In Silico Study of Rett Syndrome Treatment-Related Genes, MECP2, CDKL5, and FOXG1, by Evolutionary Classification and Disordered Region Assessment

 , and
, and
Abstract
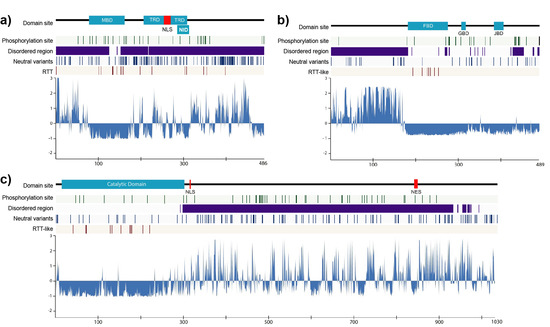
{kind=link}
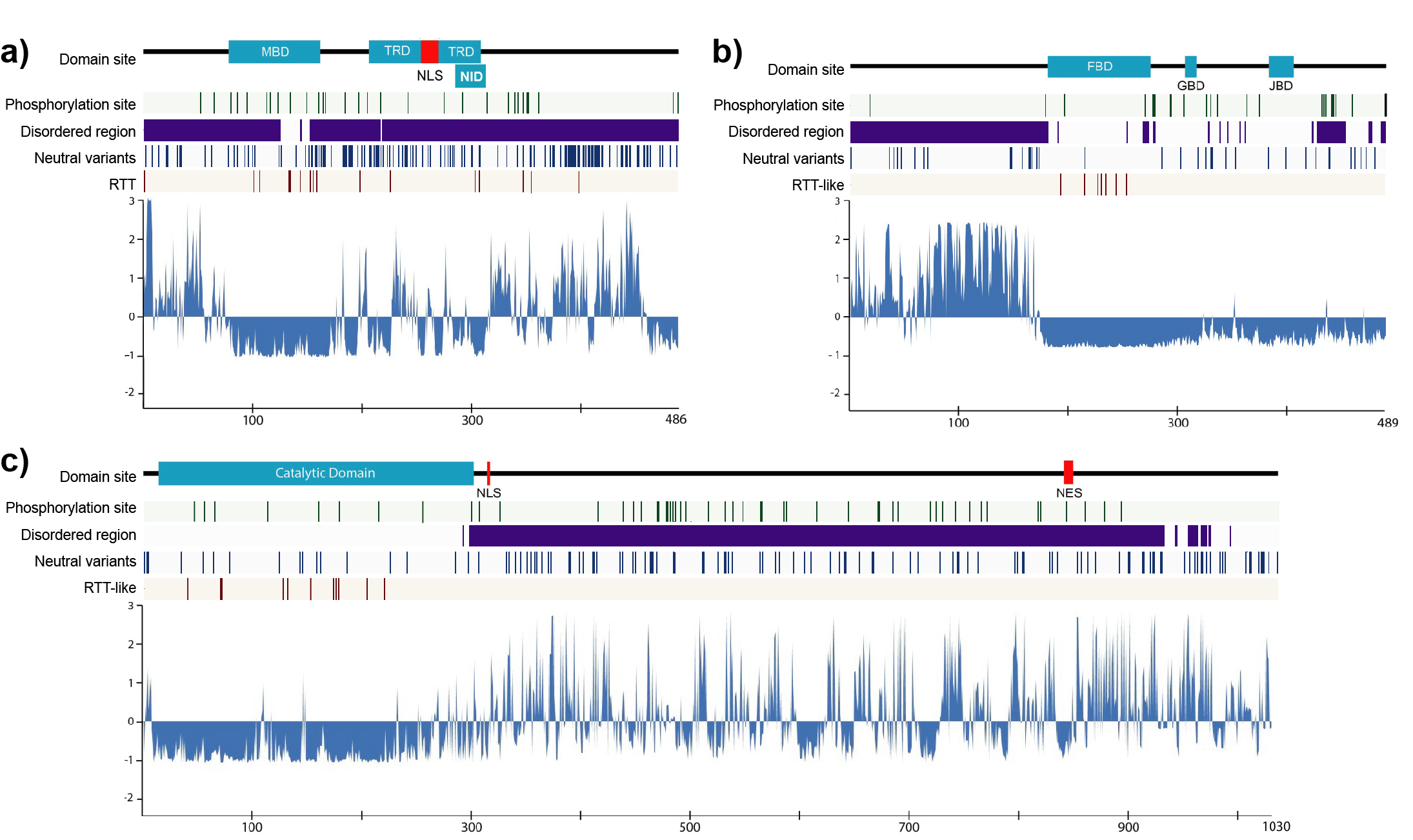
{kind=link}
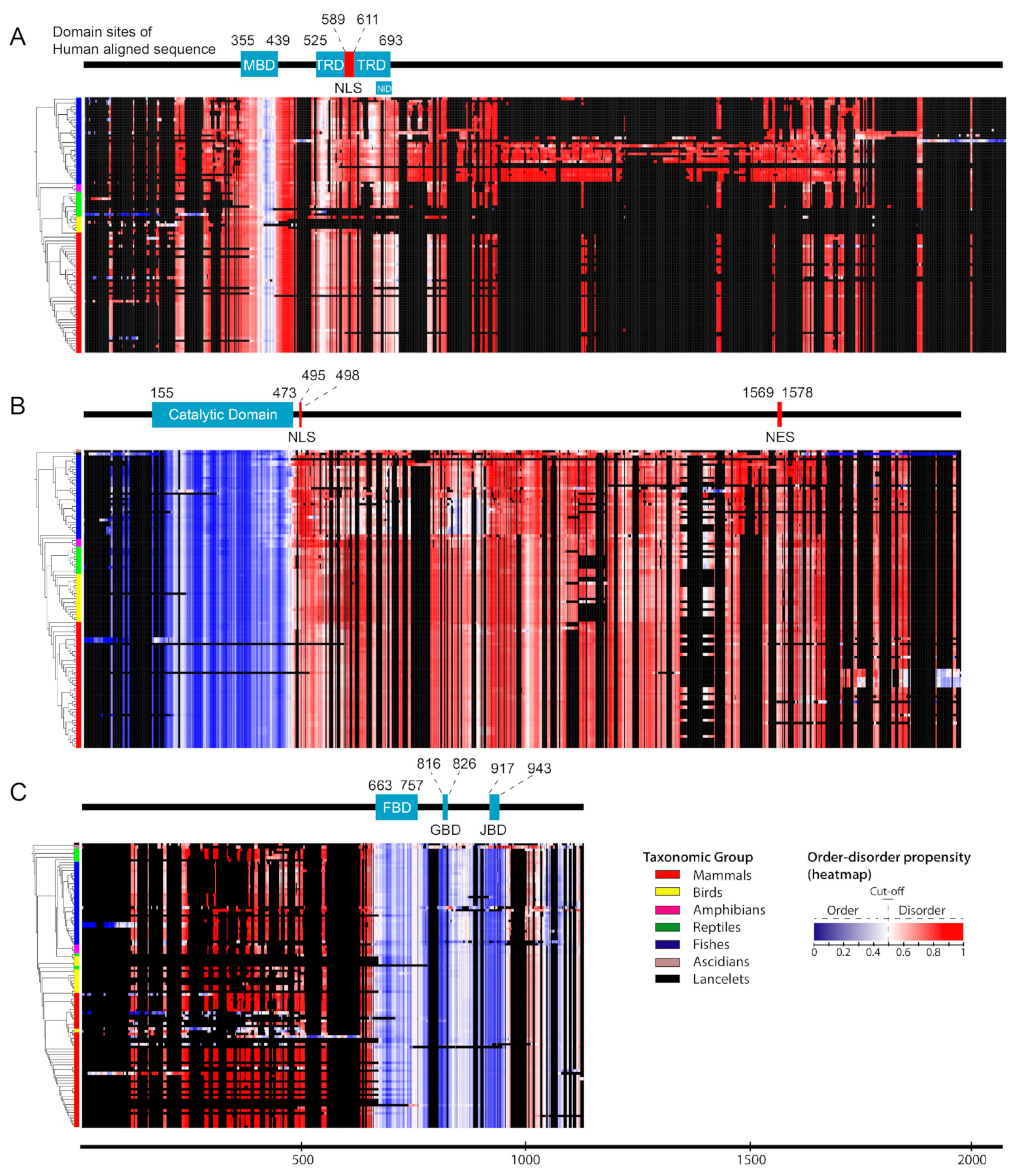
{kind=link}
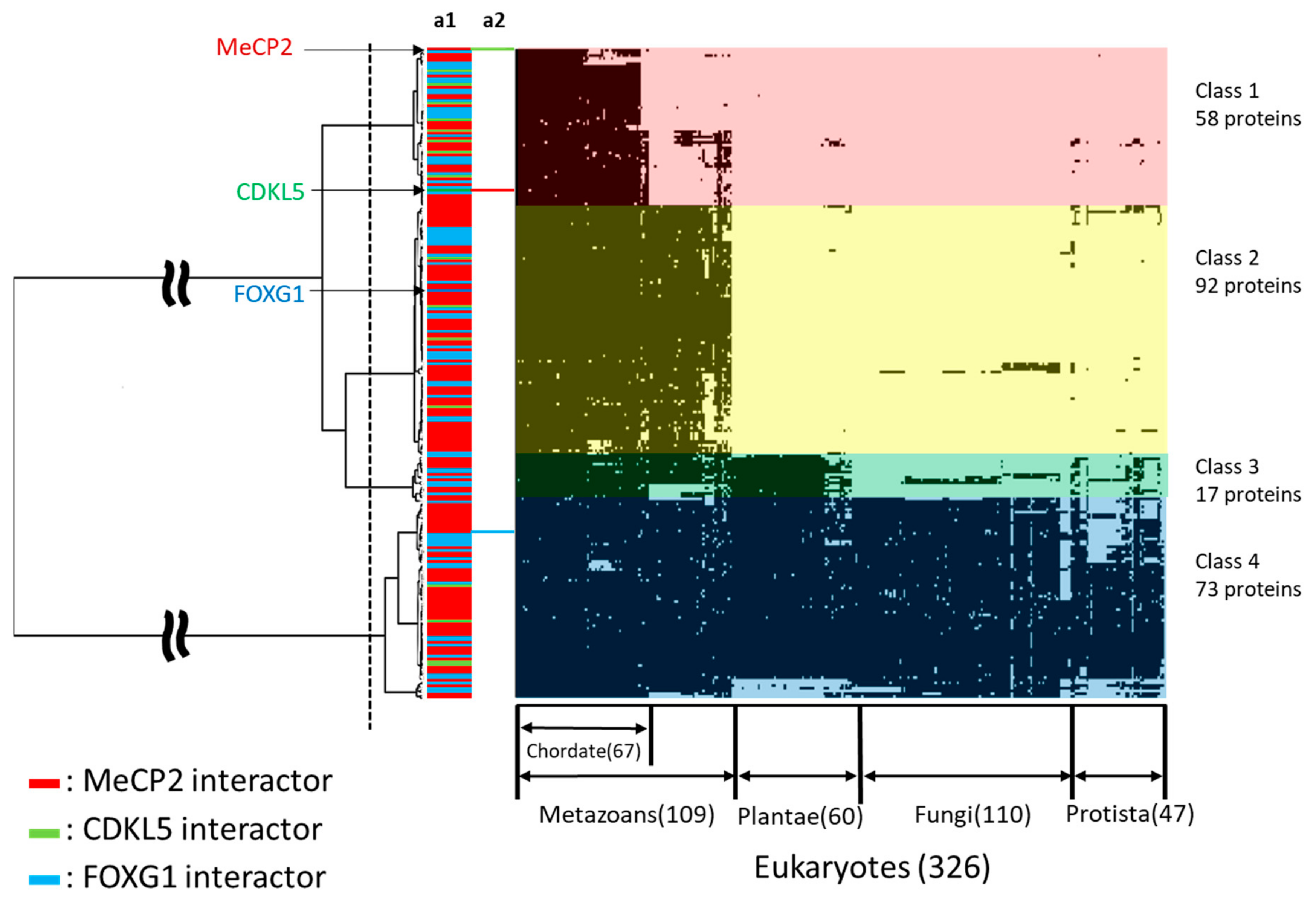{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Structural Order–Disorder Properties of RTT and RTT-like Causing Proteins during Chordate Evolution
2.2. Rate of Evolution per Site in RTT and RTT-like Causing Proteins
2.3. Post-Translational Modifications (PTMs)
2.4. Disease-Associated Missense Mutation Distribution in the Sequence of RTT and RTT-like Causing Proteins
2.5. Phylogenetic Profiling of RTT and RTT-like Causing Proteins and Their Interaction Partners
2.6. Subcellular Localization and Gene Ontology (GO) Analysis
2.7. Tissue and Organ Localization
3. Discussion
4. Materials and Methods
4.1. Sequence Retrieval, Alignment, and Phylogenetic Analysis of MeCP2, CDKL5, and FOXG1 Proteins
4.2. Structural Order–Disorder Prediction and Secondary Structure Predictions
4.3. Rate of Evolution per Site
4.4. PTM Prediction
4.5. Point Mutations in MeCP2, CDKL5, and FOXG1
4.6. Phylogenetic Profiling and Cluster Analyses of Human MeCP2, CDKL5, and FOXG1 and Their Interacting Proteins
4.7. Protein Expression in Human Tissues
4.8. GO Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| a.a | Amino acids |
| AKAP8 | A-kinase anchor protein 8 |
| APOE | Apolipoprotein E |
| BioGRID | Biological General Repository for Interaction Datasets |
| CDKL5 | Cyclin-dependent kinase-like 5 |
| CIPRES | Cyberinfrastructure for Phylogenetic Research |
| CK1 | Casein kinase 1 |
| DOI | Digital object identifier |
| FBD | Forkhead box domain |
| FOXG1 | Forkhead box protein G1 |
| GBD | Groucho-binding domain |
| GluD1 | Glutamate dehydrogenase 1 |
| GO | Gene Ontology |
| HDAC1 | Histone deacetylase 1 |
| HIPK2 | Homeodomain-interacting protein kinase 2 |
| IDDs | Intrinsically disordered domains |
| IDPs | Intrinsically disordered proteins |
| IDRs | Intrinsically disordered regions |
| iPSC | Induced pluripotent stem cell |
