Inducible Slc7a7 Knockout Mouse Model Recapitulates Lysinuric Protein Intolerance Disease

 , ,
, ,
Abstract
1. Introduction
2. Results
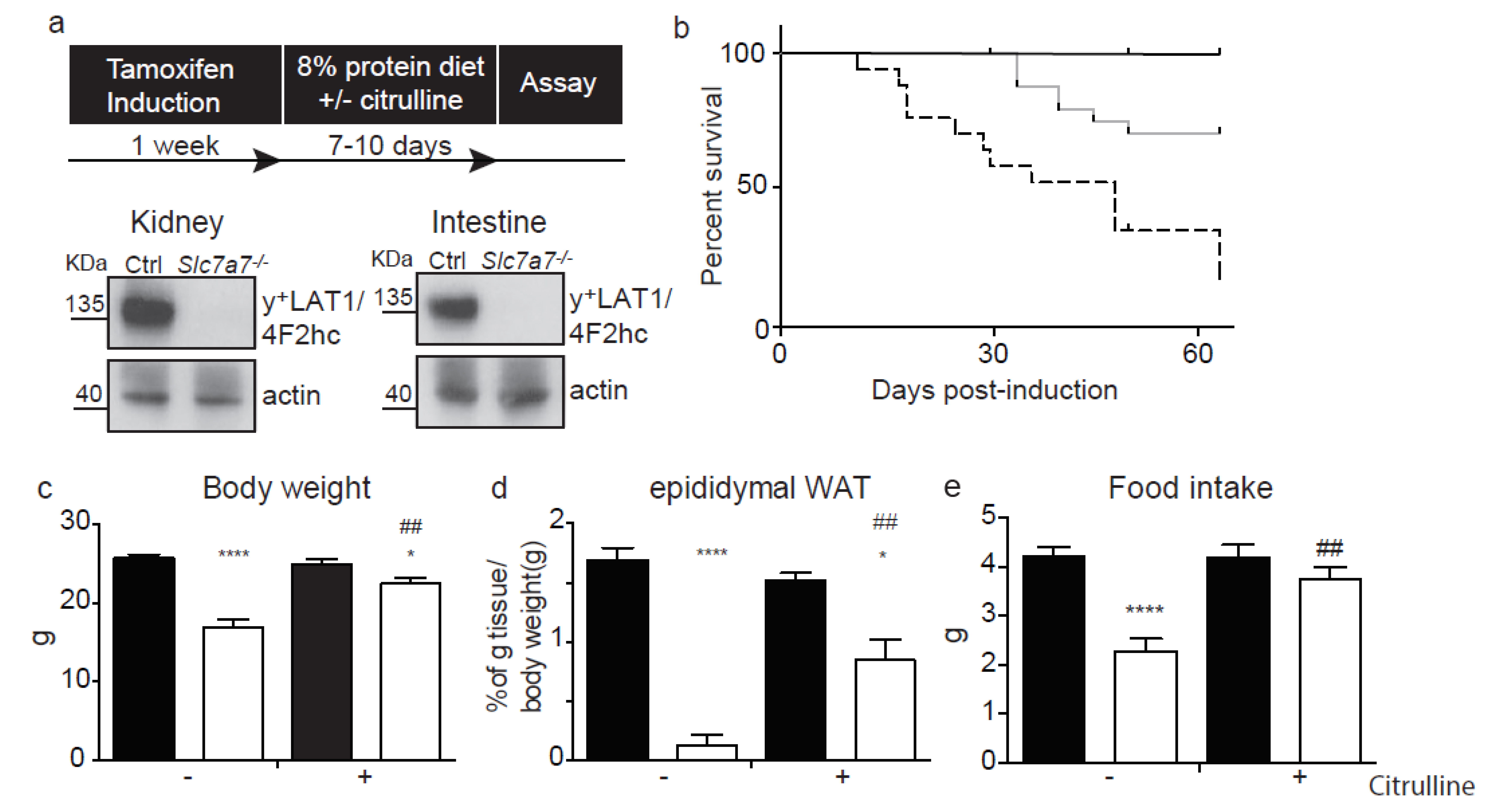
2.1. The Slc7a7−/− Mouse Model
2.2. Renal and Intestinal Phenotype
2.3. Hyperammonemia and Neurological Symptoms
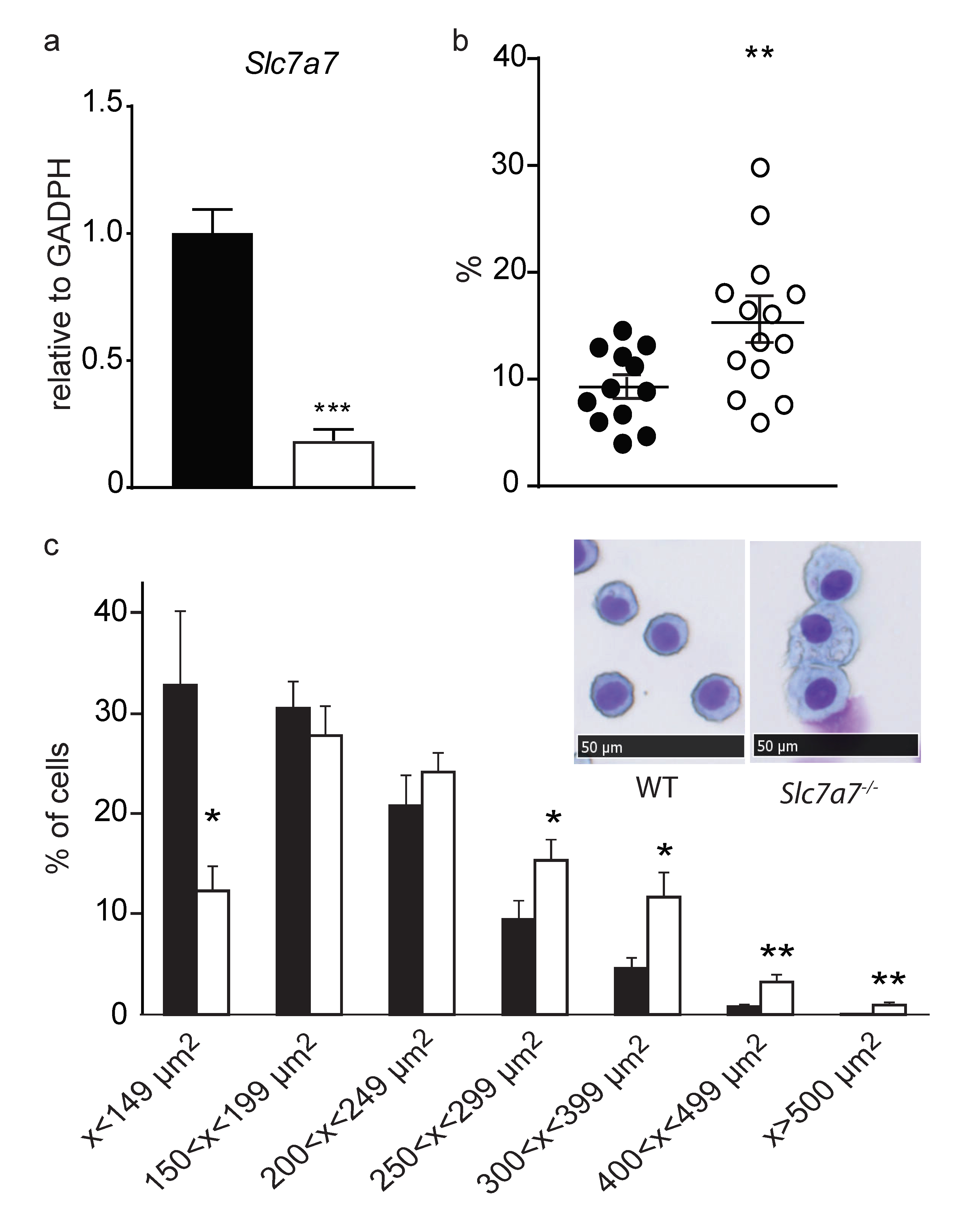
2.4. Lung Involvement
3. Discussion
4. Materials and Methods
4.1. Animal Care, Generation of Animal Model and Diet Treatments
4.2. Genotyping
4.3. Metabolic Studies
4.4. Oral Gavage
4.5. Amino Acid and Creatinine Analysis
4.6. Brain Water Content
4.7. Protein Analysis
4.8. Histological Sample Preparation and Analysis
4.9. Panoptic Staining of Cells from Bronchoalveolar Lavage
4.10. Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AM | Alveolar macrophages |
| CAA | Cationic amino acids |
| eWAT | Epydidimal white adipose tissue |
| GFAP | Glial fibrillary acidic protein |
| GFR | Glomerular filtration rate |
| GM-CSF | Granulocyte-macrophage-colony stimulating factor |
| HHH | Hyperornithinemia hyperammonemia homocitrullinuria |
| LPI | Lysinuric protein intolerance |
| PAP | Pulmonary alveolar proteinosis |
| PAS | Periodic acid–Schiff staining |
| TR | Tubular reabsorption |
References
- Torrents, D.; Mykkänen, J.; Pineda, M.; Feliubadaló, L.; Estévez, R.; de Cid, R.; Sanjurjo, P.; Zorzano, A.; Nunes, V.; Huoponen, K.; et al. Identification of SLC7A7, encoding y+LAT-1, as the lysinuric protein intolerance gene. Nat. Genet. 1999, 21, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Borsani, G.; Bassi, M.T.; Sperandeo, M.P.; De Grandi, A.; Buoninconti, A.; Riboni, M.; Manzoni, M.; Incerti, B.; Pepe, A.; Andria, G.; et al. SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat. Genet. 1999, 21, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Palacín, M.; Nunes, V.; Font-Llitjós, M.; Jiménez-Vidal, M.; Fort, J.; Gasol, E.; Pineda, M.; Feliubadaló, L.; Chillarón, J.; Zorzano, A. The Genetics of Heteromeric Amino Acid Transporters. Physiology 2005, 20, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Rotoli, B.M.; Barilli, A.; Visigalli, R.; Ingoglia, F.; Milioli, M.; Di Lascia, M.; Riccardi, B.; Puccini, P.; Dall’Asta, V. Downregulation of SLC7A7 triggers an inflammatory phenotype in human macrophages and airway epithelial cells. Front. Immunol. 2018, 9, 508. [Google Scholar] [CrossRef] [PubMed]
