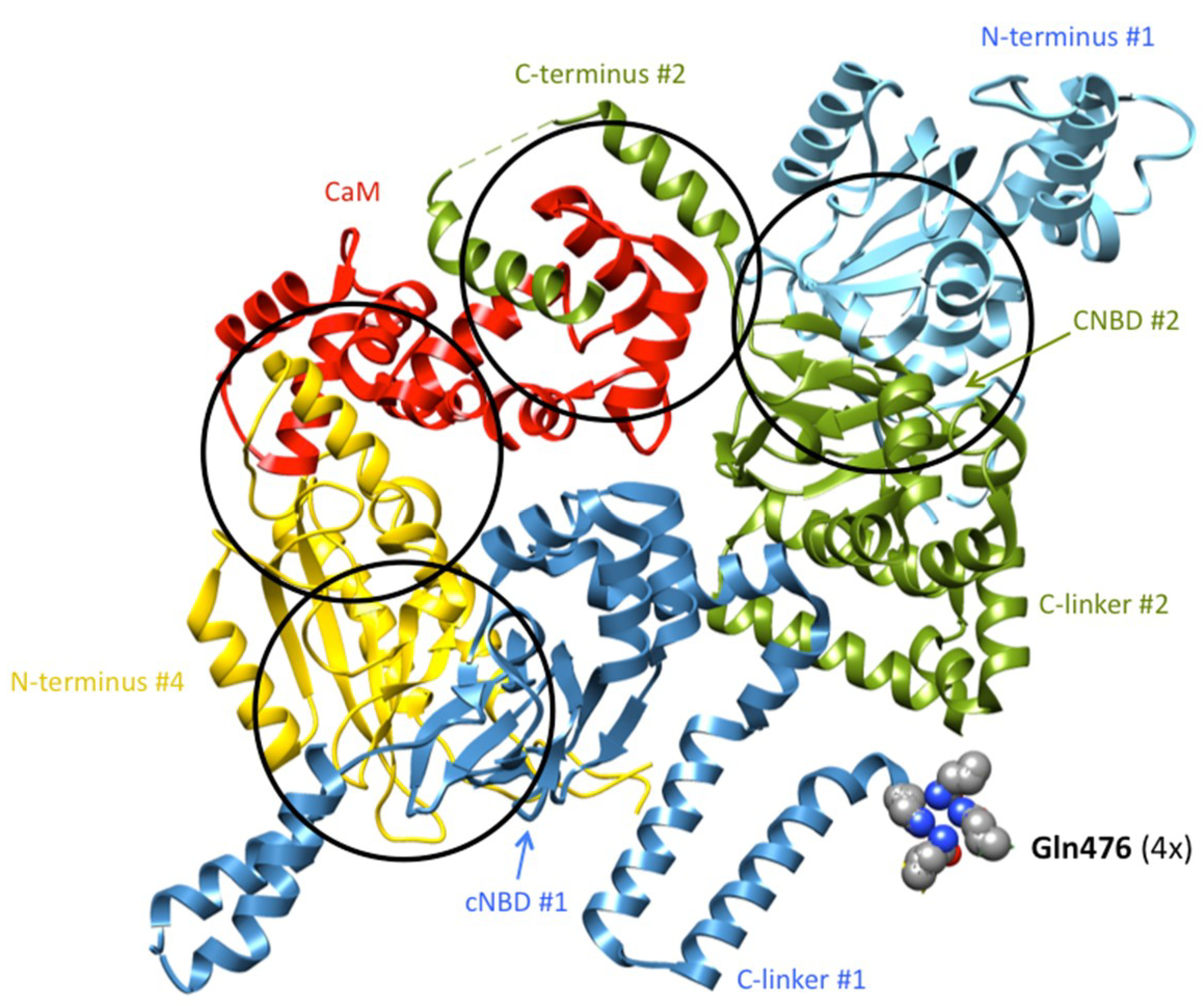
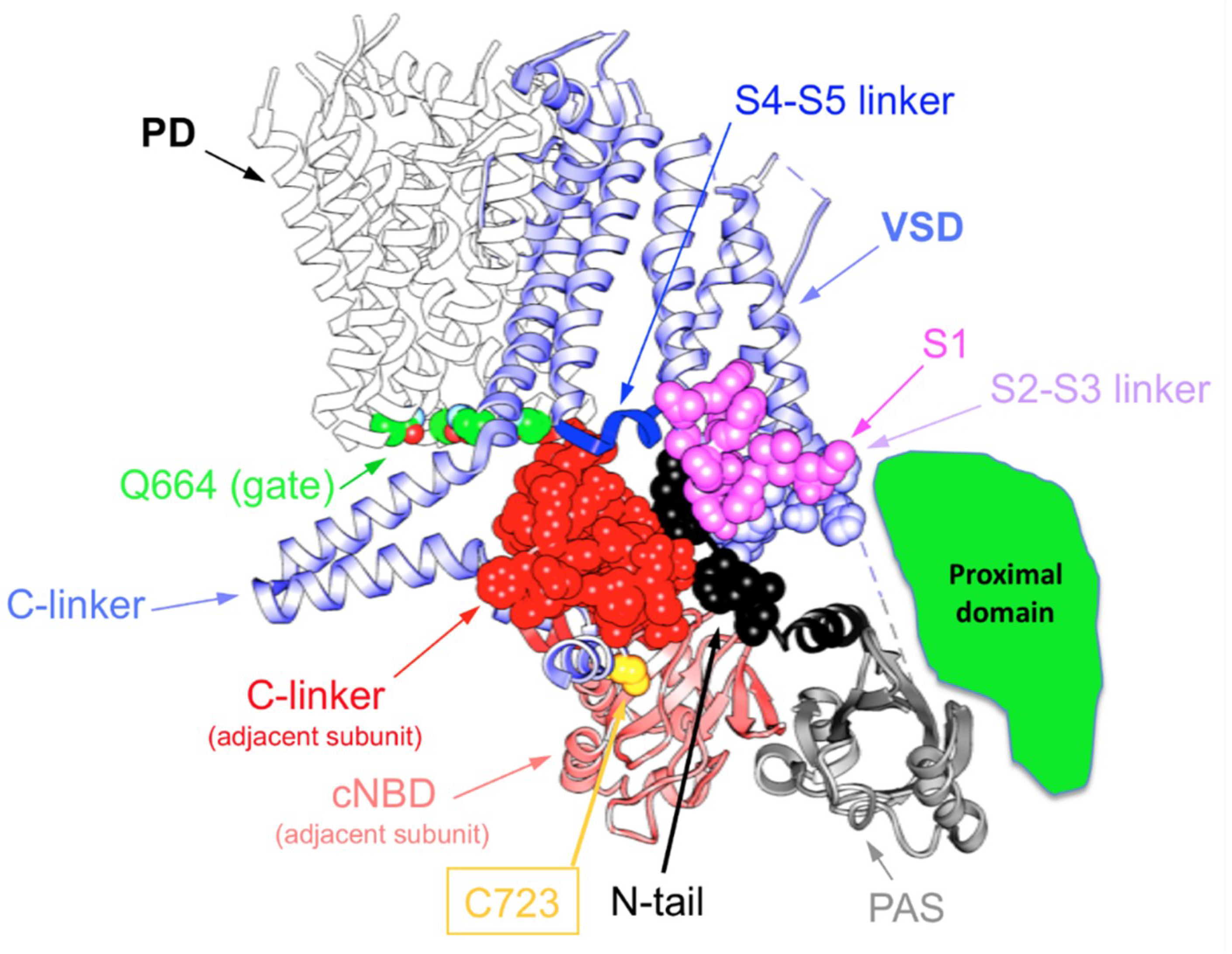
4. Allosteric Influences in the Coupling Mechanisms of Domain-Swapped Kv Channels and Their Relatives
5. Allosteric/Interactional Modulation of Coupling in KCNH Family and Other Non-Domain-Swapped Kv-Like Channels
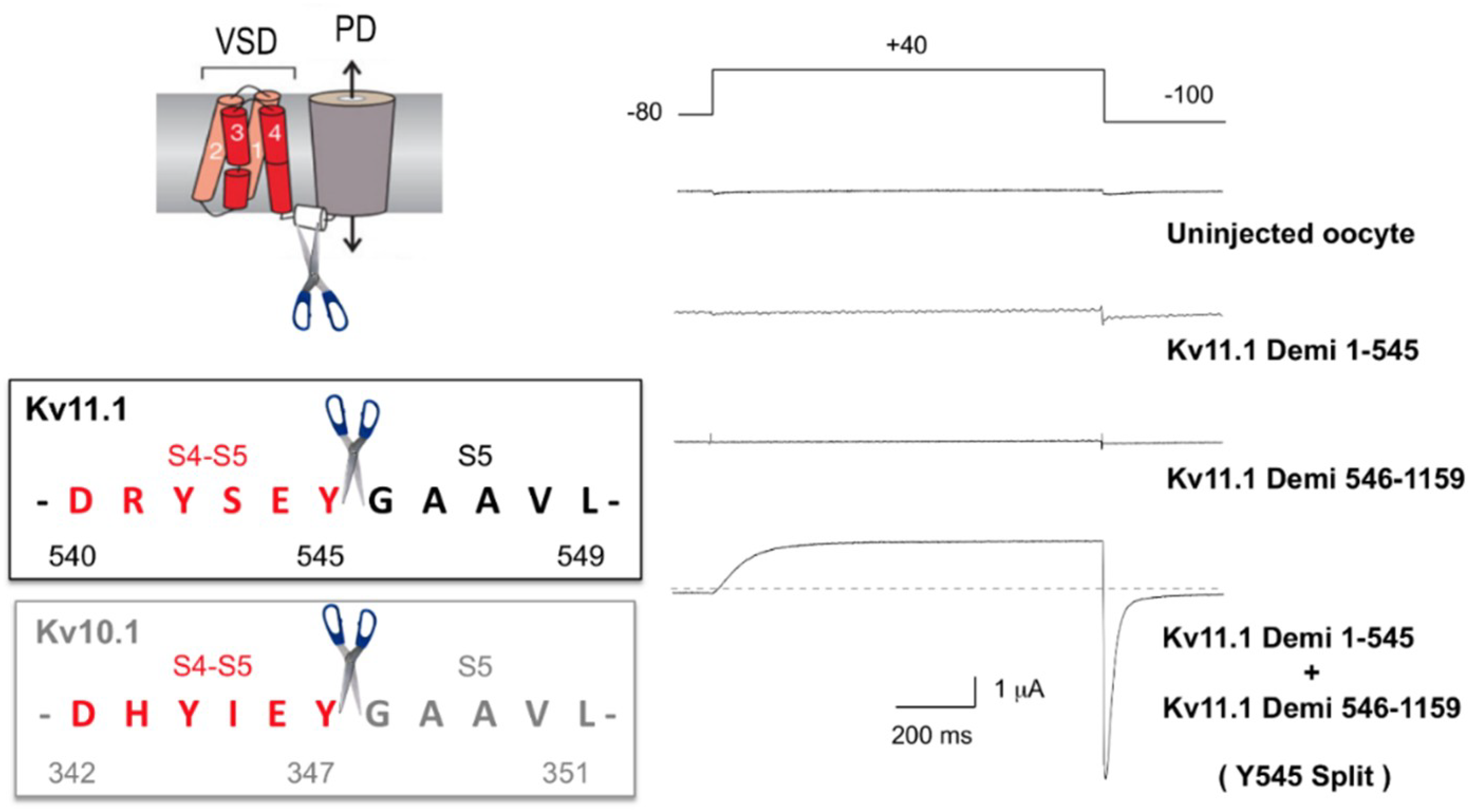
5.1. Non-Domain-Swapped Kv Channel Relatives
5.2. Evidence of Long-Range Allosteric Influences in VSD–PD Coupling of Non-Swapped Kv Channels
6. Concluding Remarks
Acknowledgements
Conflicts of Interest
References
- Miller, C. How ion channel proteins work. In Neuromodulation: The Biochemical Control of Neuronal Excitability; Kaczmarek, L.K., Levitan, I., Eds.; Oxford University Press Inc.: New York, NY, USA, 1987; pp. 39–63. ISBN 978-0195040975. [Google Scholar]
- Hering, S.; Zangerl-Plessl, E.-M.; Beyl, S.; Hohaus, A.; Andranovits, S.; Timin, E.N. Calcium channel gating. Pflüg. Arch.-Eur. J. Physiol. 2018, 470, 1291–1309. [Google Scholar] [CrossRef] [PubMed]
- Cui, J. Voltage-Dependent Gating: Novel Insights from KCNQ1 Channels. Biophys. J. 2016, 110, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.; Domínguez, P.; de la Peña, P. Cytoplasmic domains and voltage-dependent potassium channel gating. Front. Pharmacol. 2012, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stuhmer, W.; et al. International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.A. From molecule to malady. Nature 2006, 440, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Hunter, M.J.; Stewart, A.; Perozo, E.; Vandenberg, J.I. Never at rest: Insights into the conformational dynamics of ion channels from cryo-electron microscopy. J. Physiol. 2018, 596, 1107–1119. [Google Scholar] [CrossRef]
- Anderson, P.A.V.; Greenberg, R.M. Phylogeny of ion channels: Clues to structure and function. Comp. Biochem. Physiol. B 2001, 129, 17–28. [Google Scholar] [CrossRef]
- Isacoff, E.Y.; Jan, L.Y.; Minor, D.L., Jr. Conduits of life’s spark: A perspective on ion channel research since the birth of neuron. Neuron 2013, 80, 658–674. [Google Scholar] [CrossRef]
- Yu, F.H.; Catterall, W.A. The VGL-chanome: A protein super- family specialized for electrical signalling and ionic homeostasis. Sci. STKE 2004, 2004, re15. [Google Scholar] [CrossRef]
- Bichet, D.; Haass, F.A.; Jan, L.Y. Merging functional studies with structures of inward-rectifier K+ channels. Nat. Rev. Neurosci. 2003, 4, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Feliciangeli, S.; Chatelain, F.C.; Bichet, D.; Lesage, F. The family of K2P channels: Salient structural and functional properties. J. Physiol. 2015, 593, 2587–2603. [Google Scholar] [CrossRef] [PubMed]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, M.I.; Cid, L.P.; González, W.; Sepúlveda, F.V. Gating, regulation, and structure in K2P K+ channels. Mol. Pharmacol. 2016, 90, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Jegla, T.J.; Zmasek, C.M.; Batalov, S.; Nayak, S.K. Evolution of the human ion channel set. Comb. Chem. High Throughput Screen. 2009, 12, 2–23. [Google Scholar] [CrossRef] [PubMed]
- González, W.; Valdebenito, B.; Caballero, J.; Riadi, G.; Riedelsberger, J.; Gonzalo Martínez, G.; David Ramírez, D.; Zúñiga, L.; Sepúlveda, F.V.; Dreyer, I.; et al. K2P channels in plants and animals. Pflüg. Arch.-Eur. J. Physiol. 2015, 467, 1091–1104. [Google Scholar] [CrossRef]
- Tombola, F.; Ulbrich, M.H.; Isacoff, E.Y. Architecture and gating of Hv1 proton channels. J. Physiol. 2009, 587, 5325–5329. [Google Scholar] [CrossRef] [Green Version]
- DeCoursey, T.E. Voltage-gated proton channels. Comp. Physiol. 2012, 2, 1355–1385. [Google Scholar] [CrossRef]
- Castillo, K.; Pupo, A.; Baez-Nieto, D.; Contreras, G.F.; Morera, F.J.; Neely, A.; Latorre, R.; González, C. Voltage-gated proton (Hv1) channels, a singular voltage sensing domain. FEBS Lett. 2015, 589, 3471–3478. [Google Scholar] [CrossRef]
- Villalba-Galea, C.A. Voltage-controlled enzymes: The new Janus Bifrons. Front. Pharmacol. 2012, 3, 161. [Google Scholar] [CrossRef]