| iTOL | Interactive Tree of Life |
| IUPred | Prediction of Intrinsically Unstructured Proteins |
| JBD | JARID1B-binding domain |
| JARID1B | Histone Demethylase Jumonji AT-rich Interactive Domain |
| JTT | The Jones, Taylor, and Thornton |
| KDM1A | Lysine-specific histone demethylase 1A |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MAFFT | Modified Multiple Alignment Fast Fourier Transform |
| MBD | Methyl-CpG-binding domain |
| MeCP2 | Methyl-CpG-binding protein 2 |
| NCOR | Nuclear receptor corepressor |
| NCoR/SMRT | Nuclear receptor co-repressor/silencing mediator of retinoic acid and thyroid hormone receptor |
| NES | Nuclear export signal |
| NLS | Nuclear localization signal |
| NID | NCoR/SMRT interaction domain |
| OMIM | Online Mendelian Inheritance in Man |
| PTM | Post-translational modification |
| RAxML-HPC2 | Randomized Axelerated Maximum Likelihood for High-Performance Computing 2 |
| RTT | Rett syndrome |
| RettBASE | Rett syndrome Variation Database |
| SALL1 | Spalt-like transcription factor 1 |
| SATB2 | Special AT-rich sequence-binding protein 2 |
| SIN3A | SIN3 transcription regulator family member A |
| SLiMs | Short linear motifs |
| SMARCA5 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 |
| SOX2 | SRY-box transcription factor 2 |
| SSDB | Sequence Similarity DataBase |
| TRD | Transcriptional repression domain |
| TPM | Transcripts per million |
| ZNF483 | zinc finger protein (ZNF)483 |
References
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar]
- Hanefeld, F. The clinical pattern of the Rett syndrome. Brain Dev. 1985, 7, 320–325. [Google Scholar] [CrossRef]
- Laurvick, C.L.; De Klerk, N.; Bower, C.; Christodoulou, J.; Ravine, D.; Ellaway, C.; Williamson, S.; Leonard, H. Rett syndrome in Australia: A review of the epidemiology. J. Pediatr. 2006, 148, 347–352. [Google Scholar] [CrossRef]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.; et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Trappe, R.; Laccone, F.; Cobilanschi, J.; Meins, M.; Huppke, P.; Hanefeld, F.; Engel, W. MECP2. mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 2001, 68, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Gecz, J.; et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef]
