Novel Crizotinib–GnRH Conjugates Revealed the Significance of Lysosomal Trapping in GnRH-Based Drug Delivery Systems

 , , ,
, , , 
Abstract
:
1. Introduction
2. Results
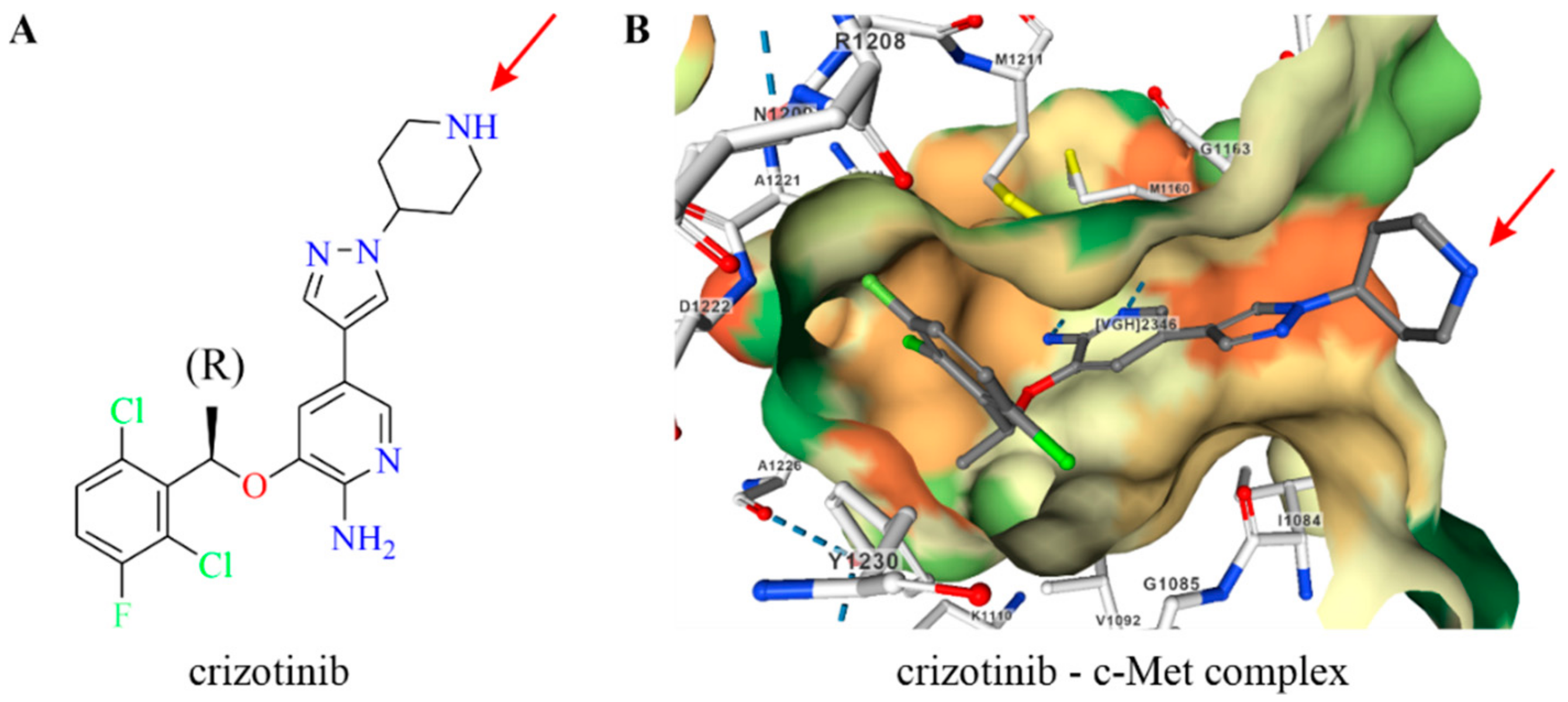
2.1. Design of Crizotinib–GNRH Conjugates
2.2. Synthesis of Crizotinib Analogues and Crizotinib–GNRH Conjugates
2.3. GnRHR Expression and GnRH Uptake of EBC-1 NSCLC Cells
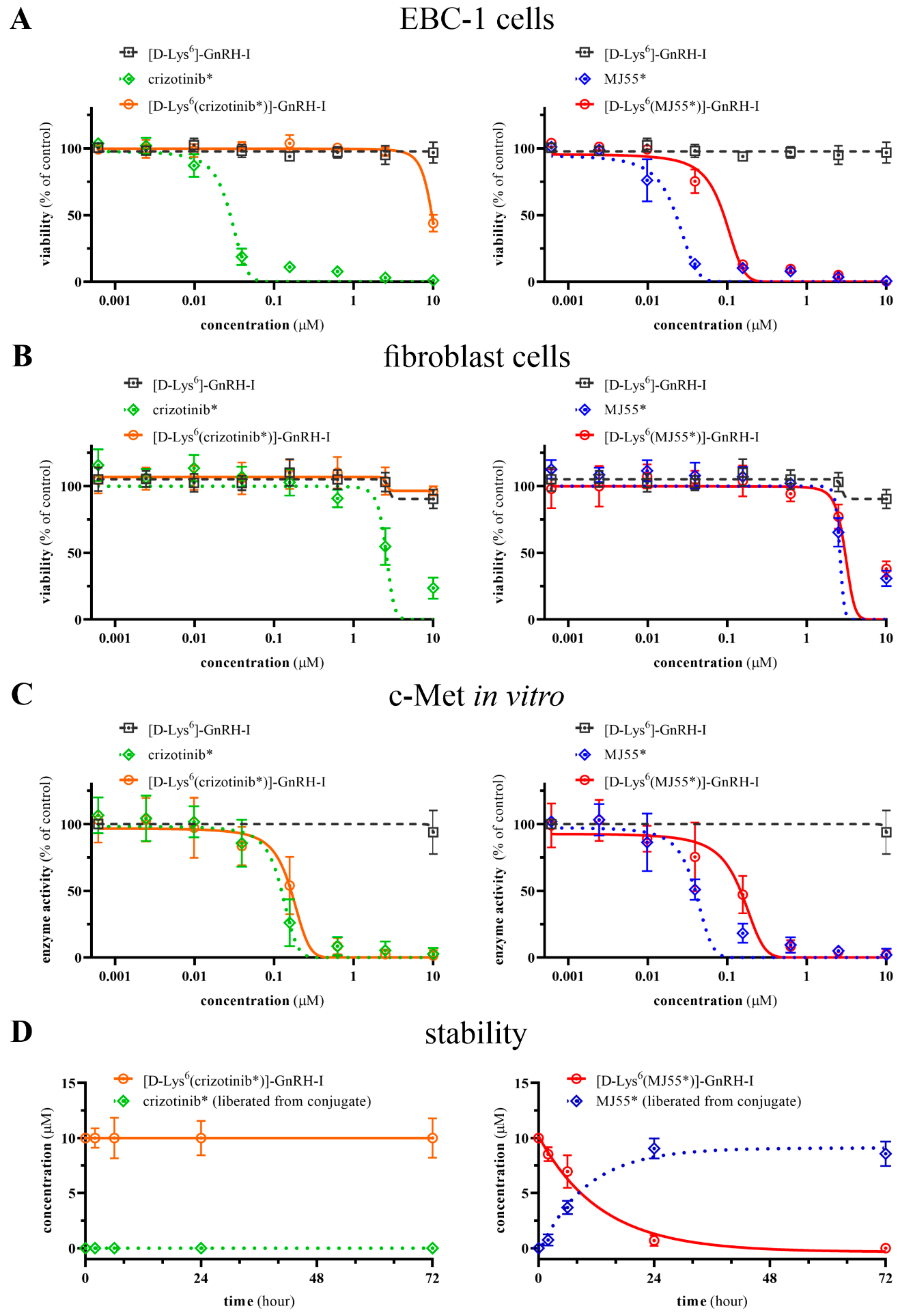
2.4. Viability Inhibition Efficacy of Compounds on EBC-1 Cells and Primary Skin Fibroblast Cells
2.5. In Vitro c-Met Inhibition Efficacy of Compounds
2.6. Stability of Compounds in Cell Culture Medium
2.7. GnRHR-Binding Affinity of Conjugates
2.8. c-Met Inhibition of Compounds in EBC-1 Cells
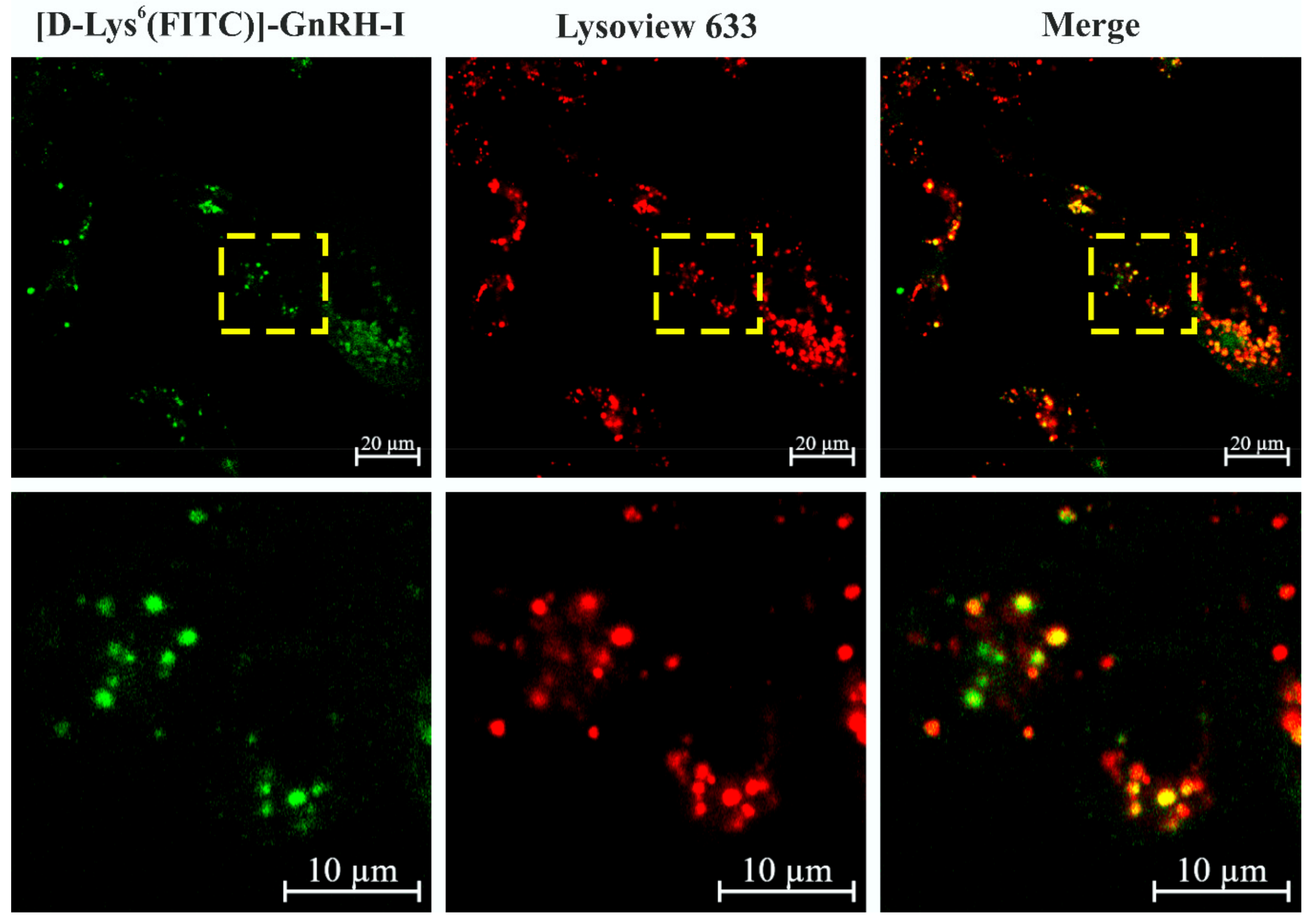
2.9. Colocalization of [d-Lys6(FITC)]–GnRH-I and Lysosomes
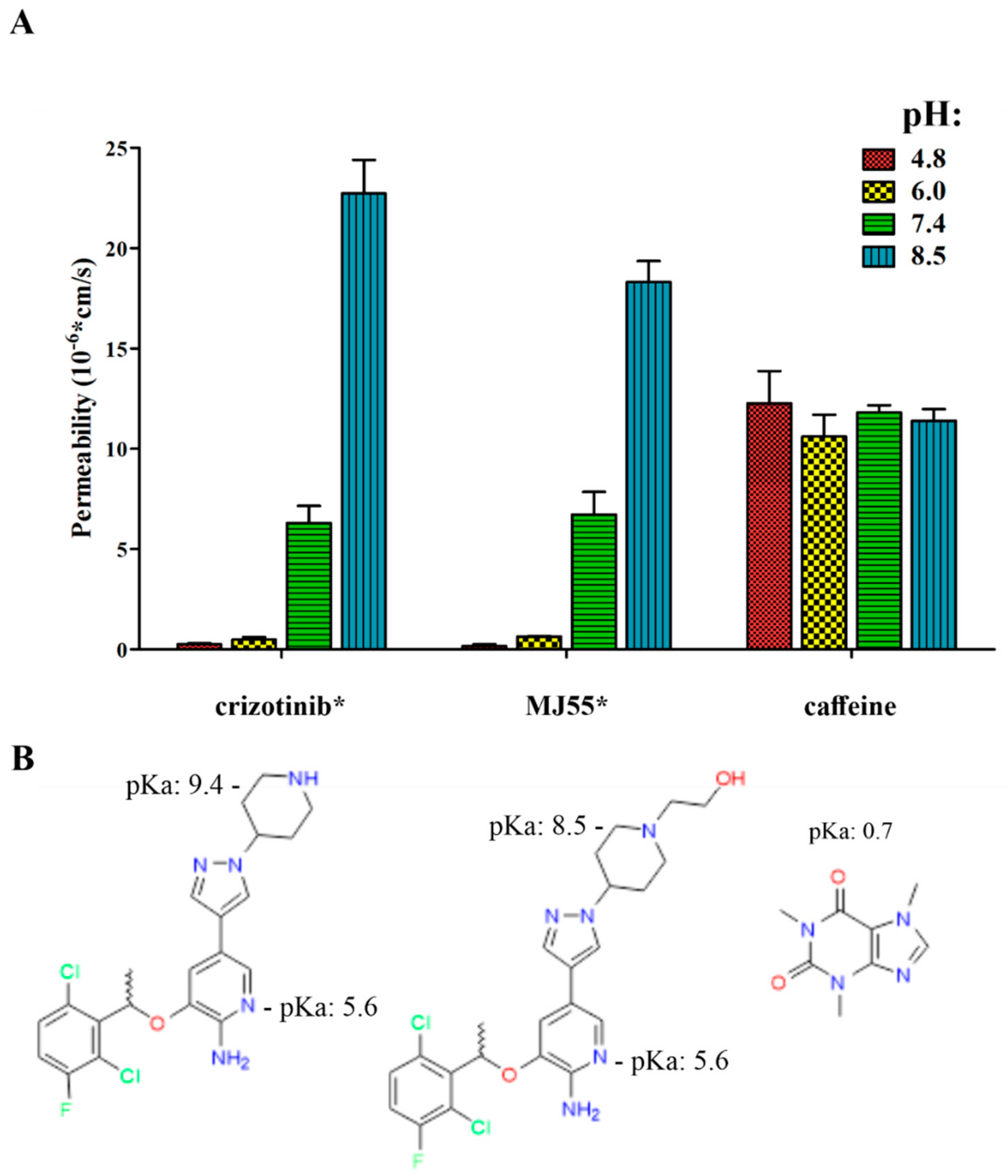
2.10. The pH-Dependent Permeability of Crizotinib* and MJ55*
2.11. [d-Lys6(crizotinib*)]–GnRH-I Resulted in Concentration-Dependent Lysosomal Membrane Permeabilization
3. Discussion
4. Materials and Methods
4.1. Synthesis of Compounds
4.2. Cell Cultures
4.3. Western Blot
4.4. Immunofluorescence Staining of GnRHR
4.5. Intracellular Localization of [d-Lys6(FITC)]–GnRH-I
4.6. Quantitative Analysis of [d-Lys6(FITC)]–GnRH-I in EBC-1 Cells
- x time (h)
- y concentration (µM)
4.7. Cell Viability Assay
4.8. In Vitro Inhibition of Recombinant c-Met Kinase
4.9. Stability Experiment
4.10. PAMPA
4.11. Galectin Puncta Assay
4.12. Radioligand-Binding Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALK | anaplastic lymphoma kinase |
| BOC | tert-butyloxycarbonyl-protecting group |
| CLSM | confocal laser scanning microscopy |
| c-Met | hepatocyte growth factor receptor |
| COMU | (1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate |
| crizotinib | (R)-crizotinib, (Xalkori®, Pfizer) |
| crizotinib* | racemic mixture of (R)-crizotinib and (S)-crizotinib |
| EMEM | Eagle’s minimum essential medium |