- Tringham, M.; Kurko, J.; Tanner, L.; Tuikkala, J.; Nevalainen, O.S.; Niinikoski, H.; Näntö-Salonen, K.; Hietala, M.; Simell, O.; Mykkänen, J. Exploring the transcriptomic variation caused by the Finnish founder mutation of lysinuric protein intolerance (LPI). Mol. Genet. Metab. 2012, 105, 408–415. [Google Scholar] [CrossRef]
- Koizumi, A.; Shoji, Y.; Nozaki, J.; Noguchi, A.; E, X.; Dakeishi, M.; Ohura, T.; Tsuyoshi, K.; Yasuhiko, W.; Manabe, M.; et al. A cluster of lysinuric protein intolerance (LPI) patients in a northern part of Iwate, Japan due to a founder effect. Hum. Mutat. 2000, 16, 270–271. [Google Scholar] [CrossRef]
- Noguchi, A.; Nakamura, K.; Murayama, K.; Yamamoto, S.; Komatsu, H.; Kizu, R.; Takayanagi, M.; Okuyama, T.; Endo, F.; Takasago, Y.; et al. Clinical and genetic features of lysinuric protein intolerance in Japan. Pediatr. Int. 2016, 58, 979–983. [Google Scholar] [CrossRef]
- Palacín, M.; Borsani, G.; Sebastio, G. The molecular bases of cystinuria and lysinuric protein intolerance. Curr. Opin. Genet. Dev. 2001, 11, 328–335. [Google Scholar] [CrossRef]
- Font-Llitjós, M.; Rodríguez-Santiago, B.; Espino, M.; Sillué, R.; Mañas, S.; Gómez, L.; Pérez-Jurado, L.A.; Palacín, M.; Nunes, V. Novel SLC7A7 large rearrangements in lysinuric protein intolerance patients involving the same AluY repeat. Eur. J. Hum. Genet. 2009, 17, 71–79. [Google Scholar] [CrossRef]
- Güzel-Ozantürk, A.; Özgül, R.K.; Ünal, Ö.; Hişmi, B.; Aydın, H.İ.; Sivri, S.; Tokatlı, A.; Coşkun, T.; Aksöz, E.; Dursun, A. Molecular and clinical evaluation of Turkish patients with lysinuric protein intolerance. Gene 2013, 521, 293–295. [Google Scholar] [CrossRef]
- Ünal, Ö.; Coşkun, T.; Orhan, D.; Tokatl, A.; Dursun, A.; Hişmi, B.; Özyüncü, Ö.; Sivri, S.H.K. Pregnancy and Lactation Outcomes in a Turkish Patient with Lysinuric Protein Intolerance. In JIMD Reports; Wiley-Blackwell: Hoboken, NJ, USA, 2013; Volume 13, pp. 33–36. [Google Scholar]
- Sperandeo, M.P.; Annunziata, P.; Ammendola, V.; Fiorito, V.; Pepe, A.; Soldovieri, M.V.; Taglialatela, M.; Andria, G.; Sebastio, G. Lysinuric protein intolerance: Identification and functional analysis of mutations of the SLC7A7 gene. Hum. Mutat. 2005, 25, 410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Cao, L. New mutations in the SLC7A7 gene of two chinese sisters with lysinuric protein intolerance. Pediatr. Pulmonol. 2017, 52, E94–E96. [Google Scholar] [CrossRef] [PubMed]
