Meta-iAVP: A Sequence-Based Meta-Predictor for Improving the Prediction of Antiviral Peptides Using Effective Feature Representation

 ,
, 
Abstract
:1. Introduction
2. Results
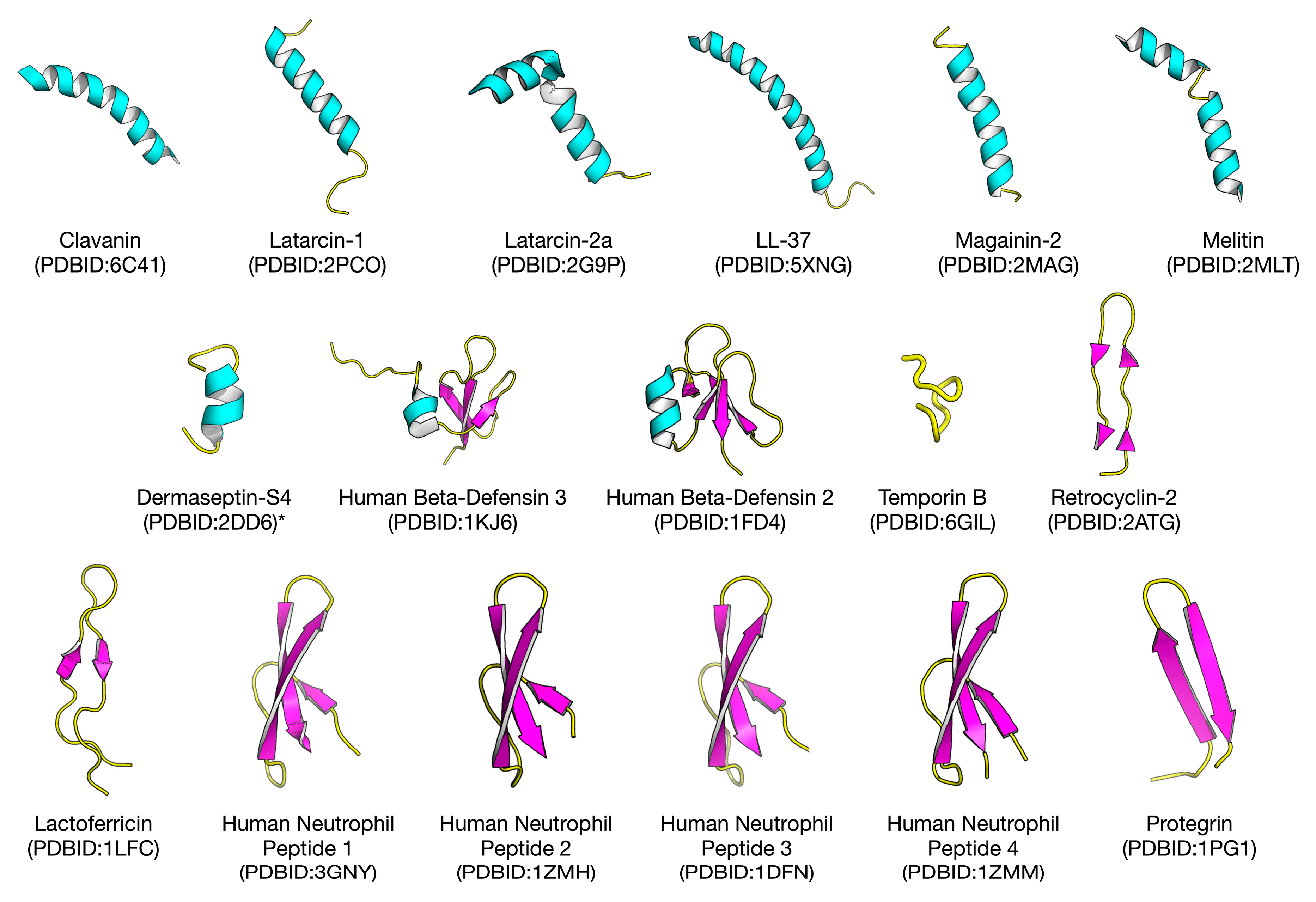
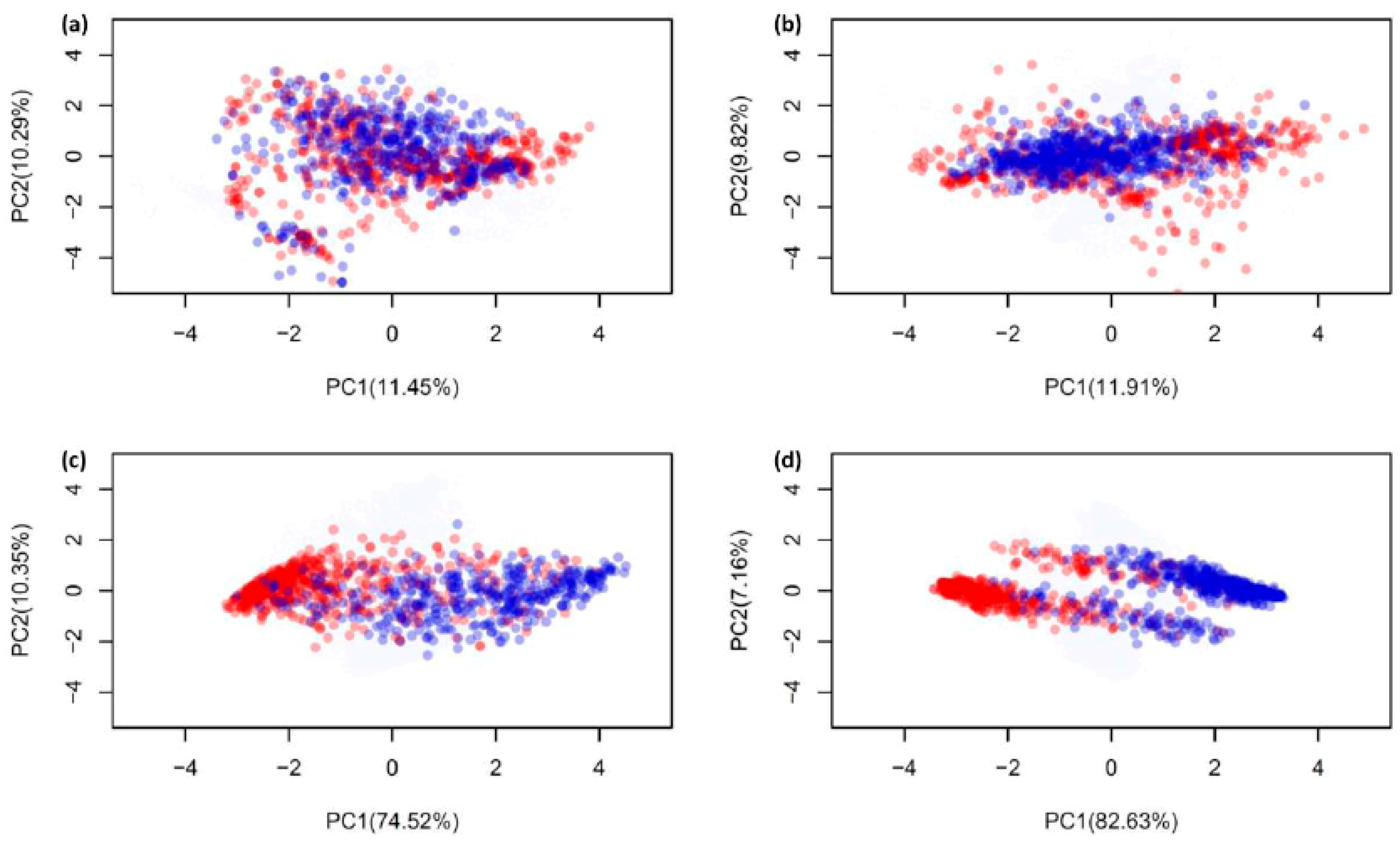
2.1. Biological Space of Antiviral Peptides
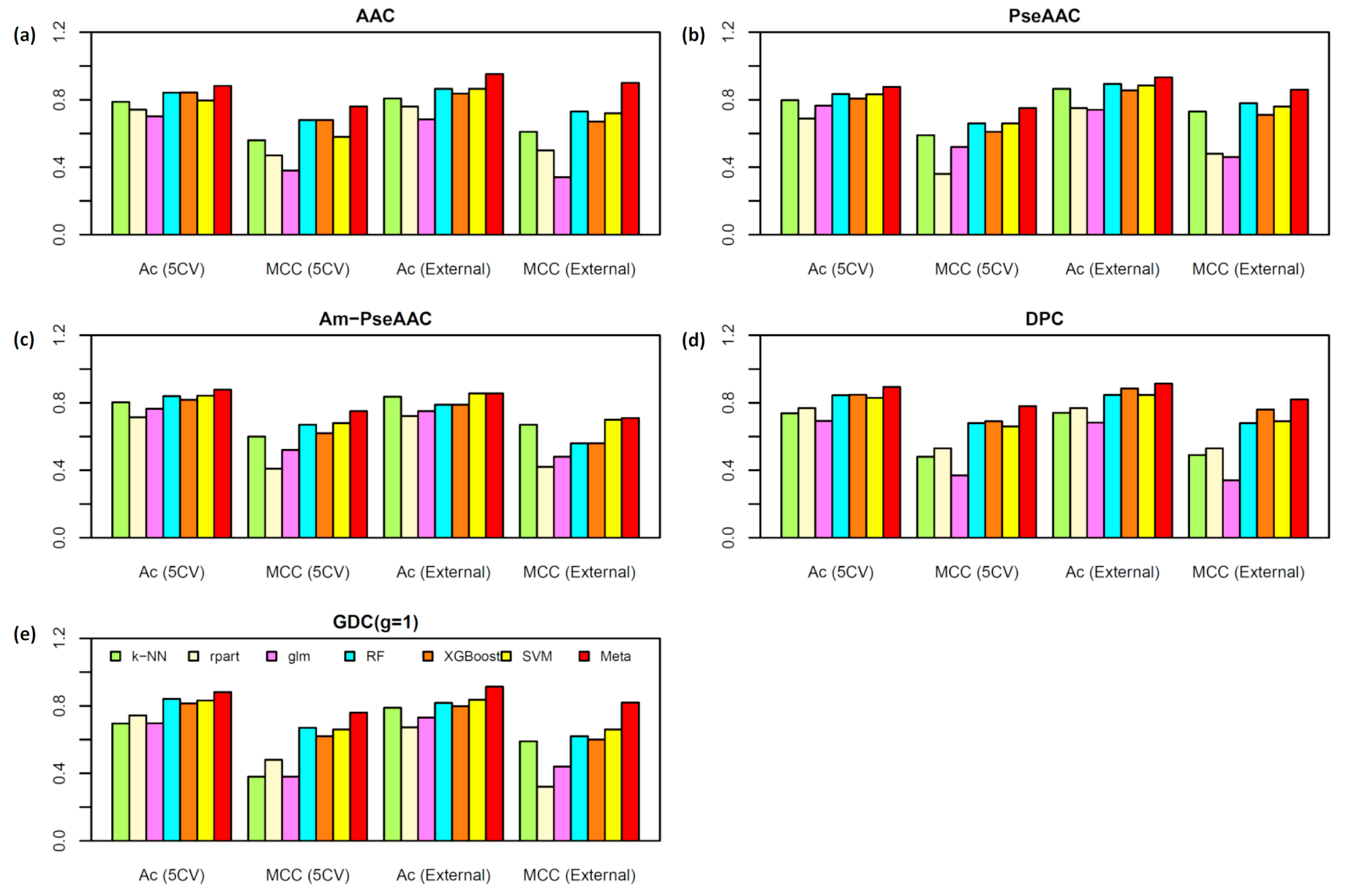
2.2. Performance Comparison of Various Types of Features
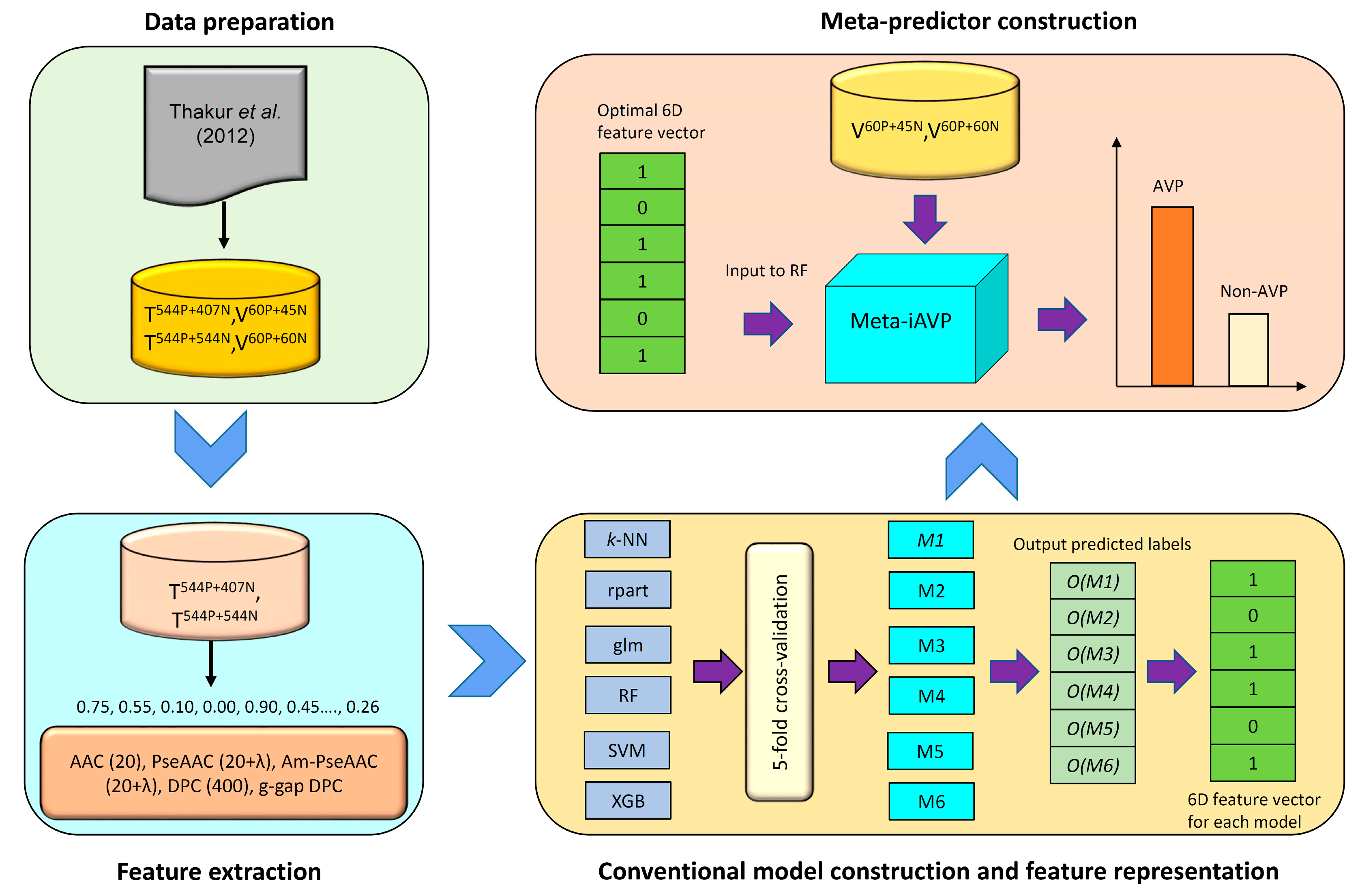
2.3. Construction of the Meta-iAVP Model
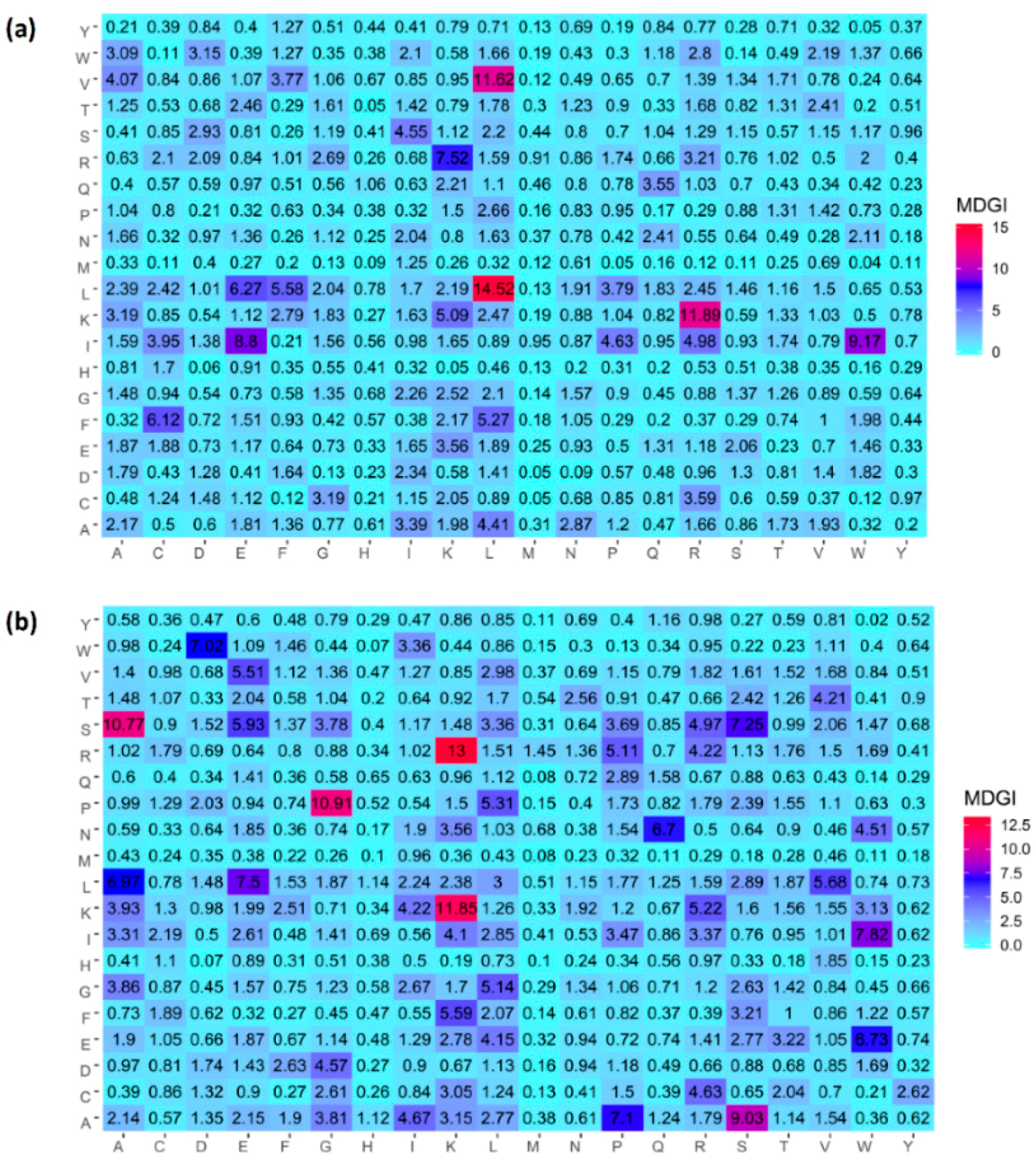
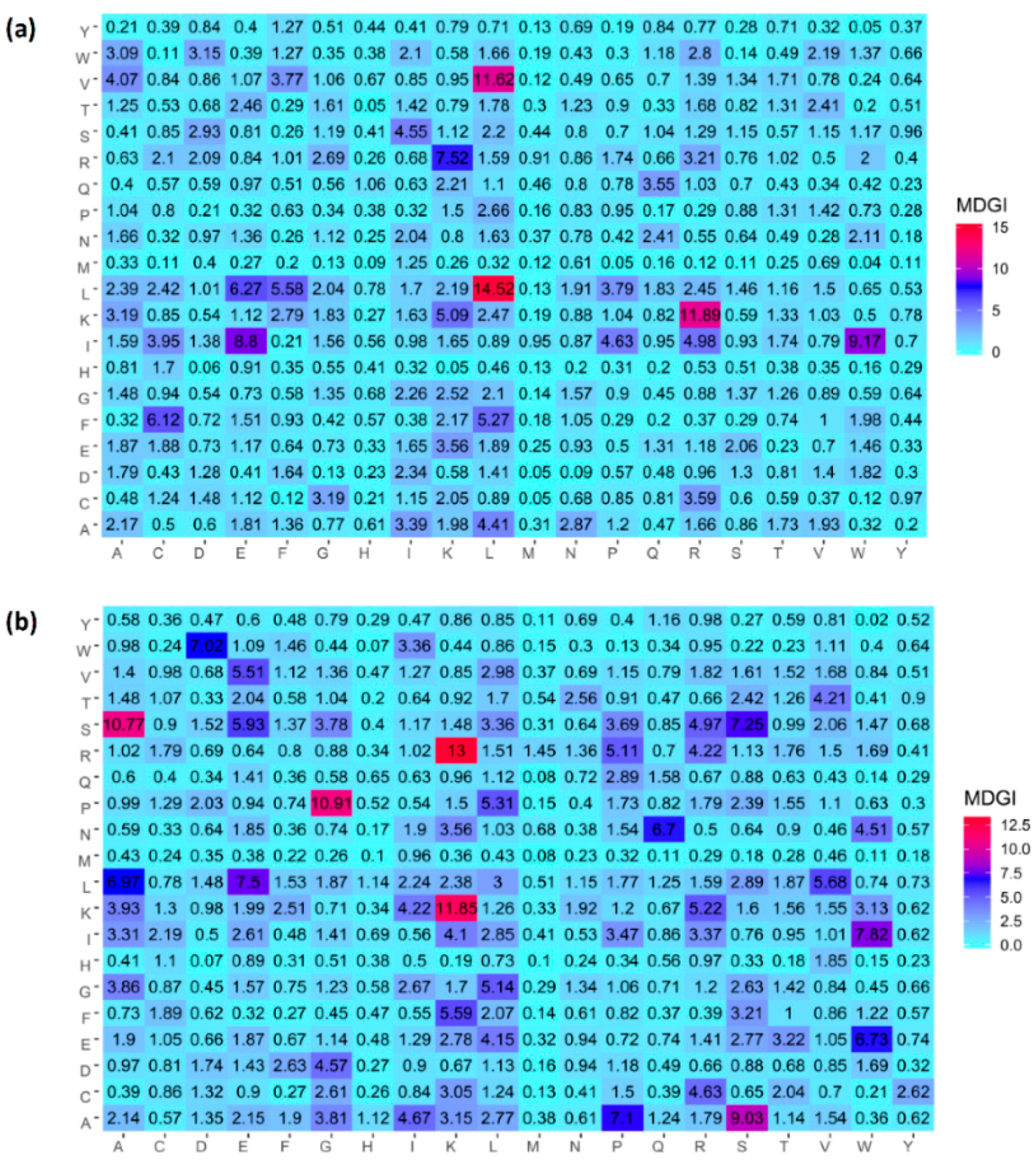