| FBS | Fetal bovine serum |
| fibroblast cells | human primary human skin fibroblast cells |
| galectin-1 | lectin galactoside binding soluble 1 |
| galectin-3 | lectin galactoside binding soluble 3 |
| HRMS | high resolution mass spectrometry |
| LMP | lysosomal membrane permeabilization |
| MJ55* | racemic mixture of (R)-MJ55 and (S)-MJ55 |
| NSCLC | non-small cell lung cancer |
| Pd(dppf)Cl2 | [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) |
| ROS-1 | ROS proto-oncogene 1, receptor tyrosine kinase |
| RTK | receptor tyrosine kinase |
| SD | standard deviation |
| TEA | triethylamine |
| TFEB | transcription factor EB |
| THF | tetrahydrofuran |
References
- Maggi, R.; Cariboni, A.M.; Marelli, M.M.; Moretti, R.M.; Andre, V.; Marzagalli, M.; Limonta, P. GnRH and GnRH receptors in the pathophysiology of the human female reproductive system. Hum. Reprod. Update 2016, 22, 358–381. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Montagnani, M.M.; Mai, S.; Motta, M.; Martini, L.; Moretti, R.M. GnRH receptors in cancer: From cell biology to novel targeted therapeutic strategies. Endocr. Rev. 2012, 33, 784–811. [Google Scholar] [CrossRef] [PubMed]
- Mezo, G.; Manea, M. Receptor-mediated tumor targeting based on peptide hormones. Expert Opin. Drug Deliv. 2010, 7, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Ndinguri, M.W.; Solipuram, R.; Wakamatsu, N.; Hammer, R.P.; Ingram, D.; Hansel, W. [DLys(6)]-luteinizing hormone releasing hormone-curcumin conjugate inhibits pancreatic cancer cell growth in vitro and in vivo. Int. J. Cancer 2011, 129, 1611–1623. [Google Scholar] [CrossRef]
- Argyros, O.; Karampelas, T.; Asvos, X.; Varela, A.; Sayyad, N.; Papakyriakou, A.; Davos, C.H.; Tzakos, A.G.; Fokas, D.; Tamvakopoulos, C. Peptide-Drug Conjugate GnRH-Sunitinib Targets Angiogenesis Selectively at the Site of Action to Inhibit Tumor Growth. Cancer Res. 2016, 76, 1181–1192. [Google Scholar] [CrossRef]