- Lu, Z.; Klem, A.M.; Ramu, Y. Ion conduction pore is conserved among potassium channels. Nature 2001, 413, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Syeda, R.; Santos, J.S.; Montal, M.; Bayley, H. Tetrameric assembly of KvLm K+ channels with defined numbers of voltage sensors. Proc. Natl. Acad. Sci. USA 2012, 109, 16917–16922. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, C.; Schroeder, I.; Romani, G.; Van Etten, J.L.; Thiel, G.; Moroni, A. The voltage-sensing domain of a phosphatase gates the pore of a potassium channel. J. Gen. Physiol. 2013, 141, 389–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorinczi, E.; Gomez-Posada, J.C.; de la Peña, P.; Tomczak, A.P.; Fernandez-Trillo, J.; Leipscher, U.; Stuhmer, W.; Barros, F.; Pardo, L.A. Voltage-dependent gating of KCNH potassium channels lacking a covalent link between voltage-sensing and pore domains. Nat. Commun. 2015, 6, 6672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomczak, A.P.; Fernández-Trillo, J.; Bharill, S.; Papp, F.; Panyi, G.; Stühmer, W.; Isacoff, E.Y.; Pardo, L.A. A new mechanism of voltage-dependent gating exposed by Kv10.1 channels interrupted between voltage sensor and pore. J. Gen. Physiol. 2017, 149, 577–593. [Google Scholar] [CrossRef] [PubMed]
- De la Peña, P.; Dominguez, P.; Barros, F. Gating mechanism of Kv11.1 (hERG) K+ channels without covalent connection between voltage sensor and pore domains. Pflüg. Arch.-Eur. J. Physiol. 2018, 470, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.E.; Zagotta, W.N. Insights into the molecular mechanism for hyperpolarization-dependent activation of HCN channels. Proc. Natl. Acad. Sci. USA 2018, 115, E8086–E8095. [Google Scholar] [CrossRef]
- Jian, Y.; Lee, A.; Chen, J.; Ruta, V.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a voltage-dependent K+ channel. Nature 2003, 423, 33–41. [Google Scholar] [CrossRef]
- Long, S.B.; Campbell, E.B.; MacKinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–902. [Google Scholar] [CrossRef]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef]
- Gaudet, R. Divide and conquer: High resolution structural information on TRP channel fragments. J. Gen. Physiol. 2009, 133, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Biggin, P.C.; Roosild, T.; Choe, S. Potassium channel structure: Domain by domain. Curr. Opin. Struct. Biol. 2000, 10, 456–461. [Google Scholar] [CrossRef]
- Higgins, M.K.; Lea, S.M. On the state of crystallography at the dawn of the electron microscopy revolution. Curr. Opin. Struct. Biol. 2017, 46, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Venien-Bryan, C.; Li, Z.; Vuillard, L.; Boutin, J.A. Cryo-electron microscopy and X-ray crystallography: Complementary approaches to structural biology and drug Discovery. Acta Crystallogr. F 2017, 73, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Wang, J.-W. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32–39. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Single particle electron cryo-microscopy of a mammalian ion cannel. Curr. Opin. Struct. Biol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Grigorieff, N.; Penczek, P.A.; Walz, T. A primer to single-particle cryo-Electron microscopy. Cell 2015, 161, 438–449. [Google Scholar] [CrossRef]
- Cheng, Y. Single-particle cryo-EM at crystallographic resolution. Cell 2015, 161, 450–457. [Google Scholar] [CrossRef]
- Cheng, Y.; Glaeser, R.M.; Nogales, E. How cryo-EM became so hot. Cell 2017, 171, 1229–1231. [Google Scholar] [CrossRef]
- Earl, L.A.; Falconieri, V.; Milne, J.L.S.; Subramaniam, S. Cryo-EM: Beyond the microscope. Curr. Opin. Struct. Biol. 2017, 46, 71–78. [Google Scholar] [CrossRef]
- Mio, K.; Sato, C. Lipid environment of membrane proteins in cryo-EM based structural analysis. Biophys. Rev. 2017, 10, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, H.E.; Ignatiou, A.; Clare, D.K.; Orlova, E.V. Structural study of heterogeneous biological samples by cryoelectron microscopy and image processing. BioMed Res. Int. 2017, 2017, 1032432. [Google Scholar] [CrossRef]
- Long, S.B.; Campbell, E.B.; MacKinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef]