- Clayton-Smith, J.; Watson, P.; Ramsden, S.; Black, G.C.M. Somatic mutation in MECP2 as a non-fatal neurodevelopmental disorder in males. Lancet 2000, 356, 830–832. [Google Scholar] [CrossRef]
- Ben Zeev, B.; Yaron, Y.; Schanen, N.C.; Wolf, H.; Brandt, N.; Ginot, N.; Shomrat, R.; Orr-Urtreger, A. Rett syndrome: Clinical manifestations in males with MECP2 mutations. J. Child. Neurol. 2002, 17, 20–24. [Google Scholar] [CrossRef]
- Neul, J.L. The relationship of Rett syndrome and MECP2 disorders to autism. Dialogues Clin. Neurosci. 2012, 14, 253–262. [Google Scholar]
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Hector, R.D.; Kalscheuer, V.M.; Hennig, F.; Leonard, H.; Downs, J.; Clarke, A.; Benke, T.A.; Armstrong, J.; Pineda, M.; Bailey, M.E.S.; et al. CDKL5 variants: Improving our understanding of a rare neurologic disorder. Neurol. Genet. 2017, 3, e200. [Google Scholar] [CrossRef] [PubMed]
- Kortum, F.; Das, S.; Flindt, M.; Morris-Rosendahl, D.J.; Stefanova, I.; Goldstein, A.; Horn, D.; Klopocki, E.; Kluger, G.; Martin, P.; et al. The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J. Med. Genet. 2011, 48, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Bird, A. Rett syndrome: A complex disorder with simple roots. Nat. Rev. Genet. 2015, 16, 261–275. [Google Scholar] [CrossRef]
- Tillotson, R.; Selfridge, J.; Koerner, M.V.; Gadalla, K.K.E.; Guy, J.; De Sousa, D.; Hector, R.D.; Cobb, S.R.; Bird, A. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 2017, 550, 398–401. [Google Scholar] [CrossRef]
- Ghosh, R.P.; Nikitina, T.; Horowitz-Scherer, R.A.; Gierasch, L.M.; Uversky, V.N.; Hite, K.; Hansen, J.C.; Woodcock, C.L. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 2010, 49, 4395–4410. [Google Scholar] [CrossRef]
- Toth-Petroczy, A.; Palmedo, P.; Ingraham, J.; Hopf, T.A.; Berger, B.; Sander, C.; Marks, D.S. Structured States of Disordered Proteins from Genomic Sequences. Cell 2016, 167, 158–170. [Google Scholar] [CrossRef]