- Karampelas, T.; Argyros, O.; Sayyad, N.; Spyridaki, K.; Pappas, C.; Morgan, K.; Kolios, G.; Millar, R.P.; Liapakis, G.; Tzakos, A.G.; et al. GnRH-Gemcitabine conjugates for the treatment of androgen-independent prostate cancer: Pharmacokinetic enhancements combined with targeted drug delivery. Bioconjug Chem. 2014, 25, 813–823. [Google Scholar] [CrossRef]
- Nagy, A.; Schally, A.V. Targeting of cytotoxic luteinizing hormone-releasing hormone analogs to breast, ovarian, endometrial, and prostate cancers. Biol. Reprod. 2005, 73, 851–859. [Google Scholar] [CrossRef]
- Fabbro, D. 25 years of small molecular weight kinase inhibitors: Potentials and limitations. Mol. Pharmacol. 2015, 87, 766–775. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dube, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- Puccini, A.; Marin-Ramos, N.I.; Bergamo, F.; Schirripa, M.; Lonardi, S.; Lenz, H.J.; Loupakis, F.; Battaglin, F. Safety and Tolerability of c-MET Inhibitors in Cancer. Drug Saf. 2019, 42, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Guo, G.; Zou, P.; Cui, R.; Chen, W.; Chen, X.; Yin, C.; He, W.; Vinothkumar, R.; Yang, F.; et al. (S)-crizotinib induces apoptosis in human non-small cell lung cancer cells by activating ROS independent of MTH1. J. Exp. Clin. Cancer Res. 2017, 36, 120. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Chen, W.; Lian, W.; Chen, R.; Yang, J.; Zhang, Q.; Weng, Q.; Khan, Z.; Hu, J.; Chen, X.; et al. (S)-crizotinib reduces gastric cancer growth through oxidative DNA damage and triggers pro-survival akt signal. Cell Death Dis. 2018, 9, 660. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann-Guerrero, D.; Kahnert, K.; Kumbrink, J.; Syunyaeva, Z.; Tufman, A.; Huber, R.M. Successful Treatment of a Patient with NSCLC Harboring an EGFR Mutation and a Concomitant Met Exon 14 Skipping Mutation Combining Afatinib and Crizotinib. Clin. Lung Cancer 2019, 20, 59–62. [Google Scholar] [CrossRef]