- Sun, J.; MacKinnon, R. Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 2017, 169, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Yan, Z.; Li, Z.; Qian, X.; Lu, S.; Dong, M.; Zhou, Q.; Yan, N. Structure of the voltage-gated calcium channel Cav1.1 at 3.6 Å resolution. Nature 2016, 537, 191–196. [Google Scholar] [CrossRef]
- Shen, H.; Zhou, Q.; Pan, X.; Li, Z.; Wu, J.; Yan, N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355, eaal4326. [Google Scholar] [CrossRef]
- Sula, A.; Booker, J.; Ng, L.C.T.; Naylor, C.E.; DeCaen, P.G.; Wallace, B.A. The complete structure of an activated open sodium channel. Nat. Commun. 2017, 8, 14205. [Google Scholar] [CrossRef] [Green Version]
- Madej, M.G.; Ziegler, C.M. Dawning of a new era in TRP cannel structural biology by cryo-electron microscopy. Pflüg. Arch.-Eur. J. Physiol. 2018, 470, 213–225. [Google Scholar] [CrossRef]
- Whicher, J.R.; MacKinnon, R. Structure of the voltage-gated K+ channel eag1 reveals an alternative voltage sensing mechanism. Science 2016, 353, 664–669. [Google Scholar] [CrossRef]
- Wang, W.; MacKinnon, R. Cryo-EM structure of the open human ether-á-go-go-related K+ channel hERG. Cell 2017, 169, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Hite, R.K.; Tao, X.; MacKinnon, R. Structural basis for gating the high-conductance Ca2+-activated K+ channel. Nature 2017, 527, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Hite, R.K.; MacKinnon, R. Cryo-EM structure of the open high-conductance Ca2+-activated K+ channel. Nature 2017, 541, 46–51. [Google Scholar] [CrossRef]
- Lee, C.-H.; MacKinnon, R. Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 2018, 360, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Hite, R.K.; Yuan, P.; Li, Z.; Hsuing, Y.; Walz, T.; MacKinnon, R. Cryo-electron microscopy structure of the Slo2.2 Na+-activated K+ channel. Nature 2015, 527, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; MacKinnon, R. Structures of the human HCN1 hyperpolarization-activated channel. Cell 2017, 168, 111–120. [Google Scholar] [CrossRef]
- James, Z.M.; Borst, A.J.; Haitin, Y.; Frenz, B.; DiMaio, F.; Zagotta, W.N.; Veesler, D. CryoEM structure of a prokaryotic cyclic nucleotide-gated ion channel. Proc. Natl. Acad. Sci. USA 2017, 114, 4430–4435. [Google Scholar] [CrossRef]
- Li, M.; Zhou, X.; Wang, S.; Michailidisong, Y.; Su, D.; Li, H.; Li, X.; Yang, J. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature 2017, 542, 60–65. [Google Scholar] [CrossRef]
- Holmgren, M.; Shin, K.S.; Yellen, G. The activation gate of voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron 1998, 21, 617–621. [Google Scholar] [CrossRef]
- Del Camino, D.; Holmgren, M.; Yellen, G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature 2000, 403, 321–325. [Google Scholar] [CrossRef]
- Mitcheson, J.S.; Chen, J.; Sanguinetti, M.C. Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J. Gen. Physiol. 2000, 115, 229–240. [Google Scholar] [CrossRef] [PubMed]
- del Camino, D.; Yellen, G. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron 2001, 32, 649–656. [Google Scholar] [CrossRef]
- Witchel, H.J. The low-potency, voltage-dependent HERG blocker propafenone-molecular determinants and drug trapping. Mol. Pharmacol. 2004, 66, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.M.; del Camino, D.; Dekker, J.P.; Yellen, G. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature 2004, 428, 864–868. [Google Scholar] [CrossRef] [PubMed]
- del Camino, D.; Kanevsky, M.; Yellen, G. Status of the intracellular gate in the activated-not-open state of Shaker K+ channels. J. Gen. Physiol. 2005, 126, 419–428. [Google Scholar] [CrossRef]
- Swartz, K.J. Structure and anticipatory movements of the S6 gate in Kv channels. J. Gen. Physiol. 2005, 126, 413–417. [Google Scholar] [CrossRef]
- Boulet, I.R.; Labro, A.J.; Raes, A.L.; Snyders, D.J. Role of the S6 C-terminus in KCNQ1 channel gating. J. Physiol. 2007, 585, 325–337. [Google Scholar] [CrossRef] [Green Version]
