Ginsenoside Rb1 Blocks Ritonavir-Induced Oxidative Stress and eNOS Downregulation through Activation of Estrogen Receptor-Beta and Upregulation of SOD in Human Endothelial Cells

Abstract
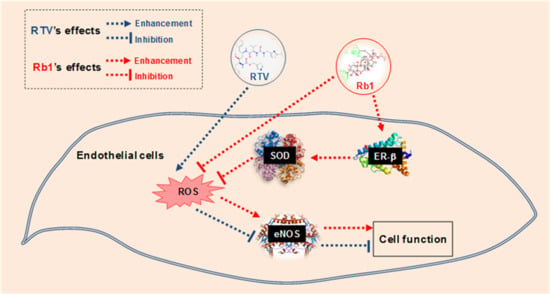
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
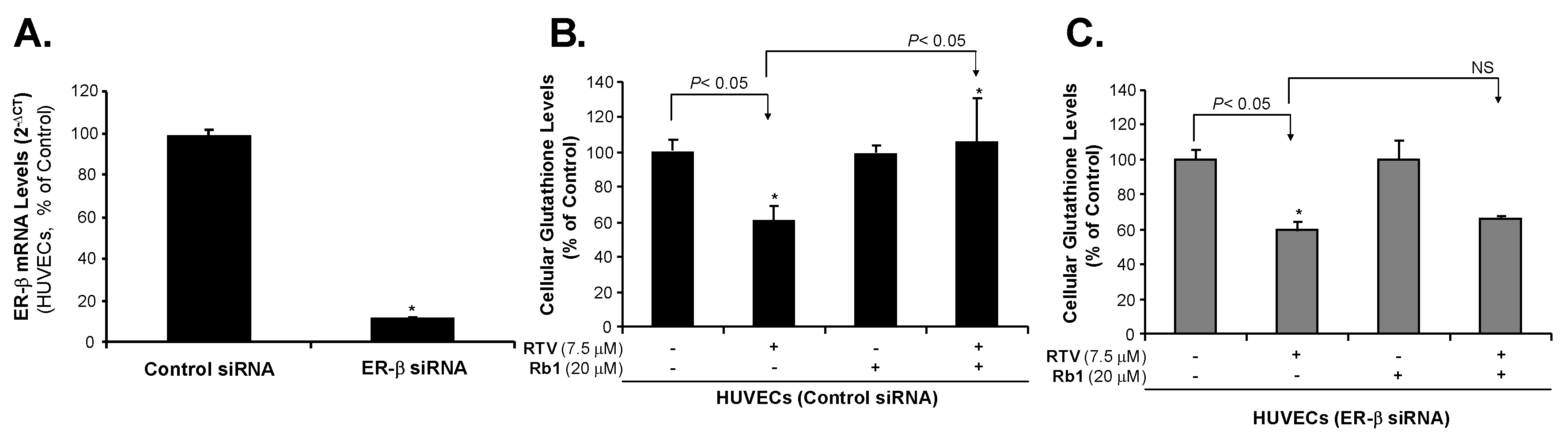
2.1. Human Endothelial Cells Express ER-β
2.2. Rb1 Blocks Ritonavir-Induced Oxidative Stress through ER-β in Human Endothelial Cells
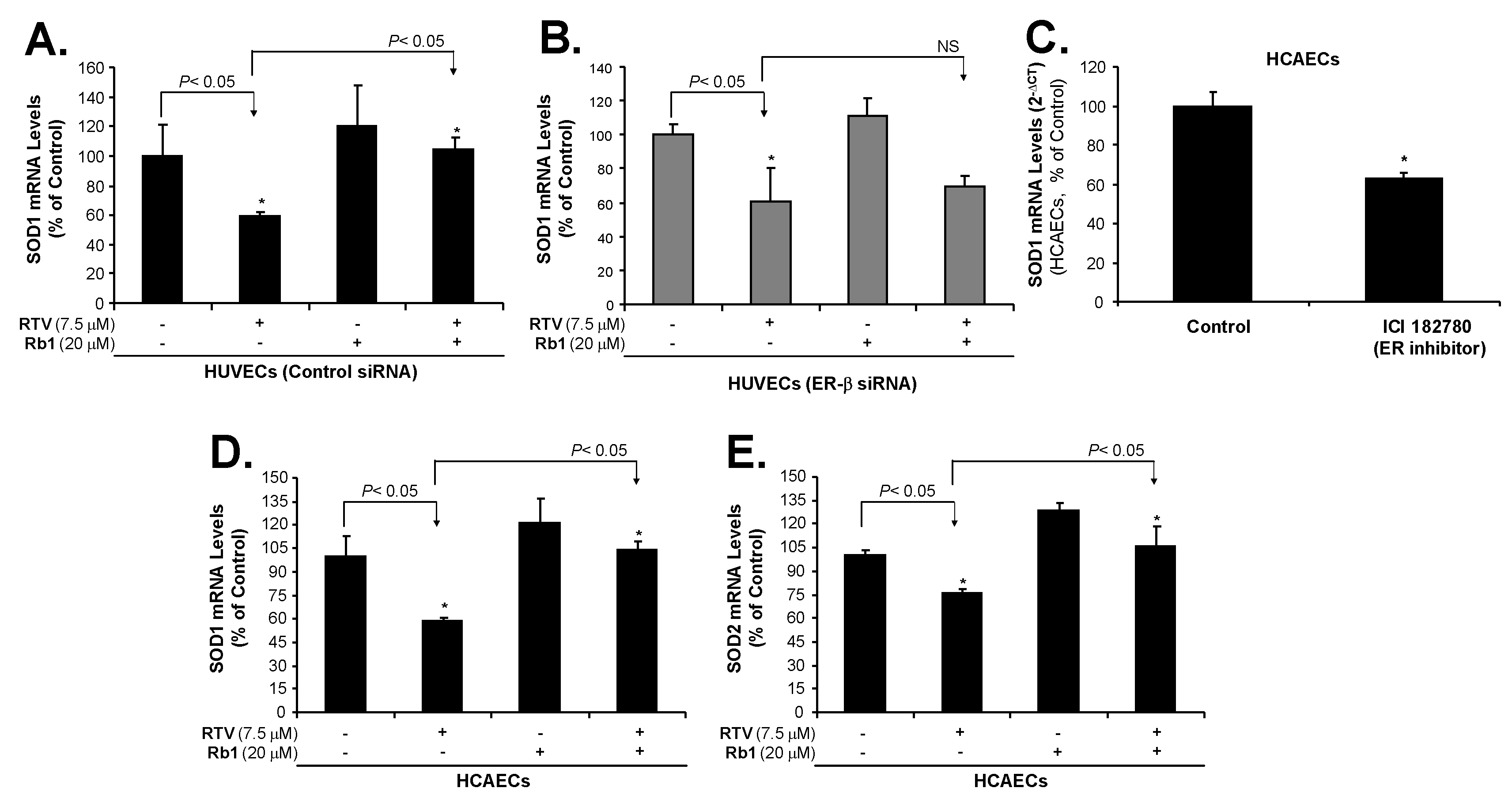