- Llanos, S.; Megias, D.; Blanco-Aparicio, C.; Hernandez-Encinas, E.; Rovira, M.; Pietrocola, F.; Serrano, M. Lysosomal trapping of palbociclib and its functional implications. Oncogene 2019, 38, 3886–3902. [Google Scholar] [CrossRef] [Green Version]
- Li, W.Q.; Sun, L.P.; Xia, Y.; Hao, S.; Cheng, G.; Wang, Z.; Wan, Y.; Zhu, C.; He, H.; Zheng, S.Y. Preoccupation of Empty Carriers Decreases Endo-/Lysosome Escape and Reduces the Protein Delivery Efficiency of Mesoporous Silica Nanoparticles. ACS Appl. Mater. Interfaces 2018, 10, 5340–5347. [Google Scholar] [CrossRef]
- Muranyi, J.; Gyulavari, P.; Varga, A.; Bokonyi, G.; Tanai, H.; Vantus, T.; Pap, D.; Ludanyi, K.; Mezo, G.; Keri, G. Synthesis, characterization and systematic comparison of FITC-labelled GnRH-I, -II and -III analogues on various tumour cells. J. Pept. Sci. 2016, 22, 552–560. [Google Scholar] [CrossRef]
- Halmos, G.; Arencibia, J.M.; Schally, A.V.; Davis, R.; Bostwick, D.G. High incidence of receptors for luteinizing hormone-releasing hormone (LHRH) and LHRH receptor gene expression in human prostate cancers. J. Urol. 2000, 163, 623–629. [Google Scholar] [CrossRef]
- Sahu, A.; Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S. Crizotinib: A comprehensive review. South Asian J. Cancer 2013, 2, 91–97. [Google Scholar]
- Engel, J.; Emons, G.; Pinski, J.; Schally, A.V. AEZS-108: A targeted cytotoxic analog of LHRH for the treatment of cancers positive for LHRH receptors. Expert Opin. Investig. Drugs 2012, 21, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Popovics, P.; Schally, A.V.; Szalontay, L.; Block, N.L.; Rick, F.G. Targeted cytotoxic analog of luteinizing hormone-releasing hormone (LHRH), AEZS-108 (AN-152), inhibits the growth of DU-145 human castration-resistant prostate cancer in vivo and in vitro through elevating p21 and ROS levels. Oncotarget 2014, 5, 4567–4578. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Schally, A.V.; Armatis, P.; Szepeshazi, K.; Halmos, G.; Kovacs, M.; Zarandi, M.; Groot, K.; Miyazaki, M.; Jungwirth, A.; et al. Cytotoxic analogs of luteinizing hormone-releasing hormone containing doxorubicin or 2-pyrrolinodoxorubicin, a derivative 500–1000 times more potent. Proc. Natl. Acad. Sci. USA 1996, 93, 7269–7273. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Siahaan, T.J. Peptide-mediated targeted drug delivery. Med. Res. Rev. 2012, 32, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, M.; Berger, S.; Poethko, T.; Schwaiger, M.; Wester, H.J. Development of novel 68Ga- and 18F-labeled GnRH-I analogues with high GnRHR-targeting efficiency. Bioconjug. Chem. 2008, 19, 1256–1268. [Google Scholar] [CrossRef]
- Finch, A.R.; Caunt, C.J.; Armstrong, S.P.; McArdle, C.A. Plasma membrane expression of gonadotropin-releasing hormone receptors: Regulation by peptide and nonpeptide antagonists. Mol. Endocrinol. 2010, 24, 423–435. [Google Scholar] [CrossRef]
- denFinch, A.R.; Sedgley, K.R.; Armstrong, S.P.; Caunt, C.J.; McArdle, C.A. Trafficking and signalling of gonadotrophin-releasing hormone receptors: An automated imaging approach. Br. J. Pharmacol. 2010, 159, 751–760. [Google Scholar]
- Hao, D.; Sun, L.; Hu, X.; Hao, X. (99m)Tc-LHRH in tumor receptor imaging. Oncol. Lett. 2017, 14, 569–578. [Google Scholar] [CrossRef]
- Lu, C.; Huang, T.; Chen, W.; Lu, H. GnRH participates in the self-renewal of A549-derived lung cancer stem-like cells through upregulation of the JNK signaling pathway. Oncol. Rep. 2015, 34, 244–250. [Google Scholar] [CrossRef]
- Nagy, A.; Plonowski, A.; Schally, A.V. Stability of cytotoxic luteinizing hormone-releasing hormone conjugate (AN-152) containing doxorubicin 14-O-hemiglutarate in mouse and human serum in vitro: Implications for the design of preclinical studies. Proc. Natl. Acad. Sci. USA 2000, 97, 829–834. [Google Scholar] [CrossRef]