- Simell, O. Lysinuric protein intolerance and other cationic aminoacidurias. In The Metabolic and Molecular Basis of Inherited Disease; Scriver, C., Beaudet, A., Sly, W., Valle, D., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 4933–4956. [Google Scholar]
- Palacín, M.; Bertran, J.; Chillarón, J.; Estévez, R.; Zorzano, A. Lysinuric protein intolerance: Mechanisms of pathophysiology. Mol. Genet. Metab. 2004, 81, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Ogier de Baulny, H.; Schiff, M.; Dionisi-Vici, C. Lysinuric protein intolerance (LPI): A multi organ disease by far more complex than a classic urea cycle disorder. Mol. Genet. Metab. 2012, 106, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Sebastio, G.; Sperandeo, M.P.; Andria, G. Lysinuric protein intolerance: Reviewing concepts on a multisystem disease. Am. J. Med. Genet. Part C Semin. Med. Genet. 2011, 157, 54–62. [Google Scholar] [CrossRef]
- Parto, K.; Maki, J.; Pelliniemi, L.J.; Simell, O. Abnormal pulmonary macrophages in lysinuric protein intolerance: Ultrastructural, morphometric, and x-ray microanalytic study. Arch. Pathol. Lab. Med. 1994, 118, 536–541. [Google Scholar]
- Mauhin, W.; Habarou, F.; Gobin, S.; Servais, A.; Brassier, A.; Grisel, C.; Roda, C.; Pinto, G.; Moshous, D.; Ghalim, F.; et al. Update on Lysinuric Protein Intolerance, a Multi-faceted Disease Retrospective cohort analysis from birth to adulthood. Orphanet J. Rare Dis. 2017, 12, 3. [Google Scholar] [CrossRef]
- Lukkarinen, M.; Näntö-Salonen, K.; Ruuskanen, O.; Lauteala, T.; Säkö, S.; Nuutinen, M.; Simell, O. Varicella and varicella immunity in patients with lysinuric protein intolerance. J. Inherit. Metab. Dis. 1998, 21, 103–111. [Google Scholar] [CrossRef]
- Duval, M.; Fenneteau, O.; Doireau, V.; Faye, A.; Emilie, D.; Yotnda, P.; Drapier, J.C.; Schlegel, N.; Sterkers, G.; de Baulny, H.O.; et al. Intermittent hemophagocytic lymphohistiocytosis is a regular feature of lysinuric protein intolerance. J. Pediatr. 1999, 134, 236–239. [Google Scholar] [CrossRef]