- Canning, P.; Park, K.; Goncalves, J.; Li, C.; Howard, C.J.; Sharpe, T.D.; Holt, L.J.; Pelletier, L.; Bullock, A.N.; Leroux, M.R. CDKL Family Kinases Have Evolved Distinct Structural Features and Ciliary Function. Cell Rep. 2018, 22, 885–894. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: Which way to go? Cell Mol. Life Sci. 2003, 60, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Equilibrium NMR studies of unfolded and partially folded proteins. Nat. Struct. Biol. 1998, 5, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztanyi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Diella, F.; Haslam, N.; Chica, C.; Budd, A.; Michael, S.; Brown, N.P.; Trave, G.; Gibson, T.J. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front. Biosci. 2008, 13, 6580–6603. [Google Scholar] [CrossRef]
- Galea, C.A.; Wang, Y.; Sivakolundu, S.G.; Kriwacki, R.W. Regulation of cell division by intrinsically unstructured proteins: Intrinsic flexibility, modularity, and signaling conduits. Biochemistry 2008, 47, 7598–7609. [Google Scholar] [CrossRef]
- Mellios, N.; Woodson, J.; Garcia, R.I.; Crawford, B.; Sharma, J.; Sheridan, S.D.; Haggarty, S.J.; Sur, M. beta2-Adrenergic receptor agonist ameliorates phenotypes and corrects microRNA-mediated IGF1 deficits in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 9947–9952. [Google Scholar] [CrossRef]
- Tropea, D.; Giacometti, E.; Wilson, N.R.; Beard, C.; McCurry, C.; Fu, D.D.; Flannery, R.; Jaenisch, R.; Sur, M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc. Natl. Acad. Sci. USA 2009, 106, 2029–2034. [Google Scholar] [CrossRef] [PubMed]
- Carrette, L.L.G.; Wang, C.Y.; Wei, C.; Press, W.; Ma, W.; Kelleher, R.J., 3rd; Lee, J.T. A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc. Natl. Acad. Sci. USA 2018, 115, E668–E675. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R.; Bird, A.P. MeCP2 mutations: Progress towards understanding and treating Rett syndrome. Genome Med. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Mauri, F.; McNamee, L.M.; Lunardi, A.; Chiacchiera, F.; Del Sal, G.; Brodsky, M.H.; Collavin, L. Modification of Drosophila p53 by SUMO modulates its transactivation and pro-apoptotic functions. J. Biol. Chem. 2008, 283, 20848–20856. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell. Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Fahmi, M.; Ito, M. Evolutionary Approach of Intrinsically Disordered CIP/KIP Proteins. Sci. Rep. 2019, 9, 1575. [Google Scholar] [CrossRef]