2.4. Analysis of new feature representation
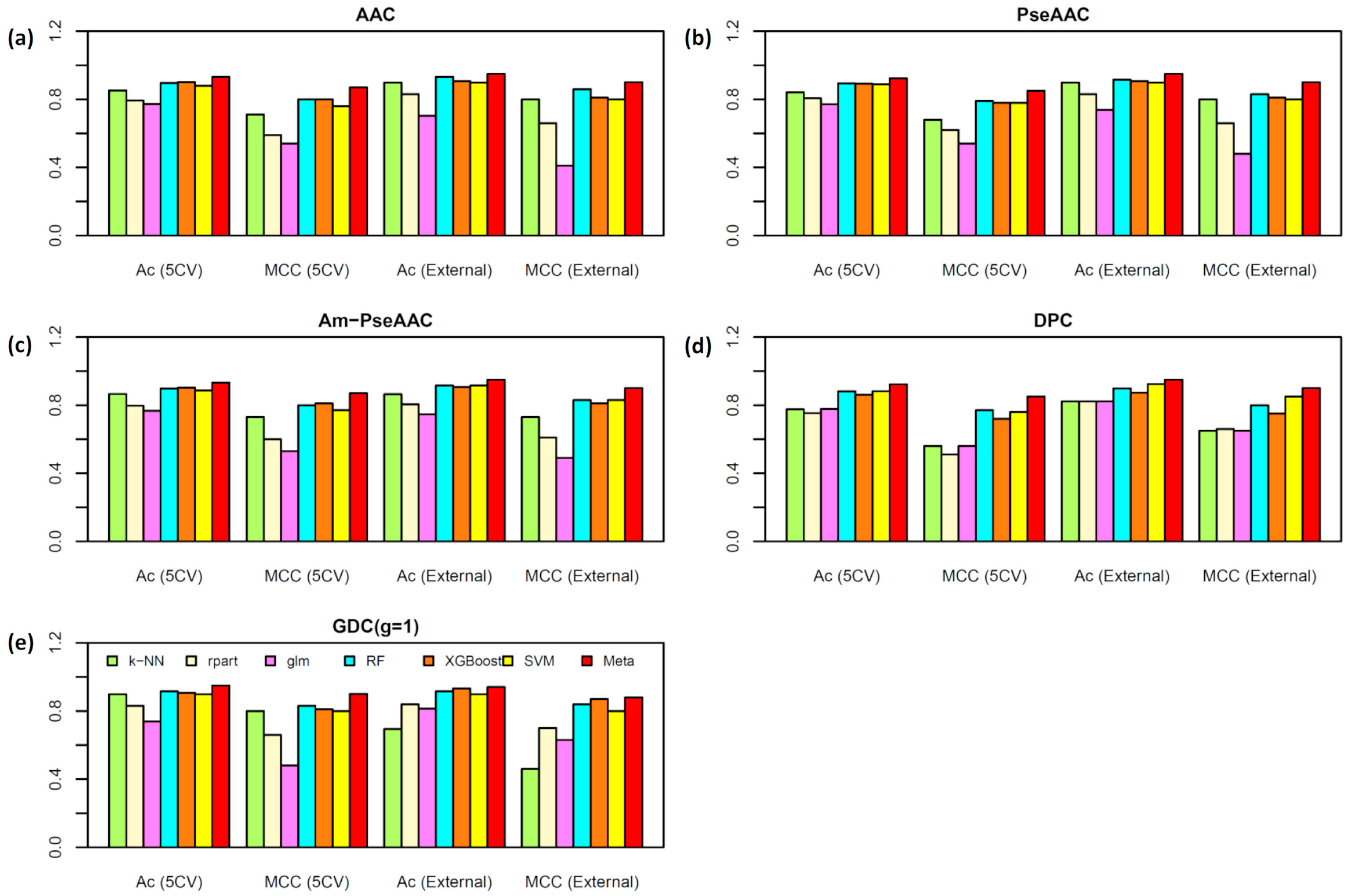
2.5. Comparison of Meta-iAVP with the State-of-Art Predictors
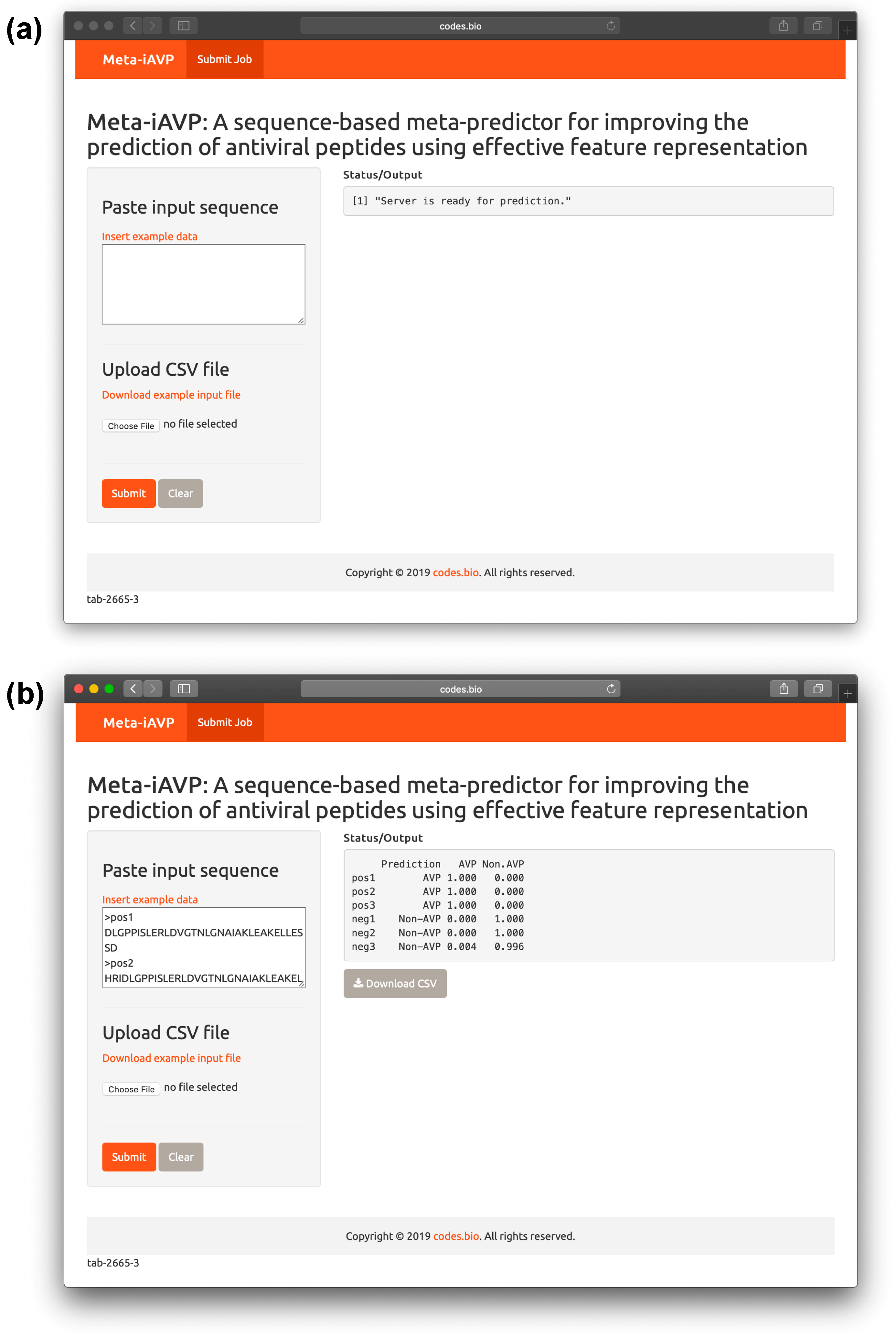
2.6. Meta-iAVP web server
- Step 1. Proceed to entering the following URL into the web browser, http://codes.bio/meta-iavp/.
- Step 2. Users have the option of either entering the query peptide sequence directly into the Input box or uploading the sequence file by clicking on the “Choose file” button (i.e., found below the “Enter your input sequence(s) in FASTA format heading”).