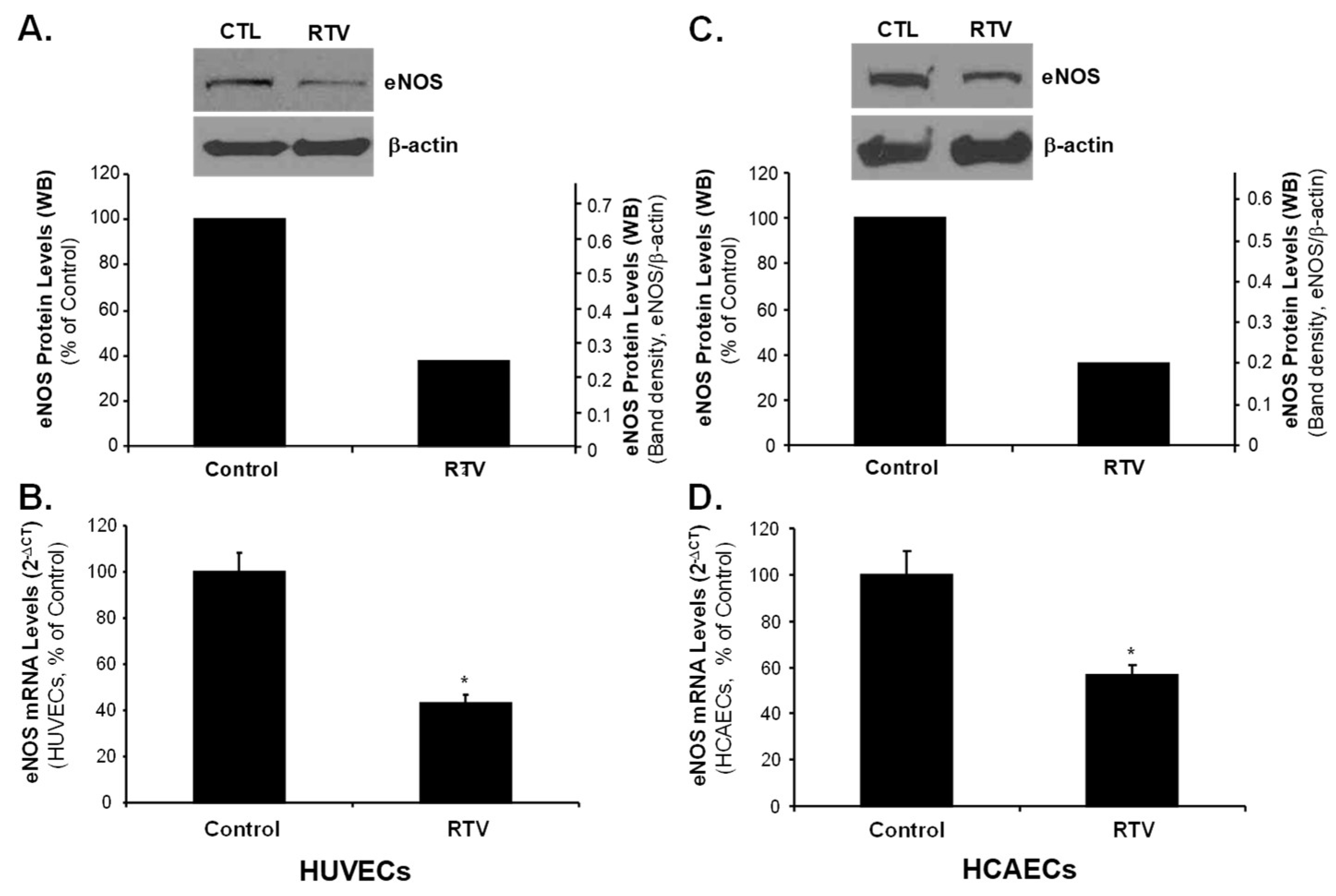
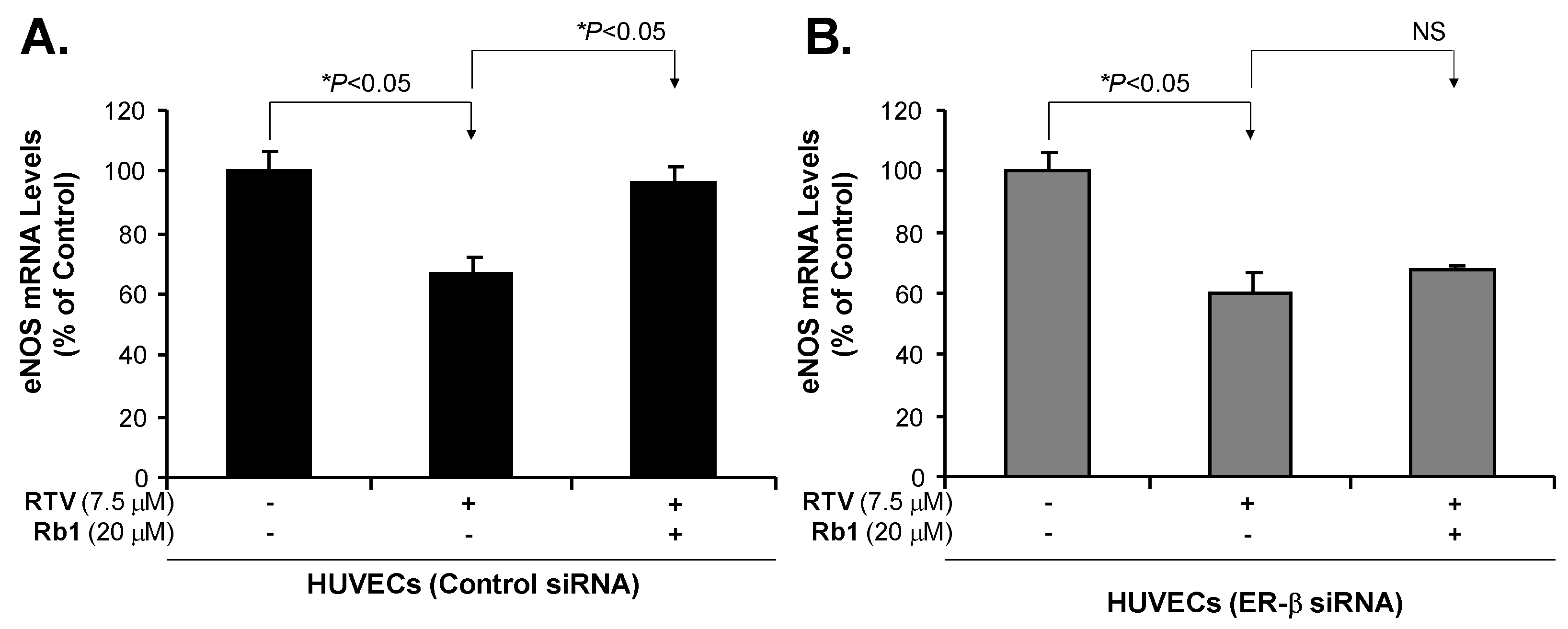
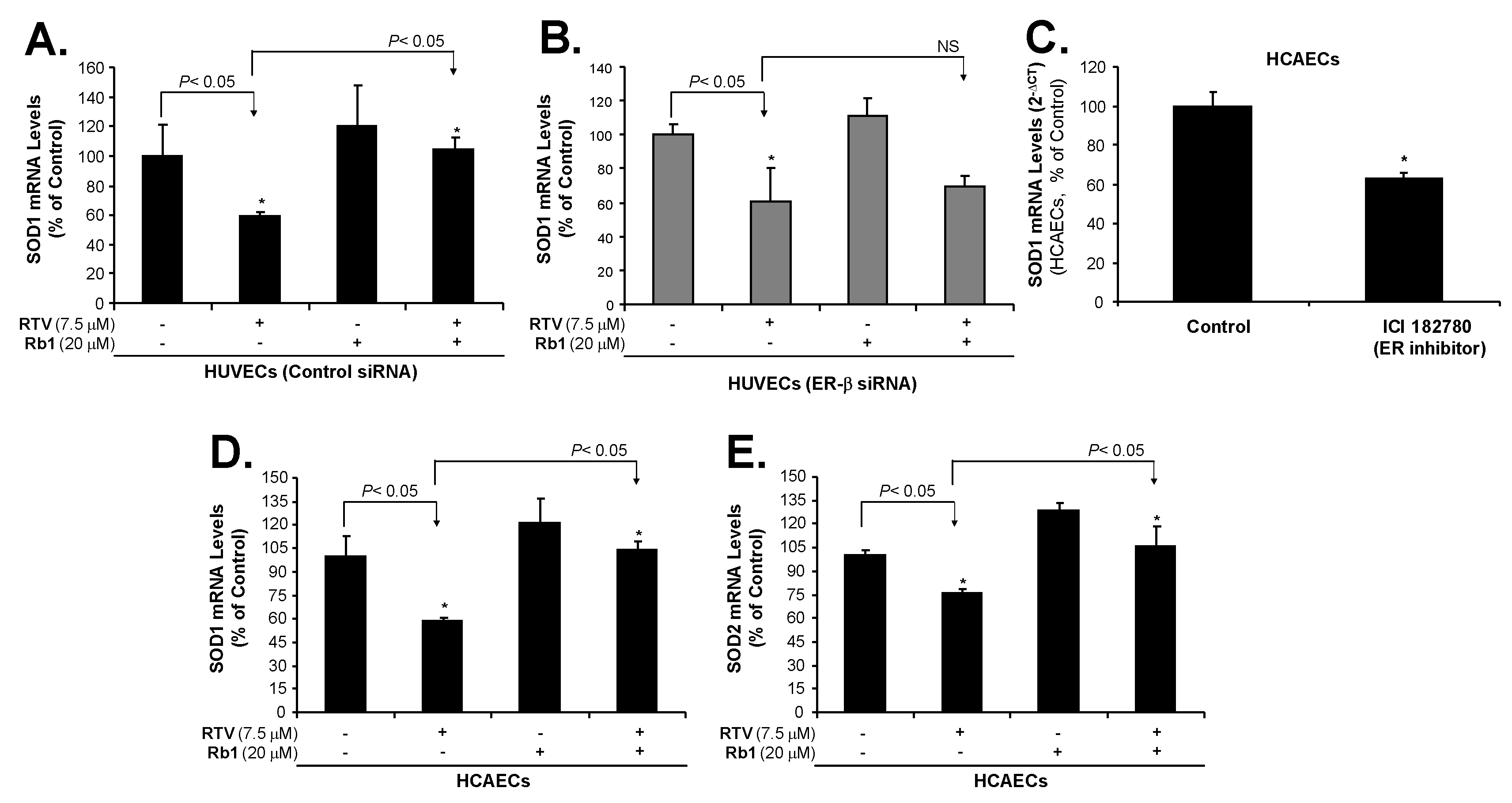
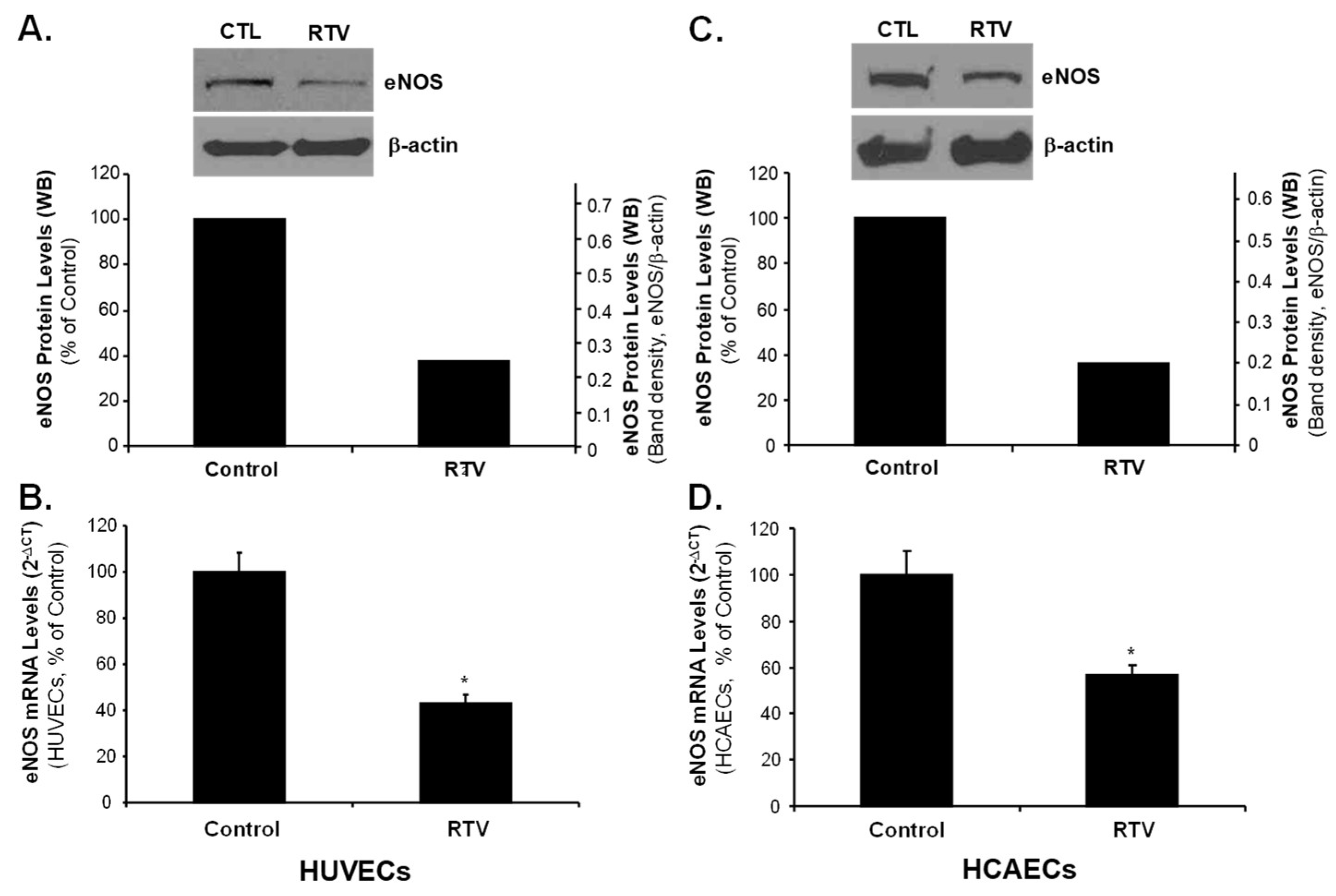
2.3. Rb1 Blocks RTV-Induced Downregulation of SOD1, SOD2 and eNOS through ER-β in Human Endothelial Cells
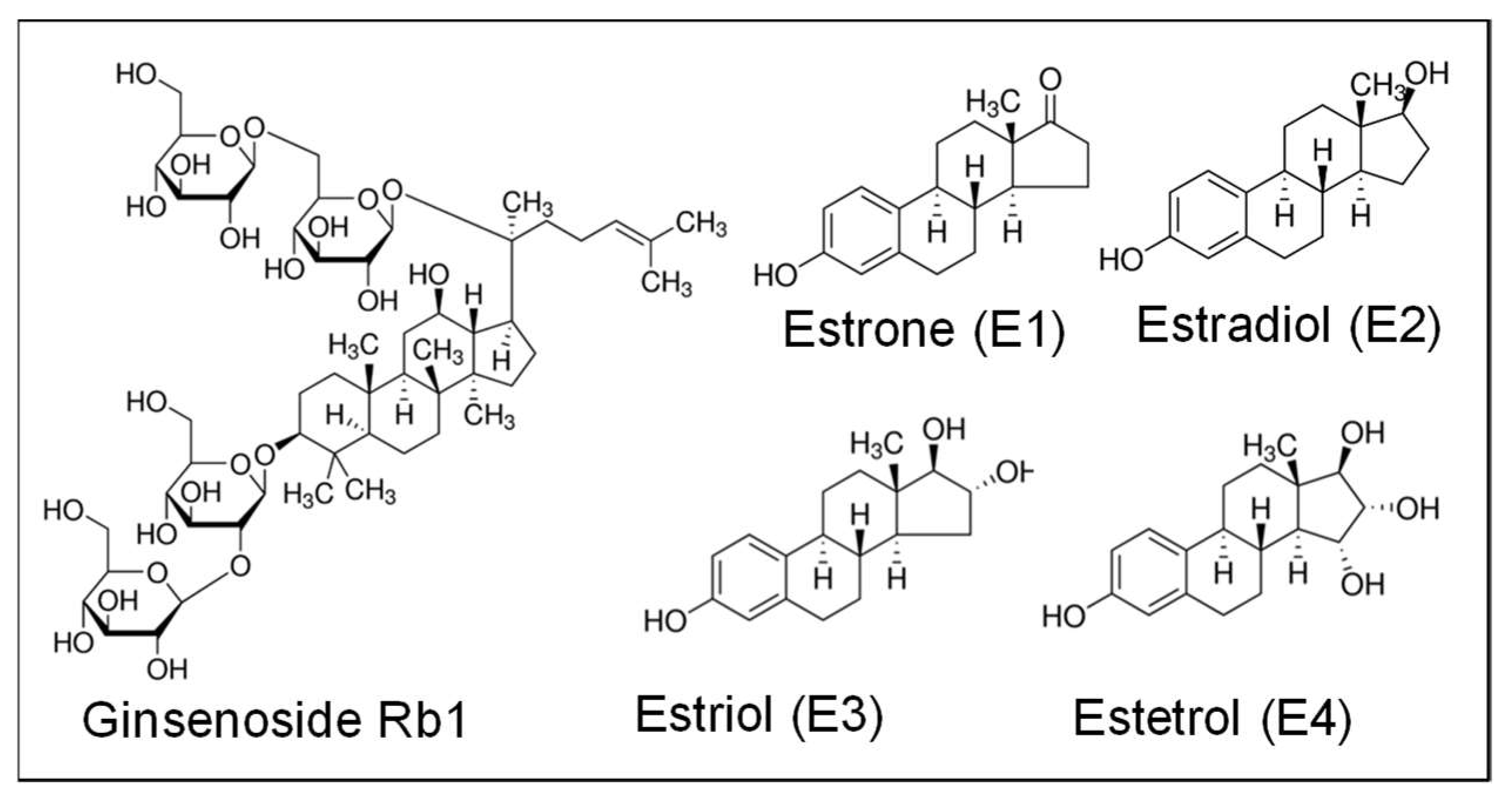
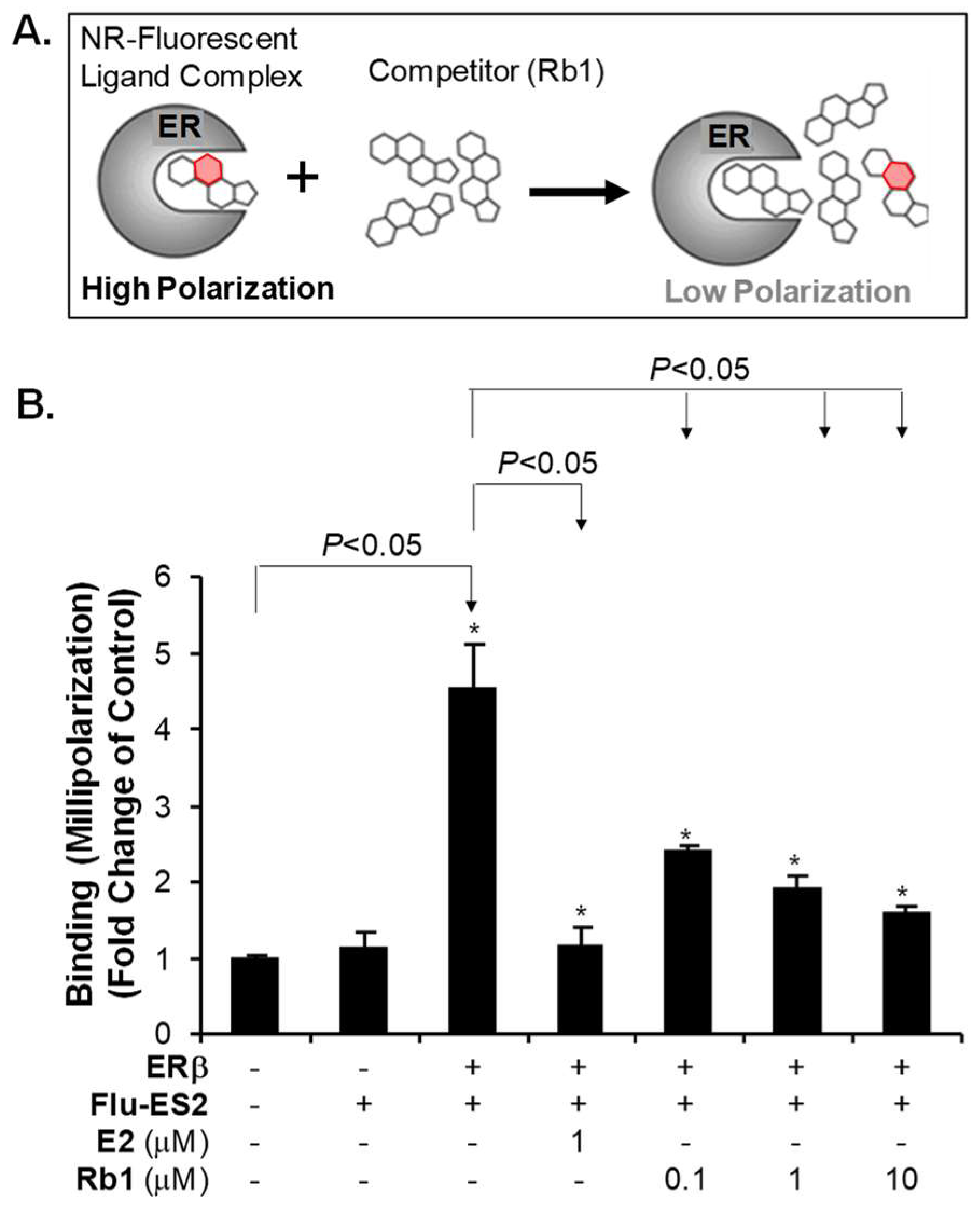
2.4. Rb1 Binds to ER-β in the Cell Free System
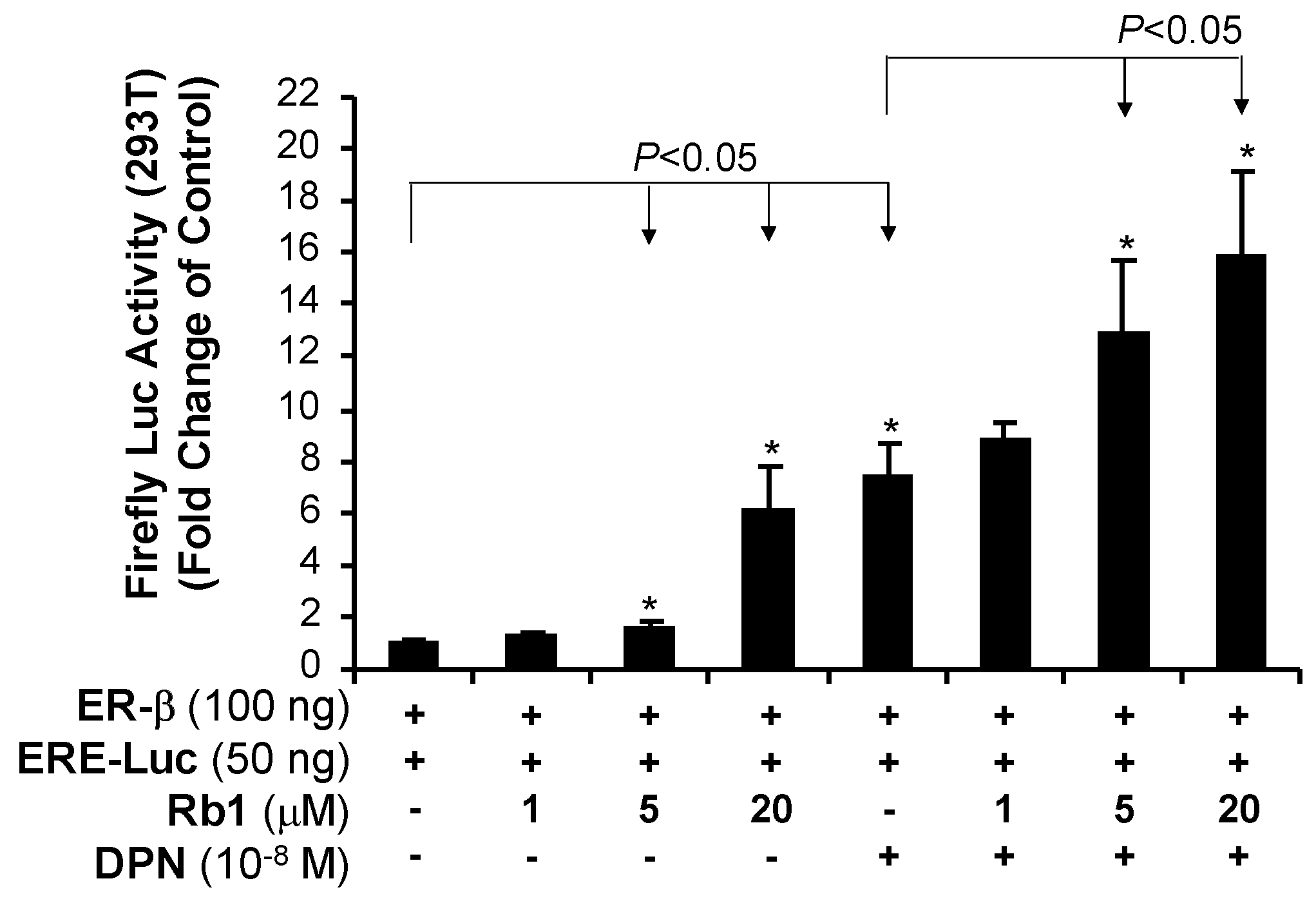