- Wynia-Smith, S.L.; Gillian-Daniel, A.L.; Satyshur, K.A.; Robertson, G.A. hERG gating microdomains defined by S6 mutagenesis and molecular modeling. J. Gen. Physiol. 2008, 132, 507–520. [Google Scholar] [CrossRef]
- Thouta, S.; Sokolov, S.; Abe, Y.; Clark, S.J.; Cheng, Y.M.; Claydon, T.W. Proline scan of the HERG channel S6 helix reveals the location of the intracellular pore gate. Biophys. J. 2014, 106, 1057–1069. [Google Scholar] [CrossRef]
- Cox, D.H.; Hoshi, T. Where’s the gate? Gating in the deep pore of the BKCa channel. J. Gen. Physiol. 2011, 138, 133–136. [Google Scholar] [CrossRef]
- Labro, A.J.; Raes, A.L.; Grottesi, A.; Van Hoorick, D.; Sansom, M.S.; Snyders, D.J. Kv channel gating requires a compatible S4–S5 linker and bottom part of S6, constrained by non-interacting residues. J. Gen. Physiol. 2008, 132, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, V.; Pongs, O. Coupling of voltage sensors to the channel pore: A comparative view. Front. Pharmacol. 2012, 3, 145. [Google Scholar] [CrossRef] [PubMed]
- Blunck, R.; Batulan, Z. Mechanism of electromechanical coupling in voltage-gated potassium channels. Front. Pharmacol. 2012, 3, 166. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.O.; Jogini, V.; Borhani, D.W.; Leffler, A.E.; Dror, R.O.; Shaw, D.E. Mechanism of voltage gating in potassium channels. Science 2012, 336, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Chowdury, S.; Haehnel, B.M.; Chanda, B. Interfacial gating triad is crucial for electromechanical transduction in voltage-activated potassium channels. J. Gen. Physiol. 2014, 144, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Kalstrup, T.; Blunck, R. S4–S5 linker movement during activation and inactivation in voltage-gated K+ channels. Proc. Natl. Acad. Sci. USA 2018, 115, E6751–E6759. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, H.; Cui, J.; Lingle, C.J. Threading the biophysics of mammalian Slo1 channels onto structures of an invertebrate Slo1 channel. J. Gen. Physiol. 2017, 149, 985–1007. [Google Scholar] [CrossRef] [Green Version]
- James, Z.M.; Zagotta, W.N. Structural insights into the mechanisms of CNBD channel function. J. Gen. Physiol. 2018, 150, 225–244. [Google Scholar] [CrossRef]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.-Y. Structure of the cold- and mentol-sensing ion channel TRPM8. Science 2018, 359, 237–241. [Google Scholar] [CrossRef]
- Singh, A.K.; Saotome, K.; Sobolevsky, A.I. Swapping of transmembrane domains in the epithelial calcium channel TRPV6. Sci. Rep. 2017, 7, 10669. [Google Scholar] [CrossRef]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Franulic, I.; Poblete, H.; Miño-Galaz, G.; González, C.; Latorre, R. Allosterism and structure in thermally activated transient receptor potential channels. Annu. Rev. Biophys. 2016, 45, 371–398. [Google Scholar] [CrossRef]
- Montell, C. The TRP superfamily of cation channels. Sci. STKE 2005, 2005, re3. [Google Scholar] [CrossRef]
- Li, H. TRP Channel Classification. Adv. Exp. Med. Biol. 2017, 976, 1–8. [Google Scholar] [CrossRef]
- Cheng, W.; Sun, C.; Zheng, J. Heteromerization of TRP channel subunits: Extending functional Diversity. Protein Cell 2010, 1, 802–810. [Google Scholar] [CrossRef]
- Voets, T.; Owsianik, G.; Janssens, A.; Talavera, K.; Nilius, B. TRPM8 voltage sensor mutants reveal a mechanism for integrating thermal and chemicals stimuli. Nat. Chem. Biol. 2007, 3, 174–182. [Google Scholar] [CrossRef]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Kühn, F.J.P.; Winking, M.; Kühn, C.; Hoffmann, D.C.; Lückhoff, A. Surface expression and channel function of TRPM8 are cooperatively controlled by transmembrane segments S3 and S4. Pflüg. Arch.-Eur. J. Physiol. 2013, 465, 1599–1610. [Google Scholar] [CrossRef]
- Guo, J.; She, J.; Zeng, W.; Chen, Q.; Bai, X.-C.; Jiang, Y. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 2017, 552, 205–209. [Google Scholar] [CrossRef]
- Winkler, P.A.; Huang, Y.; Sun, W.; Du, J.; Lü, W. Electro cryo-microscopy structure of a human TRPM4 channel. Nature 2017, 552, 200–204. [Google Scholar] [CrossRef]