- Hazum, E.; Koch, Y.; Liscovitch, M.; Amsterdam, A. Intracellular pathways of receptor-bound GnRH agonist in pituitary gonadotropes. Cell Tissue Res. 1985, 239, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Schvartz, I.; Hazum, E. Internalization and recycling of receptor-bound gonadotropin-releasing hormone agonist in pituitary gonadotropes. J. Biol. Chem. 1987, 262, 17046–17050. [Google Scholar] [PubMed]
- Da Silva, C.G.; Honeywell, R.J.; Dekker, H.; Peters, G.J. Physicochemical properties of novel protein kinase inhibitors in relation to their substrate specificity for drug transporters. Expert Opin. Drug Metab. Toxicol. 2015, 11, 703–717. [Google Scholar] [CrossRef] [PubMed]
- De Klerk, D.J.; Honeywell, R.J.; Jansen, G.; Peters, G.J. Transporter and Lysosomal Mediated (Multi)drug Resistance to Tyrosine Kinase Inhibitors and Potential Strategies to Overcome Resistance. Cancers 2018, 10, 503. [Google Scholar] [CrossRef]
- Zhong, Y.J.; Shao, L.H.; Li, Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review). Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef]
- Avet, C.; Garrel, G.; Denoyelle, C.; Laverriere, J.N.; Counis, R.; Cohen-Tannoudji, J.; Simon, V. SET protein interacts with intracellular domains of the gonadotropin-releasing hormone receptor and differentially regulates receptor signaling to cAMP and calcium in gonadotrope cells. J. Biol. Chem. 2013, 288, 2641–2654. [Google Scholar] [CrossRef]
- Brothers, S.P.; Janovick, J.A.; Conn, P.M. Calnexin regulated gonadotropin-releasing hormone receptor plasma membrane expression. J. Mol. Endocrinol 2006, 37, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Jardón-Valadez, E.; Aguilar-Rojas, A.; Maya-Núñez, G.; Leaños-Miranda, A.; Piñeiro, A.; Conn, P.M.; Ulloa-Aguirre, A. Conformational effects of Lys191 in the human GnRH receptor: Mutagenesis and molecular dynamics simulations studies. J. Endocrinol. 2009, 201, 297–307. [Google Scholar] [CrossRef]
- Kelsey, R. Macropinocytosis for proliferation. Nat. Rev. Urol. 2018, 15, 336–337. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Yunaev, A.; Kreiserman, R.; Kaplan, A.; Stark, M.; Assaraf, Y.G. Lysosomotropic drugs activate TFEB via lysosomal membrane fluidization and consequent inhibition of mTORC1 activity. Cell Death Dis. 2018, 90, 1191. [Google Scholar] [CrossRef]