- Gsponer, J.; Babu, M.M. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009, 99, 94–103. [Google Scholar] [CrossRef]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Ahrens, J.; Rahaman, J.; Siltberg-Liberles, J. Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains. Genes 2018, 11, 553. [Google Scholar] [CrossRef]
- Wakefield, R.I.; Smith, B.O.; Nan, X.; Free, A.; Soteriou, A.; Uhrin, D.; Bird, A.P.; Barlow, P.N. The solution structure of the domain from MeCP2 that binds to methylated DNA. J. Mol. Biol. 1999, 291, 1055–1065. [Google Scholar] [CrossRef]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: II. functions of conserved disorder. J. Proteome Res. 2006, 5, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Grimmler, M.; Wang, Y.; Mund, T.; Cilensek, Z.; Keidel, E.M.; Waddell, M.B.; Jakel, H.; Kullmann, M.; Kriwacki, R.W.; Hengst, L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 2007, 128, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38, D142–D148. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Guy, J.; Alexander-Howden, B.; FitzPatrick, L.; DeSousa, D.; Koerner, M.V.; Selfridge, J.; Bird, A. A mutation-led search for novel functional domains in MeCP2. Hum. Mol. Genet. 2018, 27, 2531–2545. [Google Scholar] [CrossRef]
- Ballestar, E.; Yusufzai, T.M.; Wolffe, A.P. Effects of Rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA. Biochemistry 2000, 39, 7100–7106. [Google Scholar] [CrossRef]
- Yusufzai, T.M.; Wolffe, A.P. Functional consequences of Rett syndrome mutations on human MeCP2. Nucleic Acids Res. 2000, 28, 4172–4179. [Google Scholar] [CrossRef]
- Mnatzakanian, G.N.; Lohi, H.; Munteanu, I.; Alfred, S.E.; Yamada, T.; MacLeod, P.J.; Jones, J.R.; Scherer, S.W.; Schanen, N.C.; Friez, M.J.; et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 2004, 36, 339. [Google Scholar] [CrossRef]