- Step 3. Click on the “Submit” button in order to start the prediction process.
- Step 4. Once predictions are made, the results output are shown in the grey box found below the “Status/Output” heading. The prediction process requires only a few seconds to process. After predictions are made, the prediction output can be conveniently downloaded as a CSV file by pressing on the “Download CSV button”.
3. Materials and Methods
3.1. Dataset Preparation
3.2. Feature Extraction of Peptides
3.3. Machine Learning Algorithms
3.4. Feature Importance Analysis
3.5. Performance Evaluation
3.6. Feature Representation Learning
3.6.1. Constructing Initial Features
3.6.2. Constructing a New Feature Representation
3.6.3. Learning a New Feature for Meta-Predictor Representation
3.7. Development of the Meta-iAVP Web Server
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Hydrophobicity [81] | Hydrophilicity [81] |
|---|---|---|
| A-Ala | 0.62 | −0.50 |
| C-Cys | 0.29 | −1.00 |
| D-Asp | −0.90 | 3.00 |
| E-Glu | −0.74 | 3.00 |
| F-Phe | 1.19 | −2.50 |
| G-Gly | 0.48 | 0.00 |
| H-His | −0.40 | −0.50 |
| I-Ile | 1.38 | −1.80 |
| K-Lys | −1.50 | 3.00 |
| L-Leu | 1.06 | −1.80 |
| M-Met | 0.64 | −1.30 |
| N-Asn | −0.78 | 0.20 |
| P-Pro | 0.12 | 0.00 |
| Q-Gln | −0.85 | 0.20 |
| R-Arg | −2.53 | 3.00 |
| S-Ser | −0.18 | 0.30 |
| T-Thr | −0.05 | −0.40 |
| V-Val | 1.08 | −1.50 |
| W-Trp | 0.81 | −3.40 |
| Y-Tyr | 0.26 | −2.30 |
References
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef] [PubMed]
- Infectious Diseases Dominate WHO’s List of 2019 Health Threats. Available online: https://www.contagionlive.com/news/infectious-diseases-dominate-whos-list-of-2019-health-threats (accessed on 23 January 2019).