- Villamil Giraldo, A.M.; Appelqvist, H.; Ederth, T.; Ollinger, K. Lysosomotropic agents: Impact on lysosomal membrane permeabilization and cell death. Biochem. Soc. Trans. 2014, 42, 1460–1464. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-E.; Lin, J.-F.; Tsai, T.-F.; Lin, Y.-C.; Chou, K.-Y.; Hwang, T.I.S. Chloroquine induces lysosomal membrane permeability mediated apoptotic cell death in bladder cancer cells. Collect. Urol. Sci. 2016, 27, S5. [Google Scholar] [CrossRef] [Green Version]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aits, S.; Kricker, J.; Liu, B.; Ellegaard, A.M.; Hamalisto, S.; Tvingsholm, S.; Corcelle-Termeau, E.; Hogh, S.; Farkas, T.; Holm Jonassen, A.; et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11, 1408–1424. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Biri-Kovacs, B.; Szeder, B.; Buday, L.; Gardi, J.; Szabo, Z.; Halmos, G.; Mezo, G. Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification. Pharmaceutics 2018, 10, 223. [Google Scholar] [CrossRef]
- Szabo, I.; Bosze, S.; Orban, E.; Sipos, E.; Halmos, G.; Kovacs, M.; Mezo, G. Comparative in vitro biological evaluation of daunorubicin containing GnRH-I and GnRH-II conjugates developed for tumor targeting. J. Pept. Sci. 2015, 21, 426–435. [Google Scholar] [CrossRef]
- Zhou, L.; Lv, F.; Liu, L.; Wang, S. Polarity Conversion of Conjugated Polymer for Lysosome Escaping. ACS Appl. Mater. Interfaces 2017, 9, 27427–27432. [Google Scholar] [CrossRef]
- Towers, C.G.; Thorburn, A. Targeting the Lysosome for Cancer Therapy. Cancer Discov. 2017, 7, 1218–1220. [Google Scholar] [CrossRef] [Green Version]
- Domagala, A.; Fidyt, K.; Bobrowicz, M.; Stachura, J.; Szczygiel, K.; Firczuk, M. Typical and Atypical Inducers of Lysosomal Cell Death: A Promising Anticancer Strategy. Int. J. Mol. Sci. 2018, 19, 2256. [Google Scholar] [CrossRef]