- Trapnell, B.C.; Carey, B.C.; Uchida, K.; Suzuki, T. Pulmonary Alveolar Proteinosis, a Primary Immunodeficiency of Impaired GM-CSF Stimulation of Macrophages; NIH Public Access: Bethesda, MD, USA, 2009; Volume 21, pp. 514–521. [Google Scholar]
- Rotoli, B.M.; Dall’Asta, V.; Barilli, A.; D’Ippolito, R.; Tipa, A.; Olivieri, D.; Gazzola, G.C.; Bussolati, O. Alveolar Macrophages from Normal Subjects Lack the NOS-Related System y + for Arginine Transport. Am. J. Respir. Cell Mol. Biol. 2007, 37, 105–112. [Google Scholar] [CrossRef]
- Barilli, A.; Rotoli, B.; Visigalli, R.; Bussolati, O.; Gazzola, G.C.; Kadija, Z.; Rodi, G.; Mariani, F.; Ruzza, M.; Luisetti, M.; et al. In Lysinuric Protein Intolerance system y+L activity is defective in monocytes and in GM-CSF-differentiated macrophages. Orphanet J. Rare Dis. 2010, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Bijarnia-Mahay, S.; Jain, V.; Bansal, R.K.; Reddy, G.M.; Häberle, J. Lysinuric protein intolerance presenting with recurrent hyperammonemic encephalopathy. Indian Pediatr. 2016, 53, 732–734. [Google Scholar] [CrossRef] [PubMed]
- Rama Rao, K.V.; Jayakumar, A.R.; Tong, X.; Alvarez, V.M.; Norenberg, M.D. Marked potentiation of cell swelling by cytokines in ammonia-sensitized cultured astrocytes. J. Neuroinflamm. 2010, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Hughes, R.D.; Keays, R.T.; Williams, R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology 1992, 15, 1060–1066. [Google Scholar] [CrossRef]
- Rossi, F.; Casano, A.M.; Henke, K.; Richter, K.; Peri, F. The SLC7A7 Transporter Identifies Microglial Precursors prior to Entry into the Brain. Cell Rep. 2015, 11, 1008–1017. [Google Scholar] [CrossRef]
- Sperandeo, M.P.; Annunziata, P.; Bozzato, A.; Piccolo, P.; Maiuri, L.; D’Armiento, M.; Ballabio, A.; Corso, G.; Andria, G.; Borsani, G.; et al. Slc7a7 disruption causes fetal growth retardation by downregulating Igf1 in the mouse model of lysinuric protein intolerance. Am. J. Physiol. Cell Physiol. 2007, 293, C191–C198. [Google Scholar] [CrossRef]
- Ruzankina, Y.; Pinzon-Guzman, C.; Asare, A.; Ong, T.; Pontano, L.; Cotsarelis, G.; Zediak, V.P.; Velez, M.; Bhandoola, A.; Brown, E.J.; et al. Deletion of the Developmentally Essential Gene ATR in Adult Mice Leads to Age-Related Phenotypes and Stem Cell Loss. Cell Stem Cell 2007, 1, 113–126. [Google Scholar] [CrossRef]