- Woolhouse, M.; Scott, F.; Hudson, Z.; Howey, R.; Chase-Topping, M. Human viruses: Discovery and emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Strassburg, M.A. The global eradication of smallpox. Am. J. Infect. Control. 1982, 10, 53–59. [Google Scholar] [CrossRef]
- Bahl, S.; Bhatnagar, P.; Sutter, R.W.; Roesel, S.; Zaffran, M. Global Polio Eradication-Way Ahead. Indian J. Pediatr. 2018, 85, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A. New vaccines: Challenges of discovery. Microb. Biotechnol. 2016, 9, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Duraffour, S.; Andrei, G.; Topalis, D.; Krecmerova, M.; Crance, J.M.; Garin, D.; Snoeck, R. Mutations conferring resistance to viral DNA polymerase inhibitors in camelpox virus give different drug-susceptibility profiles in vaccinia virus. J. Virol. 2012, 86, 7310–7325. [Google Scholar] [CrossRef]
- Musiime, V.; Kaudha, E.; Kayiwa, J.; Mirembe, G.; Odera, M.; Kizito, H.; Nankya, I.; Ssali, F.; Kityo, C.; Colebunders, R.; et al. Antiretroviral drug resistance profiles and response to second-line therapy among HIV type 1-infected Ugandan children. AIDS Res. Hum. Retrovir. 2013, 29, 449–455. [Google Scholar] [CrossRef]
- Le Page, A.K.; Jager, M.M.; Iwasenko, J.M.; Scott, G.M.; Alain, S.; Rawlinson, W.D. Clinical aspects of cytomegalovirus antiviral resistance in solid organ transplant recipients. Clin. Infect. Dis. 2013, 56, 1018–1029. [Google Scholar] [CrossRef]
- Hui, D.S.C.; Lee, N.; Chan, P.K.S. A clinical approach to the threat of emerging influenza viruses in the Asia-Pacific region. Respirology 2017, 22, 1300–1312. [Google Scholar] [CrossRef]
- Marston, B.J.; Dokubo, E.K.; van Steelandt, A.; Martel, L.; Williams, D.; Hersey, S.; Jambai, A.; Keita, S.; Nyenswah, T.G.; Redd, J.T. Ebola Response Impact on Public Health Programs, West Africa, 2014–2017. Emerg. Infect. Dis. 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Souza, W.V.; Albuquerque, M.; Vazquez, E.; Bezerra, L.C.A.; Mendes, A.; Lyra, T.M.; Araujo, T.V.B.; Oliveira, A.L.S.; Braga, M.C.; Ximenes, R.A.A.; et al. Microcephaly epidemic related to the Zika virus and living conditions in Recife, Northeast Brazil. BMC Public Health 2018, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. ACPred: A Computational Tool for the Prediction and Analysis of Anticancer Peptides. Molecules 2019, 24, 1973. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Zhe, W.; Wang, G. APD: The Antimicrobial Peptide Database. Nucleic Acids Res. 2004, 32, D590–D592. [Google Scholar]
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6, 148. [Google Scholar] [CrossRef]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B. DBAASP v. 2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2015, 44, D1104–D1112. [Google Scholar] [CrossRef]
- Singh, S.; Chaudhary, K.; Dhanda, S.K.; Bhalla, S.; Usmani, S.S.; Gautam, A.; Tuknait, A.; Agrawal, P.; Mathur, D.; Raghava, G.P. SATPdb: A database of structurally annotated therapeutic peptides. Nucleic Acids Res. 2015, 44, D1119–D1126. [Google Scholar] [CrossRef]
- Rajput, A.; Thakur, A.; Sharma, S.; Kumar, M. aBiofilm: A resource of anti-biofilm agents and their potential implications in targeting antibiotic drug resistance. Nucleic Acids Res. 2017, 46, D894–D900. [Google Scholar] [CrossRef]
- Sharma, D.; Priyadarshini, P.; Vrati, S. Unraveling the web of viroinformatics: Computational tools and databases in virus research. J. Virol. 2015, 89, 1489–1501. [Google Scholar] [CrossRef]
- Bulet, P.; Stocklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef]
- Badani, H.; Garry, R.F.; Wimley, W.C. Peptide entry inhibitors of enveloped viruses: The importance of interfacial hydrophobicity. Biochim. Biophys. Acta. 2014, 1838, 2180–2197. [Google Scholar] [CrossRef]
- Wang, C.K.; Shih, L.Y.; Chang, K.Y. Large-Scale Analysis of Antimicrobial Activities in Relation to Amphipathicity and Charge Reveals Novel Characterization of Antimicrobial Peptides. Molecules 2017, 22, 2037. [Google Scholar] [CrossRef]
- Dando, T.M.; Perry, C.M. Enfuvirtide. Drugs 2003, 63, 2755–2766. [Google Scholar] [CrossRef]
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498. [Google Scholar] [CrossRef]
- Clinical Progress of the Entry Inhibitor Myrcludex B. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&ved=2ahUKEwiD9vaQn-blAhUhCqYKHZlwAWwQFjAAegQIBRAC&url=http%3A%2F%2Fregist2.virology-education.com%2Fpresentations%2F2018%2FHBVCure%2F15_Urban.pdf&usg=AOvVaw0xSMm7DxOyIj7S5qhMFuAC (accessed on 7 November 2018).
- Castel, G.; Chteoui, M.; Heyd, B.; Tordo, N. Phage display of combinatorial peptide libraries: Application to antiviral research. Molecules 2011, 16, 3499–3518. [Google Scholar] [CrossRef]
- Henriques, S.T.; Craik, D.J. Cyclotides as templates in drug design. Drug Discov. Today 2010, 15, 57–64. [Google Scholar] [CrossRef]
- Nawae, W.; Hannongbua, S.; Ruengjitchatchawalya, M. Molecular dynamics exploration of poration and leaking caused by Kalata B1 in HIV-infected cell membrane compared to host and HIV membranes. Sci. Rep. 2017, 7, 3638. [Google Scholar] [CrossRef]
- Ngai, P.H.; Ng, T.B. Phaseococcin, an antifungal protein with antiproliferative and anti-HIV-1 reverse transcriptase activities from small scarlet runner beans. Biochem. Cell Biol. 2005, 83, 212–220. [Google Scholar] [CrossRef]
- Zhao, Z.; Hong, W.; Zeng, Z.; Wu, Y.; Hu, K.; Tian, X.; Li, W.; Cao, Z. Mucroporin-M1 inhibits hepatitis B virus replication by activating the mitogen-activated protein kinase (MAPK) pathway and down-regulating HNF4alpha in vitro and in vivo. J. Biol. Chem. 2012, 287, 30181–30190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothan, H.A.; Bahrani, H.; Rahman, N.A.; Yusof, R. Identification of natural antimicrobial agents to treat dengue infection: In vitro analysis of latarcin peptide activity against dengue virus. BMC Microbiol. 2014, 14, 140. [Google Scholar] [CrossRef]
- Quintero-Gil, C.; Parra-Suescun, J.; Lopez-Herrera, A.; Orduz, S. In-silico design and molecular docking evaluation of peptides derivatives from bacteriocins and porcine beta defensin-2 as inhibitors of Hepatitis E virus capsid protein. Virusdisease 2017, 28, 281–288. [Google Scholar] [CrossRef]
- Chiang, A.W.; Wu, W.Y.; Wang, T.; Hwang, M.J. Identification of Entry Factors Involved in Hepatitis C Virus Infection Based on Host-Mimicking Short Linear Motifs. PLoS Comput. Biol. 2017, 13, e1005368. [Google Scholar] [CrossRef] [Green Version]
- Yin, P.; Zhang, L.; Ye, F.; Deng, Y.; Lu, S.; Li, Y.P.; Zhang, L.; Tan, W. A screen for inhibitory peptides of hepatitis C virus identifies a novel entry inhibitor targeting E1 and E2. Sci. Rep. 2017, 7, 3976. [Google Scholar] [CrossRef] [Green Version]
- Nyanguile, O. Peptide antiviral strategies as an alternative to treat lower respiratory viral infections. Front. Immunol. 2019, 10, 1366. [Google Scholar] [CrossRef]