2.5. Rb1 Activates Transcriptional Activity of ER-β in 293T Cells
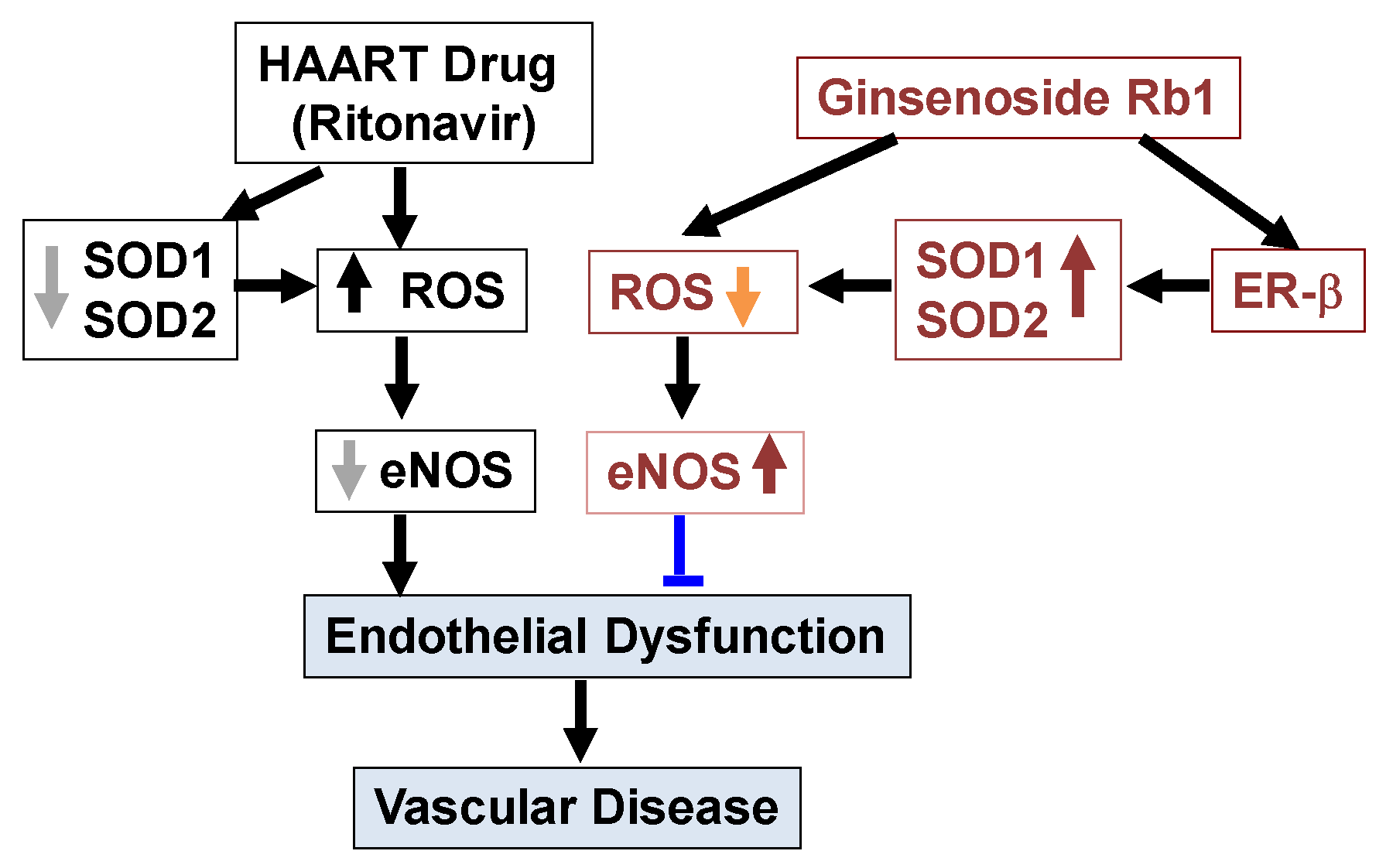
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Real Time PCR
4.4. Western Blot
4.5. Glutathione (GSH) Assay
4.6. ER-β Luciferase Reporter Assay
4.7. Rb1-ER-β Binding Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mu, H.; Chai, H.; Lin, P.H.; Yao, Q.; Chen, C. Current update on HIV-associated vascular disease and endothelial dysfunction. World J. Surg. 2007, 31, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chai, H.; Yao, Q.; Chen, C. Molecular mechanisms of HIV protease inhibitor-induced endothelial dysfunction. J. Acquir. Immune Defic. Syndr. 2007, 44, 493–499. [Google Scholar] [CrossRef]
- Dimala, C.A.; Blencowe, H. Association between highly active antiretroviral therapy and selected cardiovascular disease risk factors in sub-Saharan Africa: A systematic review and meta-analysis protocol. BMJ Open 2017, 7, e013353. [Google Scholar] [CrossRef] [PubMed]
- Ryom, L.; Lundgren, J.D.; El-Sadr, W.; Reiss, P.; Kirk, O.; Law, M.; Phillips, A.; Weber, R.; Fontas, E.; d’Arminio Monforte, A.; et al. Cardiovascular disease and use of contemporary protease inhibitors: The D: A: D international prospective multicohort study. Lancet HIV 2018, 5, e291–e300. [Google Scholar] [CrossRef]
- Global HIV & AIDS Statistics—2018 Fact Sheet. Available online: http://www.unaids.org/en/resources/fact-sheet (accessed on 21 November 2018).