- Latorre, R.; Brauchi, S.; Orta, G.; Zaelzer, C.; Vargas, G. ThermoTRP channels as modular proteins with allosteric gating. Cell Calcium 2007, 42, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Latorre, R.; Zaelzer, C.; Brauchi, S. Structure-functional intimacies of transient receptor potential channels. Q. Rev. Biophys. 2009, 42, 201–246. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Talavera, K.; Owsianik, G.; Prenen, J.; Droogmans, G.; Voets, T. Gating of TRP channels: A voltage connection? J. Physiol. 2005, 567, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, L.; Wang, H.; Zheng, W.; Philipp, S.E.; Hidalgo, P.; Cavalié, A.; Chen, X.-Z.; Beck, A.; Flockerzi, V. The S4–S5 linker–gearbox of TRP channel gating. Cell Calcium 2017, 67, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Saotome, K.; McGoldrick, L.L.; Sobolevsky, A.I. Structural bases of TRP channel TRPV6 allosteric modulation by 2-APB. Nat. Commun. 2018, 9, 2465. [Google Scholar] [CrossRef] [PubMed]
- Taberner, F.J.; López-Córdoba, A.; Fernández-Ballester, G.; Korchev, Y.; Ferrer-Montiel, A. The region adjacent to the C-end of the inner gate in transient receptor potential melastatin 8 (TRPM8) channels plays a central role in allosteric channel activation. J. Biol. Chem. 2014, 289, 28579–28594. [Google Scholar] [CrossRef] [PubMed]
- Choveau, F.S.; Rodriguez, N.; Ali, F.A.; Labro, A.J.; Rose, T.; Dahimene, S.; Boudin, H.; Le Henaff, C.; Escande, D.; Snyders, D.J.; et al. KCNQ1 channels voltage dependency through a voltage-dependent binding of the S4–S5 linker to the pore domain. J. Biol. Chem. 2011, 286, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Choveau, F.S.; Abderemane-Ali, F.; Coyan, F.C.; Es-Salah-Lamoureux, Z.; Baró, I.; Loussouarn, G. Opposite Effects of the S4–S5 Linker and PIP2 on Voltage-Gated Channel Function: KCNQ1/KCNE1 and Other Channels. Front. Pharmacol. 2012, 3, 125. [Google Scholar] [CrossRef] [PubMed]
- Chowdury, S.; Chanda, B. Thermodynamics of electromechanical coupling in voltage-gated ion channels. J. Gen. Physiol. 2012, 140, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haitin, Y.; Attali, B. The C-terminus of Kv7 channels: A multifunctional module. J. Physiol. 2008, 586, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Maljevic, S.; Wuttke, T.V.; Seebohm, G.; Lerche, H. Kav7 channelopathies. Pflüg. Arch.-Eur. J. Physiol. 2010, 460, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Zaydman, M.A.; Cui, J. PIP2 regulation of KCNQ channels: Biophysical and molecular mechanisms for lipid modulation of voltage-dependent gating. Front. Physiol. 2014, 5, 195. [Google Scholar] [CrossRef] [PubMed]
- Ledwell, J.R.; Aldrich, R.W. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 1999, 113, 389–414. [Google Scholar] [CrossRef] [PubMed]
- Pathak, M.; Kurtz, L.; Tombola, F.; Isacoff, E. The cooperative voltage sensor motion that gates a potassium channel. J. Gen. Physiol. 2005, 125, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Soler-Llavina, G.J.; Chang, T.H.; Swartz, K.J. Functional interactions at the interface between voltage-sensing and pore domains in the Shaker Kv channel. Neuron 2006, 52, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Mariño, A.I.; Harpole, T.J.; Oelstrom, K.; Delemotte, L.; Chanda, B. Gating interaction maps reveal a noncanonical electromechanical coupling mode in the Shaker K+ channel. Nat. Struct. Mol. Biol. 2018, 25, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Minor, D.L., Jr.; Lin, Y.-F.; Mobley, B.C.; Avelar, A.; Jan, Y.N.; Jan, L.Y.; Berger, J.M. The Polar T1 Interface Is Linked to Conformational Changes that Open the Voltage-Gated Potassium Channel. Cell 2000, 102, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Cushman, S.J.; Nanao, M.H.; Jahng, A.W.; DeRubeis, D.; Choe, S.; Pfaffinger, P.J. Voltage dependent activation of potassium channels is coupled to T1 domain structure. Nat. Struct. Biol. 2000, 7, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Minor, D.L., Jr. Potassium channels: Life in the post-structural world. Curr. Opin. Struct. Biol. 2001, 11, 408–414. [Google Scholar] [CrossRef]