- Williamson, S.L.; Giudici, L.; Kilstrup-Nielsen, C.; Gold, W.; Pelka, G.J.; Tam, P.P.; Grimm, A.; Prodi, D.; Landsberger, N.; Christodoulou, J. A novel transcript of cyclin-dependent kinase-like 5 (CDKL5) has an alternative C-terminus and is the predominant transcript in brain. Hum. Genet. 2012, 131, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.C.; Singh, S.; Wang, H.P.; Hsu, C.J.; Hu, S.C.; Lee, W.T. FOXG1-Related Syndrome: From Clinical to Molecular Genetics and Pathogenic Mechanisms. Int. J. Mol. Sci. 2019, 20, 4176. [Google Scholar] [CrossRef] [PubMed]
- Payankaulam, S.; Li, L.M.; Arnosti, D.N. Transcriptional repression: Conserved and evolved features. Curr. Biol. 2010, 17, R764–R771. [Google Scholar] [CrossRef] [PubMed]
- Toresson, H.; Martinez-Barbera, J.P.; Bardsley, A.; Caubit, X.; Krauss, S. Conservation of BF-1 expression in amphioxus and zebrafish suggests evolutionary ancestry of anterior cell types that contribute to the vertebrate telencephalon. Dev. Genes Evol. 1998, 208, 431–439. [Google Scholar] [CrossRef]
- Miyoshi, G.; Fishell, G. Dynamic FoxG1 expression coordinates the integration of multipolar pyramidal neuron precursors into the cortical plate. Neuron 2012, 74, 1045–1058. [Google Scholar] [CrossRef]
- Kumamoto, T.; Toma, K.; Gunadi; McKenna, W.L.; Kasukawa, T.; Katzman, S.; Chen, B.; Hanashima, C. Foxg1 coordinates the switch from nonradially to radially migrating glutamatergic subtypes in the neocortex through spatiotemporal repression. Cell Rep. 2013, 3, 931–945. [Google Scholar] [CrossRef]
- Docker, D.; Schubach, M.; Menzel, M.; Munz, M.; Spaich, C.; Biskup, S.; Bartholdi, D. Further delineation of the SATB2 phenotype. Eur. J. Hum. Genet. 2014, 22, 1034–1039. [Google Scholar] [CrossRef]
- Lee, J.S.; Yoo, Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J.H. SATB2-associated syndrome presenting with Rett-like phenotypes. Clin. Genet. 2016, 89, 728–732. [Google Scholar] [CrossRef]
- Livide, G.; Patriarchi, T.; Amenduni, M.; Amabile, S.; Yasui, D.; Calcagno, E.; Lo Rizzo, C.; De Falco, G.; Ulivieri, C.; Ariani, F.; et al. GluD1 is a common altered player in neuronal differentiation from both MECP2-mutated and CDKL5-mutated iPS cells. Eur. J. Hum. Genet. 2015, 23, 195–201. [Google Scholar] [CrossRef]