- Rothan, H.A.; Abdulrahman, A.Y.; Sasikumer, P.G.; Othman, S.; Abd Rahman, N.; Yusof, R. Protegrin-1 inhibits dengue NS2B-NS3 serine protease and viral replication in MK2 cells. BioMed Res. Int. 2012, 2012. [Google Scholar] [CrossRef]
- Thakur, N.; Qureshi, A.; Kumar, M. AVPpred: Collection and prediction of highly effective antiviral peptides. Nucleic Acids Res. 2012, 40, W199–W204. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.Y.; Yang, J.-R. Analysis and prediction of highly effective antiviral peptides based on random forests. PLoS ONE 2013, 8, e70166. [Google Scholar] [CrossRef] [Green Version]
- Zare, M.; Mohabatkar, H.; Faramarzi, F.K.; Beigi, M.M.; Behbahani, M. Using Chou’s pseudo amino acid composition and machine learning method to predict the antiviral peptides. Open Bioinform. J. 2015, 9, 16–19. [Google Scholar] [CrossRef]
- Lissabet, J.F.B.; Belén, L.H.; Farias, J.G. AntiVPP 1.0: A portable tool for prediction of antiviral peptides. Comput. Biol. Med. 2019, 107, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef] [PubMed]
- Win, T.S.; Malik, A.A.; Prachayasittikul, V.; JE, S.W.; Nantasenamat, C.; Shoombuatong, W. HemoPred: A web server for predicting the hemolytic activity of peptides. Future Med. Chem. 2017, 9, 275–291. [Google Scholar] [CrossRef]
- Win, T.S.; Schaduangrat, N.; Prachayasittikul, V.; Nantasenamat, C.; Shoombuatong, W. PAAP: A web server for predicting antihypertensive activity of peptides. Future Med. Chem. 2018, 10, 1749–1767. [Google Scholar] [CrossRef]
- Simeon, S.; Li, H.; Win, T.S.; Malik, A.A.; Kandhro, A.H.; Piacham, T.; Shoombuatong, W.; Nuchnoi, P.; Wikberg, J.E.; Gleeson, M.P. PepBio: Predicting the bioactivity of host defense peptides. RSC Adv. 2017, 7, 35119–35134. [Google Scholar] [CrossRef] [Green Version]
- Pratiwi, R.; Malik, A.A.; Schaduangrat, N.; Prachayasittikul, V.; Wikberg, J.E.; Nantasenamat, C.; Shoombuatong, W. CryoProtect: A Web Server for Classifying Antifreeze Proteins from Nonantifreeze Proteins. J. Chem. 2017, 2017, 15. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Shoombuatong, W.; Lee, H.-C.; Chaijaruwanich, J.; Huang, H.-L.; Ho, S.-Y. SCMCRYS: Predicting protein crystallization using an ensemble scoring card method with estimating propensity scores of P-collocated amino acid pairs. PLoS ONE 2013, 8, e72368. [Google Scholar] [CrossRef]
- Huang, H.L. Propensity scores for prediction and characterization of bioluminescent proteins from sequences. PLoS ONE 2014, 9, e97158. [Google Scholar] [CrossRef] [Green Version]
- Liou, Y.F.; Charoenkwan, P.; Srinivasulu, Y.; Vasylenko, T.; Lai, S.C.; Lee, H.C.; Chen, Y.H.; Huang, H.L.; Ho, S.Y. SCMHBP: Prediction and analysis of heme binding proteins using propensity scores of dipeptides. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef] [Green Version]
- Oren, Z.; Shai, Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers 1998, 47, 451–463. [Google Scholar] [CrossRef]
- Daher, K.A.; Selsted, M.E.; Lehrer, R.I. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 1986, 60, 1068–1074. [Google Scholar] [PubMed]
- Daly, N.L.; Chen, Y.K.; Rosengren, K.J.; Marx, U.C.; Phillips, M.L.; Waring, A.J.; Wang, W.; Lehrer, R.I.; Craik, D.J. Retrocyclin-2: Structural analysis of a potent anti-HIV theta-defensin. Biochemistry 2007, 46, 9920–9928. [Google Scholar] [CrossRef] [PubMed]
- Currie, S.M.; Findlay, E.G.; McHugh, B.J.; Mackellar, A.; Man, T.; Macmillan, D.; Wang, H.; Fitch, P.M.; Schwarze, J.; Davidson, D.J. The human cathelicidin LL-37 has antiviral activity against respiratory syncytial virus. PLoS ONE 2013, 8, e73659. [Google Scholar] [CrossRef] [PubMed]
- Gwyer Findlay, E.; Currie, S.M.; Davidson, D.J. Cationic host defence peptides: Potential as antiviral therapeutics. BioDrugs 2013, 27, 479–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasin, B.; Wang, W.; Pang, M.; Cheshenko, N.; Hong, T.; Waring, A.J.; Herold, B.C.; Wagar, E.A.; Lehrer, R.I. Theta defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J. Virol. 2004, 78, 5147–5156. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Lopez-Garcia, B.; Braff, M.; Dorschner, R.A.; Gallo, R.L. Postsecretory processing generates multiple cathelicidins for enhanced topical antimicrobial defense. J. Immunol. 2004, 172, 3070–3077. [Google Scholar] [CrossRef] [Green Version]
- Mandelboim, O.; Wilson, S.B.; Vales-Gomez, M.; Reyburn, H.T.; Strominger, J.L. Self and viral peptides can initiate lysis by autologous natural killer cells. Proc. Natl. Acad. Sci. USA 1997, 94, 4604–4609. [Google Scholar] [CrossRef] [Green Version]
- Jacob, T.; Van den Broeke, C.; Favoreel, H.W. Viral serine/threonine protein kinases. J. Virol. 2011, 85, 1158–1173. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.A.; Carvalho, C.M.; Florentino, L.H.; Ramos, H.J.; Fontes, E.P. Conserved threonine residues within the A-loop of the receptor NIK differentially regulate the kinase function required for antiviral signaling. PLoS ONE 2009, 4, e5781. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Knebel, A.; Botting, C.H.; Galloway, C.S.; Precious, B.L.; Jackson, D.; Elliott, R.M.; Randall, R.E. CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology 2009, 383, 6–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemonnot, B.; Cartier, C.; Gay, B.; Rebuffat, S.; Bardy, M.; Devaux, C.; Boyer, V.; Briant, L. The host cell MAP kinase ERK-2 regulates viral assembly and release by phosphorylating the p6gag protein of HIV-1. J. Biol. Chem. 2004, 279, 32426–32434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalhan, S.C.; Hanson, R.W. Resurgence of serine: An often neglected but indispensable amino Acid. J. Biol. Chem. 2012, 287, 19786–19791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scala, M.C.; Sala, M.; Pietrantoni, A.; Spensiero, A.; Di Micco, S.; Agamennone, M.; Bertamino, A.; Novellino, E.; Bifulco, G.; Gomez-Monterrey, I.M.; et al. Lactoferrin-derived Peptides Active towards Influenza: Identification of Three Potent Tetrapeptide Inhibitors. Sci. Rep. 2017, 7, 10593. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A new ensemble learning framework for improved linear B-cell epitope prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [Green Version]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. mAHTPred: A sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 2018, 35, 2757–2765. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. Meta-4mCpred: A Sequence-Based Meta-Predictor for Accurate DNA 4mC Site Prediction Using Effective Feature Representation. Mol. Ther. Nucleic Acids 2019, 16, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Shin, T.H.; Kim, M.O.; Lee, G. PIP-EL: A new ensemble learning method for improved proinflammatory peptide predictions. Front. Immunol. 2018, 9, 1783. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Shoombuatong, W.; Schaduangrat, N.; Pratiwi, R.; Nantasenamat, C. THPep: A machine learning-based approach for predicting tumor homing peptides. Comput. Biol. Chem. 2019, 80, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Laengsri, V.; Nantasenamat, C.; Schaduangrat, N.; Nuchnoi, P.; Prachayasittikul, V.; Shoombuatong, W. TargetAntiAngio: A Sequence-Based Tool for the Prediction and Analysis of Anti-Angiogenic Peptides. Int. J. Mol. Sci. 2019, 20, 2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hongjaisee, S.; Nantasenamat, C.; Carraway, T.S.; Shoombuatong, W. HIVCoR: A sequence-based tool for predicting HIV-1 CRF01_AE coreceptor usage. Comput. Biol. Chem. 2019, 80, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Kanehisa, M. AAindex: Amino acid index database. Nucleic Acids Res. 2000, 28, 374. [Google Scholar] [CrossRef]