- Wei, A.D.; Gutman, G.A.; Aldrich, R.; Chandy, K.G.; Grissmer, S.; Wulff, H. International Union of Pharmacology. LII. Nomenclature and Molecular Relationships of Calcium-Activated Potassium Channels. Pharmacol. Rev. 2005, 57, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Giráldez, T.; Rothberg, B.S. Understanding the conformational motions of RCK gating rings. J. Gen. Physiol. 2017, 149, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giráldez, T. Physiological Roles and Therapeutic Potential of Ca2+ Activated Potassium Channels in the Nervous System. Front. Mol. Neurosci. 2018, 11, 258. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, T.; Pantazis, A.; Olcese, R. Transduction of voltage and Ca2+ signals by Slo1 BK channels. Physiology 2013, 28, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Hite, R.K.; MacKinnon, R. Structural titration of Slo2.2, a Na+-dependent K+ channel. Cell 2017, 168, 390–399. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, G.; Cui, J. BK channels: Multiple sensors, one activation gate. Front. Physiol. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Latorre, R.; Castillo, K.; Carrasquel-Ursulaez, W.; Sepúlveda, R.V.; González-Nilo, F.; González, C.; Alvarez, O. Molecular determinants of BK channel functional diversity and functioning. Physiol. Rev. 2017, 97, 39–87. [Google Scholar] [CrossRef]
- Horrigan, F.T.; Aldrich, R.W. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 2002, 120, 267–305. [Google Scholar] [CrossRef]
- Jia, Z.; Yazdani, M.; Zhang, G.; Cui, J.; Chen, J. Hydrophobic gating in BK channels. Nat. Commun. 2018, 9, 3408. [Google Scholar] [CrossRef]
- Hofmann, F.; Biel, M.; Kaupp, U.B. International Union of Pharmacology. LI. Nomenclature and structure-function relationships of cyclic nucleotide-regulated channels. Pharmacol. Rev. 2005, 57, 455–462. [Google Scholar] [CrossRef]
- Marchesi, A.; Gao, X.; Adaixo, R.; Rheinberger, J.; Stahlberg, H.; Nimigean, C.; Scheuring, S. An iris diaphragm mechanism to gate a cyclic nucleotide-gated ion channel. Nat. Commun. 2018, 9, 3978. [Google Scholar] [CrossRef]
- Mazzolini, M.; Arcangeletti, M.; Marchesi, A.; Napolitano, L.M.R.; Grosa, D.; Maity, S.; Anselmi, C.; Torre, V. The gating mechanism in cyclic nucleotide-gated ion channels. Sci. Rep. 2018, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Männiko, R.; Elinder, F.; Larsson, H.P. Voltage-sensing mechanism is conserved among ion channels gated by opposite voltages. Nature 2002, 419, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Vemana, S.; Pandey, S.; Larsson, H.P. S4 movement in a mammalian HCN channel. J. Gen. Physiol. 2004, 123, 21–32. [Google Scholar] [CrossRef]
- Decher, N.; Chen, J.; Sanguinetti, M.C. Voltage-dependent gating of hyperpolarization-activated, cyclic nucleotide-gated pacemaker channels: Molecular coupling between the S4–S5 and C-linkers. J. Biol. Chem. 2004, 279, 13859–13865. [Google Scholar] [CrossRef]
- Prole, D.L.; Yellen, G. Reversal of HCN channel voltage dependence via bridging of the S4–S5 linker and Post-S6. J. Gen. Physiol. 2006, 128, 273–282. [Google Scholar] [CrossRef]
- Kwan, D.C.; Prole, D.L.; Yellen, G. Structural changes during HCN channel gating defined by high affinity metal bridges. J. Gen. Physiol. 2012, 140, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainger, B.J.; DeGennaro, M.; Santoro, B.; Siegelbaum, S.A.; Tibbs, G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 2001, 411, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Aldrich, R.W. Tissue-specific N terminus of the HCN4 channel affects channel activation. J. Biol. Chem. 2011, 286, 14209–14214. [Google Scholar] [CrossRef]
- Lu, Z.; Klem, A.M.; Ramu, Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 2002, 120, 663–676. [Google Scholar] [CrossRef]
- Hardman, R.M.; Stansfeld, P.J.; Dalibalta, S.; Sutcliffe, M.J.; Mitcheson, J.S. Activation gating of hERG potassium channels: S6 glycines are not required as gating hinges. J. Biol. Chem. 2007, 282, 31972–31981. [Google Scholar] [CrossRef] [PubMed]