- Patriarchi, T.; Amabile, S.; Frullanti, E.; Landucci, E.; Rizzo, C.L.; Ariani, F.; Costa, M.; Olimpico, F.; Hell, J.W.; Vaccarino, F.M.; et al. Imbalance of excitatory/inhibitory synaptic protein expression in iPSC-derived neurons from FOXG1(+/−) patients and in foxg1(+/−) mice. Eur. J. Hum. Genet. 2016, 24, 871–880. [Google Scholar] [CrossRef]
- Bracaglia, G.; Conca, B.; Bergo, A.; Rusconi, L.; Zhou, Z.; Greenberg, M.E.; Landsberger, N.; Soddu, S.; Kilstrup-Nielsen, C. Methyl-CpG-binding protein 2 is phosphorylated by homeodomain-interacting protein kinase 2 and contributes to apoptosis. EMBO Rep. 2009, 10, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Trazzi, S.; Torricella, R.; Viggiano, R.; De Franceschi, M.; Amendola, E.; Gross, C.; Calza, L.; Bartesaghi, R.; Ciani, E. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol. Dis. 2014, 70, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Mari, F.; Azimonti, S.; Bertani, I.; Bolognese, F.; Colombo, E.; Caselli, R.; Scala, E.; Longo, I.; Grosso, S.; Pescucci, C.; et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005, 14, 1935–1946. [Google Scholar] [CrossRef] [PubMed]
- Carouge, D.; Host, L.; Aunis, D.; Zwiller, J.; Anglard, P. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation. Neurobiol. Dis. 2010, 38, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 2008, 283, 30101–30111. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Johnson, A.K.; Dunker, A.K.; Daughdrill, G.W. Evolution and disorder. Curr. Opin. Struct. Biol. 2011, 21, 441–446. [Google Scholar] [CrossRef]
- Regad, T.; Roth, M.; Bredenkamp, N.; Illing, N.; Papalopulu, N. The neural progenitor-specifying activity of FoxG1 is antagonistically regulated by CKI and FGF. Nat. Cell Biol. 2007, 9, 531–540. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, X.; Wang, J.; Li, J.; Wu, Q.; Wen, Y.; Zhao, Y.; Zhang, X.; Yao, H.; Wu, X.; et al. Genomic mosaicism in the pathogenesis and inheritance of a Rett syndrome cohort. Genet. Med. 2019, 21, 1330–1338. [Google Scholar] [CrossRef]