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and ART. FEBS J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B. Oxidative stress in HIV patients receiving antiretroviral therapy. Curr. HIV Res. 2014, 12, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.A.; Sitole, L.J.; Meyer, D. HIV/HAART-associated oxidative stress is detectable by metabonomics. Mol. Biosyst. 2017, 13, 2202–2217. [Google Scholar] [CrossRef]
- Jiang, J.; Fu, W.; Wang, X.; Lin, P.H.; Yao, Q.; Chen, C. HIV gp120 induces endothelial dysfunction in tumour necrosis factor-alpha-activated porcine and human endothelial cells. Cardiovasc. Res. 2010, 87, 366–374. [Google Scholar] [CrossRef]
- Jamaluddin, M.S.; Lin, P.H.; Yao, Q.; Chen, C. Non-nucleoside reverse transcriptase inhibitor efavirenz increases monolayer permeability of human coronary artery endothelial cells. Atherosclerosis 2010, 208, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chai, H.; Lin, P.H.; Yao, Q.; Chen, C. Roles and mechanisms of human immunodeficiency virus protease inhibitor ritonavir and other anti-human immunodeficiency virus drugs in endothelial dysfunction of porcine pulmonary arteries and human pulmonary artery endothelial cells. Am. J. Pathol. 2009, 174, 771–781. [Google Scholar] [CrossRef]
- Haser, G.C.; Sumpio, B. Systemic and cell-specific mechanisms of vasculopathy induced by human immunodeficiency virus and highly active antiretroviral therapy. J. Vasc. Surg. 2017, 65, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Rath, G.; Dessy, C.; Feron, O. Caveolae, caveolin and control of vascular tone: Nitric oxide (NO) and endothelium derived hyperpolarizing factor (EDHF) regulation. J. Physiol. Pharmacol. 2009, 60 (Suppl. 4), 105–109. [Google Scholar] [PubMed]
- Muller, G.; Morawietz, H. Nitric oxide, NAD(P)H oxidase, and atherosclerosis. Antioxid. Redox Signal. 2009, 11, 1711–1731. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lü, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef] [PubMed]
- Fourquet, S.; Huang, M.E.; D’Autreaux, B.; Toledanol, M.B. The dual functions of thiol-based peroxidases in H2O2 scavenging and signaling. Antioxid. Redox Signal. 2008, 10, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Darley-Usmar, V.; Wiseman, H.; Halliwell, B. Nitric oxide and oxygen radicals: A question of balance. FEBS Lett. 1995, 369, 131–135. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Kobayashi, S.; Inoue, N.; Azumi, H.; Seno, T.; Hirata, K.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yokozaki, H.; Yokoyama, M. Expressional changes of the vascular antioxidant system in atherosclerotic coronary arteries. J. Atheroscler. Thromb. 2002, 9, 184–190. [Google Scholar] [CrossRef]
- Lubrano, V.; Balzan, S. Enzymatic antioxidant system in vascular inflammation and coronary artery disease. World J. Exp. Med. 2015, 5, 218–224. [Google Scholar] [CrossRef]
- Lü, J.M.; Yao, Q.; Chen, C. Ginseng compounds: An update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 2009, 7, 293–2302. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Molecular mechanisms and clinical applications of ginseng root for cardiovascular disease. Med. Sci. Monit. 2004, 10, RA187–RA192. [Google Scholar] [PubMed]
- Lee, C.H.; Kim, J.H. A review on the medicinal potentials of ginseng and ginsenosides on cardiovascular diseases. J. Ginseng Res. 2014, 38, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Ginsenoside Rb1 blocks homocysteine-induced endothelial dysfunction in porcine coronary arteries. J. Vasc. Surg. 2005, 41, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Mu, H.; Ohashi, R.; Yan, S.; Chai, H.; Yang, H.; Lin, P.; Yao, Q.; Chen, C. Adipokine resistin promotes in vitro angiogenesis of human endothelial cells. Cardiovasc. Res. 2006, 70, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kougias, P.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Adipocyte-derived cytokine resistin causes endothelial dysfunction of porcine coronary arteries. J. Vasc. Surg. 2005, 41, 691–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Jiang, J.; Lu, J.M.; Chai, H.; Wang, X.; Lin, P.H.; Yao, Q. Resistin decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H193–H201. [Google Scholar] [CrossRef] [Green Version]
- Leung, K.W.; Leung, F.P.; Mak, N.K.; Tombran-Tink, J.; Huang, Y.; Wong, R.N. Protopanaxadiol and protopanaxatriol bind to glucocorticoid and oestrogen receptors in endothelial cells. Br. J. Pharmacol. 2009, 156, 626–637. [Google Scholar] [CrossRef] [Green Version]
- Mohanan, P.; Subramaniyam, S.; Mathiyalagan, R.; Yang, D.C. Molecular signaling of ginsenosides Rb1, Rg1, and Rg3 and their mode of actions. J. Ginseng Res. 2018, 42, 123–132. [Google Scholar] [CrossRef]
- Zhang, B.; Zhou, W.J.; Gu, C.J.; Wu, K.; Yang, H.L.; Mei, J.; Yu, J.J.; Hou, X.F.; Sun, J.S.; Xu, F.Y.; et al. The ginsenoside PPD exerts anti-endometriosis effects by suppressing estrogen receptor-mediated inhibition of endometrial stromal cell autophagy and NK cell cytotoxicity. Cell Death Dis. 2018, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, S.O.; Kim, G.L.; Rhee, D.K. Estrogen receptor-β of microglia underlies sexual differentiation of neuronal protection via ginsenosides in mice brain. CNS Neurosci. Ther. 2018, 24, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.W.; Leung, F.P.; Huang, Y.; Mak, N.K.; Wong, R.N. Non-genomic effects of ginsenoside-Re in endothelial cells via glucocorticoid receptor. FEBS Lett. 2007, 581, 2423–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Eto, M.; Akishita, M.; Kaneko, A.; Ouchi, Y.; Okabe, T. Signaling pathway of nitric oxide production induced by ginsenoside Rb1 in human aortic endothelial cells: A possible involvement of androgen receptor. Biochem. Biophys. Res. Commun. 2007, 353, 764–769. [Google Scholar] [CrossRef]
- Strehlow, K.; Rotter, S.; Wassmann, S.; Adam, O.; Grohe, C.; Laufs, K.; Bohm, M.; Nickenig, G. Modulation of antioxidant enzyme expression and function by estrogen. Circ. Res. 2003, 93, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Fang, C.; Ji, X.; Liang, X.; Liu, Y.; Han, C.; Huang, L.; Zhang, Q.; Li, H.; Zhang, Y.; et al. Estrogen Receptor (ER)-α36 Is Involved in estrogen- and tamoxifen-induced neuroprotective effects in ischemic stroke models. PLoS ONE 2015, 10, e0140660. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Takemoto, T.; Ishida, A.; Yamazaki, T. Protective actions of 17β-estradiol and progesterone on oxidative neuronal injury induced by organometallic compounds. Oxid. Med. Cell. Longev. 2015, 2015, 343706. [Google Scholar] [CrossRef]