- Rahimipour, S.; Ben-Aroya, N.; Ziv, K.; Chen, A.; Fridkin, M.; Koch, Y. Receptor-mediated targeting of a photosensitizer by its conjugation to gonadotropin-releasing hormone analogues. J. Med. Chem. 2003, 46, 3965–3974. [Google Scholar] [CrossRef]
- Petho, L.; Muranyi, J.; Penzes, K.; Gurbi, B.; Brauswetter, D.; Halmos, G.; Csik, G.; Mezo, G. Suitability of GnRH Receptors for Targeted Photodynamic Therapy in Head and Neck Cancers. Int. J. Mol. Sci. 2019, 20, 5027. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 Values (nM) 1 | |
|---|---|---|
| Human Pituitary | Human Prostate Cancer | |
| [d-Lys6]GnRH-I (vehicle) | 6.44 ± 1.0 | 4.31 ± 0.8 |
| [d-Lys6(FITC)]–GnRH-I | 27.2 ± 2.8 | 18.3 ± 2.9 |
| [d-Lys6(crizotinib*)]–GnRH-I | 34.6 ± 4.2 | 39.5 ± 1.7 |
| [d-Lys6(MJ55*)]–GnRH-I | 21.7 ± 1.4 | 29.7 ± 3.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murányi, J.; Varga, A.; Gyulavári, P.; Pénzes, K.; Németh, C.E.; Csala, M.; Pethő, L.; Csámpai, A.; Halmos, G.; Peták, I.; et al. Novel Crizotinib–GnRH Conjugates Revealed the Significance of Lysosomal Trapping in GnRH-Based Drug Delivery Systems. Int. J. Mol. Sci. 2019, 20, 5590. https://doi.org/10.3390/ijms20225590
Murányi J, Varga A, Gyulavári P, Pénzes K, Németh CE, Csala M, Pethő L, Csámpai A, Halmos G, Peták I, et al. Novel Crizotinib–GnRH Conjugates Revealed the Significance of Lysosomal Trapping in GnRH-Based Drug Delivery Systems. International Journal of Molecular Sciences. 2019; 20(22):5590. https://doi.org/10.3390/ijms20225590
Chicago/Turabian StyleMurányi, József, Attila Varga, Pál Gyulavári, Kinga Pénzes, Csilla E. Németh, Miklós Csala, Lilla Pethő, Antal Csámpai, Gábor Halmos, István Peták, and et al. 2019. "Novel Crizotinib–GnRH Conjugates Revealed the Significance of Lysosomal Trapping in GnRH-Based Drug Delivery Systems" International Journal of Molecular Sciences 20, no. 22: 5590. https://doi.org/10.3390/ijms20225590