- Sato, Y.; Nakaya, A.; Shiraishi, K.; Kawashima, S.; Goto, S.; Kanehisa, M. Ssdb: Sequence similarity database in kegg. Genome Inf. 2001, 12, 230–231. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Pupko, T.; Bell, R.E.; Mayrose, I.; Glaser, F.; Ben-Tal, N. Rate4Site: An algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 2002, 18 (Suppl. 1), S71–S77. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Krishnaraj, R.; Ho, G.; Christodoulou, J. RettBASE: Rett syndrome database update. Hum. Mutat. 2017, 38, 922–931. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Pellegrini, M.; Marcotte, E.M.; Thompson, M.J.; Eisenberg, D.; Yeates, T.O. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 1999, 96, 4285–4288. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, A.; Katayama, T.; Itoh, M.; Hiranuka, K.; Kawashima, S.; Moriya, Y.; Okuda, S.; Tanaka, M.; Tokimatsu, T.; Yamanishi, Y.; et al. KEGG OC: A large-scale automatic construction of taxonomy-based ortholog clusters. Nucleic Acids Res. 2013, 41, D353–D357. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahmi, M.; Yasui, G.; Seki, K.; Katayama, S.; Kaneko-Kawano, T.; Inazu, T.; Kubota, Y.; Ito, M. In Silico Study of Rett Syndrome Treatment-Related Genes, MECP2, CDKL5, and FOXG1, by Evolutionary Classification and Disordered Region Assessment. Int. J. Mol. Sci. 2019, 20, 5593. https://doi.org/10.3390/ijms20225593
Fahmi M, Yasui G, Seki K, Katayama S, Kaneko-Kawano T, Inazu T, Kubota Y, Ito M. In Silico Study of Rett Syndrome Treatment-Related Genes, MECP2, CDKL5, and FOXG1, by Evolutionary Classification and Disordered Region Assessment. International Journal of Molecular Sciences. 2019; 20(22):5593. https://doi.org/10.3390/ijms20225593
Chicago/Turabian StyleFahmi, Muhamad, Gen Yasui, Kaito Seki, Syouichi Katayama, Takako Kaneko-Kawano, Tetsuya Inazu, Yukihiko Kubota, and Masahiro Ito. 2019. "In Silico Study of Rett Syndrome Treatment-Related Genes, MECP2, CDKL5, and FOXG1, by Evolutionary Classification and Disordered Region Assessment" International Journal of Molecular Sciences 20, no. 22: 5593. https://doi.org/10.3390/ijms20225593
APA StyleFahmi, M., Yasui, G., Seki, K., Katayama, S., Kaneko-Kawano, T., Inazu, T., Kubota, Y., & Ito, M. (2019). In Silico Study of Rett Syndrome Treatment-Related Genes, MECP2, CDKL5, and FOXG1, by Evolutionary Classification and Disordered Region Assessment. International Journal of Molecular Sciences, 20(22), 5593. https://doi.org/10.3390/ijms20225593