- Perissinotti, L.L.; De Biase, P.M.; Guo, J.; Yang, P.-C.; Lee, M.C.; Clancy, C.E.; Duff, H.J.; Noskov, S.Y. Determinants of isoform-specific gating kinetics of hERG1 channel: Combined experimental and simulation study. Front. Physiol. 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, J.I.; Torrres, A.M.; Campbell, T.J.; Kuchel, P.W. The HERG K+ channel: Progress in understanding the molecular basis of its unusual gating kinetics. Eur. Biophys. J. 2004, 33, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gustina, A.S.; Trudeau, M.C. HERG potassium channel regulation by the N-terminal eag domain. Cell. Signal. 2012, 24, 1592–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K+ channels: Structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.M.; Claydon, T.W. Voltage-dependent gating of hERG potassium channels. Front. Pharmacol. 2012, 3, 83. [Google Scholar] [CrossRef] [PubMed]
- Morais-Cabral, J.H.; Robertson, G.A. The enigmatic cytoplasmic regions of KCNH channels. J. Mol. Biol. 2015, 427, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Perry, M.D.; Ng, C.-A.; Mann, S.A.; Sadrieh, A.; Imtiaz, M.; Hill, A.P.; Vandenberg, J.I. Getting to the heart of hERG K+ channel gating. J. Physiol. 2015, 593, 2575–2585. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perozo, E.; Allen, T.W. Towards a structural view of drug binding to hERG K+ channels. Trends Pharmacol. Sci. 2017, 38, 899–907. [Google Scholar] [CrossRef]
- de la Peña, P.; Alonso-Ron, C.; Machín, A.; Fernández-Trillo, J.; Carretero, L.; Domínguez, P.; Barros, F. Demonstration of physical proximity between the amino terminus and the S4–S5 linker of the hERG potassium channel. J. Biol. Chem. 2011, 286, 19065–19075. [Google Scholar] [CrossRef]
- de la Peña, P.; Machín, A.; Fernández-Trillo, J.; Domínguez, P.; Barros, F. Mapping of interactions between the amino and carboxy termini and the channel core in hERG K+ channels. Biochem. J. 2013, 451, 463–474. [Google Scholar] [CrossRef]
- de la Peña, P.; Machín, A.; Fernández-Trillo, J.; Domínguez, P.; Barros, F. Interactions between the N-terminal tail and the gating machinery of hERG K+ channels both in closed and open/inactive states. Pflüg. Arch.-Eur. J. Physiol. 2015, 467, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Trillo, J.; Barros, F.; Machín, A.; Carretero, L.; Domínguez, P.; de la Peña, P. Molecular determinants of interactions between the N-terminal domain and the transmembrane core that modulate hERG K+ channel gating. PLoS ONE 2011, 6, e24674. [Google Scholar] [CrossRef] [PubMed]
- de la Peña, P.; Domínguez, P.; Barros, F. Functional characterization of Kv11.1 (hERG) potassium channels split in the voltage-sensing domain. Pflüg. Arch.-Eur. J. Physiol. 2018, 470, 1069–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, F.; Domínguez, P.; de la Peña, P. Relative positioning of Kv11.1 (hERG) K+ channel cytoplasmic domain-located fluorescent tags toward the plasma membrane. Sci. Rep. 2018, 8, 15494. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Goldschen-Ohm, M.P.; Morais-Cabral, J.H.; Chanda, B.; Robertson, G.A. The intrinsically liganded cyclic nucleotide-binding homology domain promotes KCNH channel activation. J. Gen. Physiol. 2017, 149, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Blunck, R. The isolated voltage sensing domain of the Shaker potassium channel forms a voltage-gated cation channel. eLife 2016, 5, e18130. [Google Scholar] [CrossRef]
- Wang, J.; Trudeau, M.C.; Zappia, A.M.; Robertson, G.A. Regulation of deactivation by an amino terminal domain in human ether-á-go-go-related gene potassium channels. J. Gen. Physiol. 1998, 112, 637–647. [Google Scholar] [CrossRef]
- Morais Cabral, J.H.; Lee, A.; Cohen, S.L.; Chait, B.; Li, M.; MacKinnon, R. Crystal structure and functional analysis of the HERG potassium channel N terminus: A eukaryotic PAS domain. Cell 1998, 95, 649–655. [Google Scholar] [CrossRef]
- Wang, J.; Myers, C.D.; Robertson, G.A. Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K+ channels. J. Gen. Physiol. 2000, 115, 749–758. [Google Scholar] [CrossRef]