- Bartoccioni, P.; Fort, J.; Zorzano, A.; Errasti-Murugarren, E.; Palacín, M. Functional characterization of the alanine-serine-cysteine exchanger of Carnobacterium sp AT7. J. Gen. Physiol. 2019, 151, 505–517. [Google Scholar] [CrossRef]
- Errasti-Murugarren, E.; Fort, J.; Bartoccioni, P.; Díaz, L.; Pardon, E.; Carpena, X.; Espino-Guarch, M.; Zorzano, A.; Ziegler, C.; Steyaert, J.; et al. L amino acid transporter structure and molecular bases for the asymmetry of substrate interaction. Nat. Commun. 2019, 10, 1807. [Google Scholar] [CrossRef]
- Yan, R.; Zhao, X.; Lei, J.; Zhou, Q. Structure of the human LAT1-4F2hc heteromeric amino acid transporter complex. Nature 2019, 568, 127–130. [Google Scholar] [CrossRef]
- Persson, P.; Fasching, A.; Teerlink, T.; Hansell, P.; Palm, F. L-citrulline, but not l-arginine, prevents diabetes mellitus-induced glomerular hyperfiltration and proteinuria in rat. Hypertension 2014, 64, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, M.E.; Brosnan, J.T. Renal Arginine Metabolism. J. Nutr. 2004, 134, 2791S–2795S. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Palacín, M. The role of amino acid transporters in inherited and acquired diseases. Biochem. J. 2011, 436, 193–211. [Google Scholar] [CrossRef] [PubMed]
- Vijay, G.K.M.; Hu, C.; Peng, J.; Martinez, I.G.; Hoque, R.; Verghis, R.M.; Ma, Y.; Mehal, W.; Shawcross, D.L.; Wen, L. Ammonia-Induced Brain Edema Requires Macrophage and T Cell Expression of Toll-Like Receptor 9. Cell. Mol. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Trapnell, B.C.; Whitsett, J.A.; Nakata, K. Pulmonary Alveolar Proteinosis. N. Engl. J. Med. 2003, 349, 2527–2539. [Google Scholar] [CrossRef]
- Tanner, L.M.; Kurko, J.; Tringham, M.; Aho, H.; Mykkänen, J.; Näntö-Salonen, K.; Niinikoski, H.; Lukkarinen, H. Inhaled Sargramostim Induces Resolution of Pulmonary Alveolar Proteinosis in Lysinuric Protein Intolerance. In JIMD Reports; Wiley-Blackwell: Hoboken, NJ, USA, 2016; pp. 97–104. [Google Scholar]
- Valimahamed-Mitha, S.; Berteloot, L.; Ducoin, H.; Ottolenghi, C.; de Lonlay, P.; de Blic, J. Lung involvement in children with lysinuric protein intolerance. J. Inherit. Metab. Dis. 2015, 38, 257–263. [Google Scholar] [CrossRef]