- Xia, Y.; Krukoff, T.L. Estrogen induces nitric oxide production via activation of constitutive nitric oxide synthases in human neuroblastoma cells. Endocrinology 2004, 145, 4550–4557. [Google Scholar] [CrossRef]
- Nickenig, G.; Baumer, A.T.; Grohe, C.; Kahlert, S.; Strehlow, K.; Rosenkranz, S.; Stablein, A.; Beckers, F.; Smits, J.F.; Daemen, M.J.; et al. Estrogen modulates AT1 receptor gene expression in vitro and in vivo. Circulation 1998, 97, 2197–2201. [Google Scholar] [CrossRef]
- Zhang, Q.G.; Raz, L.; Wang, R.; Han, D.; De Sevilla, L.; Yang, F.; Vadlamudi, R.K.; Brann, D.W. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J. Neurosci. 2009, 29, 13823–13836. [Google Scholar] [CrossRef]
- Weiser, M.J.; Foradori, C.D.; Handa, R.J. Estrogen receptor beta in the brain: From form to function. Brain Res. Rev. 2008, 57, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Couse, J.F.; Lindzey, J.; Grandien, K.; Gustafsson, J.A.; Korach, K.S. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERα) and estrogen receptor-beta (ERβ) messenger ribonucleic acid in the wild-type and ERα-knockout mouse. Endocrinology 1997, 138, 4613–4621. [Google Scholar] [CrossRef] [PubMed]
- Gargett, C.E.; Bucak, K.; Zaitseva, M.; Chu, S.; Taylor, N.; Fuller, P.J.; Rogers, P.A. Estrogen receptor-α and -β expression in microvascular endothelial cells and smooth muscle cells of myometrium and leiomyoma. Mol. Hum. Reprod. 2002, 8, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Paech, K.; Webb, P.; Kuiper, G.G.; Nilsson, S.; Gustafsson, J.; Kushner, P.J.; Scanlan, T.S. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science 1997, 277, 1508–1510. [Google Scholar] [CrossRef]
- Dechering, K.; Boersma, C.; Mosselman, S. Estrogen receptors alpha and beta: Two receptors of a kind? Curr. Med. Chem. 2000, 7, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Sastre, J.; Pallardo, F.V.; Gambini, J.; Borras, C. Modulation of longevity-associated genes by estrogens or phytoestrogens. Biol. Chem. 2008, 389, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.A.; Chai, H.; Fu, W.; Ramaswami, G.; Cox, M.W.; Conklin, B.S.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Estrogen blocks homocysteine-induced endothelial dysfunction in porcine coronary arteries (1,2). J. Surg. Res. 2004, 118, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.M.; Weakley, S.M.; Yang, Z.; Hu, M.; Yao, Q.; Chen, C. Ginsenoside Rb1 directly scavenges hydroxyl radical and hypochlorous acid. Curr. Pharm. Des. 2012, 18, 6339–6347. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharm. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Morales Pantoja, I.E.; Hu, C.L.; Perrone-Bizzozero, N.I.; Zheng, J.; Bizzozero, O.A. Nrf2-dysregulation correlates with reduced synthesis and low glutathione levels in experimental autoimmune encephalomyelitis. J. Neurochem. 2016, 139, 640–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Wang, X.; Weakley, S.M.; Kougias, P.; Lin, P.H.; Yao, Q.; Chen, C. The soybean isoflavonoid equol blocks ritonavir-induced endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells. J. Nutr. 2010, 140, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Zhou, W.; Lin, P.; Lumsden, A.; Yao, Q.; Chen, C. Ginsenosides block HIV protease inhibitor ritonavir-induced vascular dysfunction of porcine coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2965–H2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liao, D.; Lin, P.H.; Yao, Q.; Chen, C. Highly active antiretroviral therapy drugs inhibit in vitro cholesterol efflux from human macrophage-derived foam cells. Lab. Investig. 2009, 89, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conklin, B.S.; Fu, W.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. HIV protease inhibitor ritonavir decreases endothelium-dependent vasorelaxation and increases superoxide in porcine arteries. Cardiovasc. Res. 2004, 63, 168–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.; Chai, H.; Yao, Q.; Chen, C. Effects of HIV protease inhibitor ritonavir on vasomotor function and endothelial nitric oxide synthase expression. J. Acquir. Immune Defic. Syndr. 2005, 39, 152–158. [Google Scholar]
- Chai, H.; Yang, H.; Yan, S.; Li, M.; Lin, A.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Effects of 5 HIV protease inhibitors on vasomotor function and superoxide anion production in porcine coronary arteries. J. Acquir. Immune Defic. Syndr. 2005, 40, 12–19. [Google Scholar] [CrossRef]
- Chen, C.; Lu, X.H.; Yan, S.; Chai, H.; Yao, Q. HIV protease inhibitor ritonavir increases endothelial monolayer permeability. Biochem. Biophys. Res. Commun. 2005, 335, 874–882. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species: Key regulators in vascular health and diseases. Br. J. Pharmacol. 2018, 175, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.F., Jr. Atherosclerosis: From lesion formation to plaque activation and endothelial dysfunction. Mol. Asp. Med. 2000, 21, 99–166. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Metodiewa, D. The reaction of superoxide with reduced glutathione. Arch. Biochem. Biophys. 1994, 314, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2008, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Toth, B.; Saadat, G.; Geller, A.; Scholz, C.; Schulze, S.; Friese, K.; Jeschke, U. Human umbilical, vascular endothelial cells express estrogen receptor β (ERβ) and progesterone receptor A (PR-A), but not ERα and PR-B. Histochem. Cell Biol. 2008, 130, 399–405. [Google Scholar] [CrossRef]
- Fortini, F.; Vieceli Dalla Sega, F.; Caliceti, C.; Aquila, G.; Pannella, M.; Pannuti, A.; Miele, L.; Ferrari, R.; Rizzo, P. Estrogen receptor β-dependent Notch1 activation protects vascular endothelium against tumor necrosis factor α (TNFα)-induced apoptosis. J. Biol. Chem. 2017, 292, 18178–18191. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Weil, B.; Abarbanell, A.; Herrmann, J.; Tan, J.; Kelly, M.; Meldrum, D.R. Estrogen receptor beta mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R972–R978. [Google Scholar] [CrossRef]