- Muskett, F.W.; Thouta, S.; Thomson, S.J.; Bowen, A.; Stansfeld, P.J.; Mitcheson, J.S. Mechanistic insight into human ether-á-go-go-related gene (hERG) K+ channel deactivation gating from the solution structure of the EAG domain. J. Biol. Chem. 2011, 286, 6184–6191. [Google Scholar] [CrossRef]
- Ng, C.A.; Hunter, M.J.; Perry, M.D.; Mobli, M.; Ke, Y.; Kuchel, P.W.; King, G.F.; Stock, D.; Vandenberg, J.I. The N-terminal tail of hERG contains an amphipathic a-helix that regulates channel deactivation. PLoS ONE 2011, 6, e16191. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; James, Z.M.; Zagotta, W.N. Dynamic rearrangement of the intrinsic ligand regulates KCNH potassium channels. J. Gen. Physiol. 2018, 150, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C.; Nomura, H.; Tsuda, T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature 2004, 432, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, C.R.D. Ion pump in the movies. Nature 2004, 432, 286–287. [Google Scholar] [CrossRef] [PubMed]
- Moller, J.V.; Nissen, P.; Sorensen, T.L.-M.; le Maire, M. Transport mechanism of the sarcoplasmic reticulum Ca2+-ATPase pump. Curr. Opin. Struct. Biol. 2005, 15, 387–393. [Google Scholar] [CrossRef]
- Olesen, C.; Picard, M.; Winther, A.-M.L.; Gyrup, C.; Morth, J.P.; Oxvig, C.; Moller, J.V.; Nissen, P. The structural basis of calcium transport by the calcium pump. Nature 2007, 450, 1036–1042. [Google Scholar] [CrossRef]
- Toyoshima, C. Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch. Biochem. Biophys. 2008, 476, 3–11. [Google Scholar] [CrossRef]
- Moller, J.V.; Olesen, C.; Winther, A.-M.L.; Nissen, P. What Can Be Learned About the Function of a Single Protein from Its Various X-Ray Structures: The Example of the Sarcoplasmic Calcium Pump. Methods Mol. Biol. 2010, 654, 119–140. [Google Scholar] [CrossRef]
- Rubinstein, J.L. Cryo-EM Captures the Dynamics of Ion Channel Opening. Cell 2017, 168, 341–343. [Google Scholar] [CrossRef]
- Wang, L.; Sigworth, F.J. Structure of the BK potassium channel in a lipid membrane from electron cryomicroscopy. Nature 2009, 461, 292–295. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Sigworth, F.J. Automatic cryo-EM particle selection for membrane proteins in spherical liposomes. J. Struct. Biol. 2014, 185, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L. Random spherically constrained single-particle (RSC) method to study voltage-gated ion channels. Methods Mol. Biol. 2018, 1684, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Beyl, S.; Depil, K.; Hohaus, A.; Stary-Weinzinger, A.; Linder, T.; Timin, E.; Hering, S. Neutralisation of a single voltage sensor affects gating determinants in all four pore-forming S6 segments of Ca(V)1.2: A cooperative gating model. Pflüg. Arch.-Eur. J. Physiol. 2012, 464, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, C.; Rohaim, A.; Shaya, D.; Findeisen, F.; Stein, R.A.; Nurva, S.R.; Mishra, S.; Mchaourab, H.S.; Minor, D.L., Jr. Unfolding of a temperature-sensitive domain controls voltage-gated channel activation. Cell 2016, 164, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhou, Q.; Wang, L.; Wu, J.; Zhao, Y.; Huang, G.; Peng, W.; Shen, H.; Lei, J.; Yan, N. Structure of the Nav1.4-β1 complex from electric eel. Cell 2017, 170, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barros, F.; Pardo, L.A.; Domínguez, P.; Sierra, L.M.; De la Peña, P. New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question. Int. J. Mol. Sci. 2019, 20, 248. https://doi.org/10.3390/ijms20020248
Barros F, Pardo LA, Domínguez P, Sierra LM, De la Peña P. New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question. International Journal of Molecular Sciences. 2019; 20(2):248. https://doi.org/10.3390/ijms20020248
Chicago/Turabian StyleBarros, Francisco, Luis A. Pardo, Pedro Domínguez, Luisa Maria Sierra, and Pilar De la Peña. 2019. "New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question" International Journal of Molecular Sciences 20, no. 2: 248. https://doi.org/10.3390/ijms20020248