- Perheentupa, J.; Visakorpi, J.K. Protein intolerance with deficient transport of basic aminoacids. Another Inborn Error of Metabolism. Lancet 1965, 2, 813–816. [Google Scholar] [CrossRef]
- Bender, D.A. Amino Acid Metabolism, 3rd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2012; ISBN 9780470661512. [Google Scholar]
- De Jonge, W.J.; Hallemeesch, M.M.; Kwikkers, K.L.; Ruijter, J.M.; De Vries, C.G.D.; Van Roon, M.A.; Meijer, A.J.; Marescau, B.; De Deyn, P.P.; Deutz, N.E.P.; et al. Overexpression of arginase I in enterocytes of transgenic mice elicits a selective arginine deficiency and affects skin, muscle, and lymphoid development. Am. J. Clin. Nutr. 2002, 76, 128–140. [Google Scholar] [CrossRef]
- Davis, T.A.; Fiorotto, M.L.; Reeds, P.J. Amino Acid Compositions of Body and Milk Protein Change during the Suckling Period in Rats. J. Nutr. 1993, 123, 947–956. [Google Scholar] [CrossRef]
- de Jonge, W.J.; Dingemanse, M.A.; de Boer, P.A.J.; Lamers, W.H.; Moorman, A.F.M. Arginine-Metabolizing Enzymes in the Developing Rat Small Intestine. Pediatr. Res. 1998, 43, 442–451. [Google Scholar] [CrossRef]
- Richir, M.C.; Siroen, M.P.C.; van Elburg, R.M.; Fetter, W.P.F.; Quik, F.; Nijveldt, R.J.; Heij, H.A.; Smit, B.J.; Teerlink, T.; van Leeuwen, P.A.M. Low plasma concentrations of arginine and asymmetric dimethylarginine in premature infants with necrotizing enterocolitis. Br. J. Nutr. 2007, 97, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Erez, A.; Nagamani, S.C.S.; Shchelochkov, O.A.; Premkumar, M.H.; Campeau, P.M.; Chen, Y.; Garg, H.K.; Li, L.; Mian, A.; Bertin, T.K.; et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 2011, 17, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.Y.; Ballantyne, L.L.; Mukherjee, K.; St Amand, T.; Kyriakopoulou, L.; Schulze, A.; Funk, C.D. Inducible arginase 1 deficiency in mice leads to hyperargininemia and altered amino acid metabolism. PLoS ONE 2013, 8, e80001. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, F.; Brancaccio, G.; Parenti, G.; Francalanci, P.; Squitieri, C.; Sebastio, G.; Dionisi-Vici, C.; D’Argenio, P.; Andria, G.; Parisi, F. Recurrent fatal pulmonary alveolar proteinosis after heart-lung transplantation in a child with lysinuric protein intolerance. J. Pediatr. 