- Jazbutyte, V.; Arias-Loza, P.A.; Hu, K.; Widder, J.; Govindaraj, V.; von Poser-Klein, C.; Bauersachs, J.; Fritzemeier, K.H.; Hegele-Hartung, C.; Neyses, L.; et al. Ligand-dependent activation of ERβ lowers blood pressure and attenuates cardiac hypertrophy in ovariectomized spontaneously hypertensive rats. Cardiovasc. Res. 2008, 77, 774–781. [Google Scholar] [CrossRef]
- Vina, J.; Borras, C.; Gambini, J.; Sastre, J.J.; Pallardo, F.V. Why females live longer than males? Importance of the upregulation of longevity-associated genes by oestrogenic compounds. FEBS Lett. 2005, 579, 2541–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.H.; Han, H.; Hu, X.D.; Shi, L.L. Protective effect of ginsenoside Rb1 on beta-amyloid protein(1-42)-induced neurotoxicity in cortical neurons. Neurol. Res. 2009, 31, 663–667. [Google Scholar] [CrossRef]
- Zhu, D.; Wu, L.; Li, C.R.; Wang, X.W.; Ma, Y.J.; Zhong, Z.Y.; Zhao, H.B.; Cui, J.; Xun, S.F.; Huang, X.L.; et al. Ginsenoside Rg1 protects rat cardiomyocyte from hypoxia/reoxygenation oxidative injury via antioxidant and intracellular calcium homeostasis. J. Cell. Biochem. 2009, 108, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Attele, A.S.; Wu, J.A.; Yuan, C.S. Ginseng pharmacology: Multiple constituents and multiple actions. Biochem. Pharmacol. 1999, 58, 1685–1693. [Google Scholar] [CrossRef]
- Paik, N.H.; Park, M.K.; Choi, K.J.; Cho, Y.H. Isolation of ginsenosides Rb1, Rb2, Rc, Rd, Re, Rf and Rg1 from ginseng root by high performance liquid chromatography. Arch. Pharmacal. Res. 1982, 5, 7–12. [Google Scholar] [CrossRef]
- Hashimoto, R.; Yu, J.; Koizumi, H.; Ouchi, Y.; Okabe, T. Ginsenoside Rb1 Prevents MPP(+)-Induced Apoptosis in PC12 Cells by Stimulating Estrogen Receptors with Consequent Activation of ERK1/2, Akt and Inhibition of SAPK/JNK, p38 MAPK. Evid.-Based Complement. Altern. Med. 2012, 2012, 693717. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.P.; Jeong, H.G. Ginsenoside Rb1 protects against 6-hydroxydopamine-induced oxidative stress by increasing heme oxygenase-1 expression through an estrogen receptor-related PI3K/Akt/Nrf2-dependent pathway in human dopaminergic cells. Toxicol. Appl. Pharmacol. 2010, 242, 18–28. [Google Scholar] [CrossRef]
- Cho, J.; Park, W.; Lee, S.; Ahn, W.; Lee, Y. Ginsenoside-Rb1 from Panax ginseng C.A. Meyer activates estrogen receptor-alpha and -beta, independent of ligand binding. J. Clin. Endocrinol. Metab. 2004, 89, 3510–3515. [Google Scholar] [CrossRef]
- Lee, A.V.; Weng, C.N.; Jackson, J.G.; Yee, D. Activation of estrogen receptor-mediated gene transcription by IGF-I in human breast cancer cells. J. Endocrinol. 1997, 152, 39–47. [Google Scholar] [CrossRef]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef]
- Karas, R.H.; Gauer, E.A.; Bieber, H.E.; Baur, W.E.; Mendelsohn, M.E. Growth factor activation of the estrogen receptor in vascular cells occurs via a mitogen-activated protein kinase-independent pathway. J. Clin. Investig. 1998, 101, 2851–2861. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.W.; Cheung, L.W.; Pon, Y.L.; Wong, R.N.; Mak, N.K.; Fan, T.P.; Au, S.C.; Tombran-Tink, J.; Wong, A.S. Ginsenoside Rb1 inhibits tube-like structure formation of endothelial cells by regulating pigment epithelium-derived factor through the oestrogen β receptor. Br. J. Pharmacol. 2007, 152, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Cvoro, A.; Paruthiyil, S.; Jones, J.O.; Tzagarakis-Foster, C.; Clegg, N.J.; Tatomer, D.; Medina, R.T.; Tagliaferri, M.; Schaufele, F.; Scanlan, T.S.; et al. Selective activation of estrogen receptor-β transcriptional pathways by an herbal extract. Endocrinology 2007, 148, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jin, Y.; Lim, W.; Ji, S.; Choi, S.; Jang, S.; Lee, S. A ginsenoside-Rh1, a component of ginseng saponin, activates estrogen receptor in human breast carcinoma MCF-7 cells. J. Steroid Biochem. Mol. Biol. 2003, 84, 463–468. [Google Scholar] [CrossRef]
- Ohashi, R.; Yan, S.; Mu, H.; Chai, H.; Yao, Q.; Lin, P.H.; Chen, C. Effects of homocysteine and ginsenoside Rb1 on endothelial proliferation and superoxide anion production. J. Surg. Res. 2006, 133, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Yan, S.; Lin, P.; Lumsden, A.B.; Yao, Q.; Chen, C. Curcumin blocks HIV protease inhibitor ritonavir-induced vascular dysfunction in porcine coronary arteries. J. Am. Coll. Surg. 2005, 200, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Dhadwal, A.K.; Wang, X.; Annambhotla, S.; Lin, P.H.; Yao, Q.; Chen, C. Capsaicin blocks HIV protease inhibitor ritonavir-induced vascular dysfunction in porcine pulmonary arteries. Med. Sci. Monit. 2009, 15, BR1–BR5. [Google Scholar]
- Lu, J.M.; Nurko, J.; Jiang, J.; Weakley, S.M.; Lin, P.H.; Yao, Q.; Chen, C. Nordihydroguaiaretic acid (NDGA) inhibits ritonavir-induced endothelial dysfunction in porcine pulmonary arteries. Med. Sci. Monit. 2011, 17, BR312–BR318. [Google Scholar] [CrossRef] [Green Version]
- Weakley, S.M.; Jiang, J.; Lu, J.; Wang, W.; Lin, L.H.; Yao, Q.; Chen, C. Natural antioxidant dihydroxybenzyl alcohol blocks ritonavir-induced endothelial dysfunction in porcine pulmonary arteries and human endothelial cells. Med. Sci. Monit. 2011, 17, BR235–BR241. [Google Scholar] [CrossRef] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lü, J.-M.; Jiang, J.; Jamaluddin, M.S.; Liang, Z.; Yao, Q.; Chen, C. Ginsenoside Rb1 Blocks Ritonavir-Induced Oxidative Stress and eNOS Downregulation through Activation of Estrogen Receptor-Beta and Upregulation of SOD in Human Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 294. https://doi.org/10.3390/ijms20020294
Lü J-M, Jiang J, Jamaluddin MS, Liang Z, Yao Q, Chen C. Ginsenoside Rb1 Blocks Ritonavir-Induced Oxidative Stress and eNOS Downregulation through Activation of Estrogen Receptor-Beta and Upregulation of SOD in Human Endothelial Cells. International Journal of Molecular Sciences. 2019; 20(2):294. https://doi.org/10.3390/ijms20020294
Chicago/Turabian StyleLü, Jian-Ming, Jun Jiang, Md Saha Jamaluddin, Zhengdong Liang, Qizhi Yao, and Changyi Chen. 2019. "Ginsenoside Rb1 Blocks Ritonavir-Induced Oxidative Stress and eNOS Downregulation through Activation of Estrogen Receptor-Beta and Upregulation of SOD in Human Endothelial Cells" International Journal of Molecular Sciences 20, no. 2: 294. https://doi.org/10.3390/ijms20020294