2004, 145, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Sempere, A.; Arias, A.; Farré, G.; García-Villoria, J.; Rodríguez-Pombo, P.; Desviat, L.R.; Merinero, B.; García-Cazorla, A.; Vilaseca, M.A.; Ribes, A.; et al. Study of inborn errors of metabolism in urine from patients with unexplained mental retardation. J. Inherit. Metab. Dis. 2010, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Boiadjieva, E.; Vilches, C.; Bodoy, S.; Oparija, L.; Jando, J.; Nunes, V.; Verrey, F.; Palacin, M. Cooperation of basolateral epithelial amino acid transporters TAT1 and LAT2 investigated in a double knockout mouse model. Amino Acids 2015, 47, 1627. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Control | Slc7a7-/- | Control + Cit | Slc7a7-/- + Cit |
|---|---|---|---|---|
| ARG | 99.5 ± 0.6 | 53.2 ± 13.4 ** | 97.6 ± 0.7 # | −213.3 ± 112.5 *# |
| ORN | 99.1 ± 0.1 | 50.6 ± 11.7 ** | 99.1 ± 0.2 | −51.5 ± 38.1 **# |
| LYS | 99.7 ± 0.1 | 98.4 ± 0.4 * | 99.6 ± 0.1 | 94.5 ± 1.5 **# |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodoy, S.; Sotillo, F.; Espino-Guarch, M.; Sperandeo, M.P.; Ormazabal, A.; Zorzano, A.; Sebastio, G.; Artuch, R.; Palacín, M. Inducible Slc7a7 Knockout Mouse Model Recapitulates Lysinuric Protein Intolerance Disease. Int. J. Mol. Sci. 2019, 20, 5294. https://doi.org/10.3390/ijms20215294
Bodoy S, Sotillo F, Espino-Guarch M, Sperandeo MP, Ormazabal A, Zorzano A, Sebastio G, Artuch R, Palacín M. Inducible Slc7a7 Knockout Mouse Model Recapitulates Lysinuric Protein Intolerance Disease. International Journal of Molecular Sciences. 2019; 20(21):5294. https://doi.org/10.3390/ijms20215294
Chicago/Turabian StyleBodoy, Susanna, Fernando Sotillo, Meritxell Espino-Guarch, Maria Pia Sperandeo, Aida Ormazabal, Antonio Zorzano, Gianfranco Sebastio, Rafael Artuch, and Manuel Palacín. 2019. "Inducible Slc7a7 Knockout Mouse Model Recapitulates Lysinuric Protein Intolerance Disease" International Journal of Molecular Sciences 20, no. 21: 5294. https://doi.org/10.3390/ijms20215294
APA StyleBodoy, S., Sotillo, F., Espino-Guarch, M., Sperandeo, M. P., Ormazabal, A., Zorzano, A., Sebastio, G., Artuch, R., & Palacín, M. (2019). Inducible Slc7a7 Knockout Mouse Model Recapitulates Lysinuric Protein Intolerance Disease. International Journal of Molecular Sciences, 20(21), 5294. https://doi.org/10.3390/ijms20215294