- Akbar, S.; Hayat, M.; Iqbal, M.; Jan, M.A. iACP-GAEnsC: Evolutionary genetic algorithm based ensemble classification of anticancer peptides by utilizing hybrid feature space. Artif. Intell. Med. 2017, 79, 62–70. [Google Scholar] [CrossRef]
- Hajisharifi, Z.; Piryaiee, M.; Beigi, M.M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou′ s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol. 2014, 341, 34–40. [Google Scholar] [CrossRef]
- Chou, K.-C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2004, 21, 10–19. [Google Scholar] [CrossRef]
- Kim, J.-H. Estimating classification error rate: Repeated cross-validation, repeated hold-out and bootstrap. Comput. Stat. Data Anal. 2009, 53, 3735–3745. [Google Scholar] [CrossRef]
- Chou, K.-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Feng, P.; Lin, H.; Chou, K.-C. iACP: A sequence-based tool for identifying anticancer peptides. Oncotarget 2016, 7, 16895. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.-C. Impacts of bioinformatics to medicinal chemistry. Med. Chem. 2015, 11, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Towards understanding aromatase inhibitory activity via QSAR modeling. EXCLI J. 2018, 17, 688. [Google Scholar] [PubMed]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Unraveling the bioactivity of anticancer peptides as deduced from machine learning. EXCLI J. 2018, 17, 734. [Google Scholar] [PubMed]
- Shoombuatong, W.; Mekha, P.; Waiyamai, K.; Cheevadhanarak, S.; Chaijaruwanicha, J. Prediction of human leukocyte antigen gene using k-nearest neighbour classifier based on spectrum kernel. ScienceAsia 2013, 39, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Shoombuatong, W.; Mekha, P.; Chaijaruwanich, J. Sequence based human leukocyte antigen gene prediction using informative physicochemical properties. Int. J. Data Min. Bioinform. 2015, 13, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Shoombuatong, W.; Huang, H.-L.; Chaijaruwanich, J.; Charoenkwan, P.; Lee, H.-C.; Ho, S.-Y. Predicting protein crystallization using a simple scoring card method. Proceedings of 2013 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Singapore, 16–19 April 2013; pp. 23–30. [Google Scholar]
- Simeon, S.; Shoombuatong, W.; Anuwongcharoen, N.; Preeyanon, L.; Prachayasittikul, V.; Wikberg, J.E.; Nantasenamat, C. osFP: A web server for predicting the oligomeric states of fluorescent proteins. J. Cheminformatics 2016, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Xiao, N.; Cao, D.-S.; Zhu, M.-F.; Xu, Q.-S. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef] [Green Version]
- Shoombuatong, W.; Prachayasittikul, V.; Anuwongcharoen, N.; Songtawee, N.; Monnor, T.; Prachayasittikul, S.; Prachayasittikul, V.; Nantasenamat, C. Navigating the chemical space of dipeptidyl peptidase-4 inhibitors. Drug Des. Dev. Ther. 2015, 9, 4515–4549. [Google Scholar]
- Shoombuatong, W.; Prachayasittikul, V.; Prachayasittikul, V.; Nantasenamat, C. Prediction of aromatase inhibitory activity using the efficient linear method (ELM). EXCLI J. 2015, 14, 452–464. [Google Scholar]
- Anuwongcharoen, N.; Shoombuatong, W.; Tantimongcolwat, T.; Prachayasittikul, V.; Nantasenamat, C. Exploring the chemical space of influenza neuraminidase inhibitors. PeerJ 2016, 4, e1958. [Google Scholar] [CrossRef] [Green Version]
- Prachayasittikul, V.; Worachartcheewan, A.; Shoombuatong, W.; Prachayasittikul, V.; Nantasenamat, C. Classification of P-glycoprotein-interacting compounds using machine learning methods. EXCLI J. 2015, 14, 958. [Google Scholar] [PubMed]
- Shoombuatong, W.; Prathipati, P.; Prachayasittikul, V.; Schaduangrat, N.; Ahmad Malik, A.; Pratiwi, R.; Wanwimolruk, S.; ES Wikberg, J.; Paul Gleeson, M.; Spjuth, O. Towards predicting the cytochrome P450 modulation: From QSAR to proteochemometric modeling. Curr. Drug Metab. 2017, 18, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Mandi, P.; Shoombuatong, W.; Phanus-Umporn, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V.; Bülow, L.; Nantasenamat, C. Exploring the origins of structure–oxygen affinity relationship of human haemoglobin allosteric effector. Mol. Simul. 2015, 41, 1283–1291. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Nabu, S.; Simeon, S.; Prachayasittikul, V.; Lapins, M.; Wikberg, J.E.; Nantasenamat, C. Extending proteochemometric modeling for unraveling the sorption behavior of compound–soil interaction. Chemom. Intell. Lab. Syst. 2016, 151, 219–227. [Google Scholar] [CrossRef]
- Nava Lara, R.A.; Aguilera-Mendoza, L.; Brizuela, C.A.; Peña, A.; Del Rio, G. Heterologous Machine Learning for the Identification of Antimicrobial Activity in Human-Targeted Drugs. Molecules 2019, 24, 1258. [Google Scholar] [CrossRef] [Green Version]
- Rajput, A.; Gupta, A.K.; Kumar, M. Prediction and analysis of quorum sensing peptides based on sequence features. PLoS ONE 2015, 10, e0120066. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Classification and Regression Trees; Boca Raton: New York, NY, USA, 2017. [Google Scholar]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 1189–1232. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Bibliometrics: San Francisco, CA, USA, 2016; pp. 785–794. [Google Scholar]
- Wang, H.; Liu, C.; Deng, L. Enhanced prediction of hot spots at protein-protein interfaces using extreme gradient boosting. Sci. Rep. 2018, 8, 14285. [Google Scholar] [CrossRef]
- Vapnik, V. The Nature of Statistical Learning Theory; Springer: New York, NY, USA, 2013. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Vapnik, V.N. An overview of statistical learning theory. IEEE Trans. Neural Netw. 1999, 10, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Shoombuatong, W.; Hongjaisee, S.; Barin, F.; Chaijaruwanich, J.; Samleerat, T. HIV-1 CRF01_AE coreceptor usage prediction using kernel methods based logistic model trees. Comput. Biol. Med. 2012, 42, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Rajput, A.; Kumar, M. Anti-flavi: A web platform to predict inhibitors of flaviviruses using QSAR and peptidomimetic approaches. Front. Microbiol. 2018, 9, 3121. [Google Scholar] [CrossRef] [PubMed]
- Karatzoglou, A.; Smola, A.; Hornik, K.; Zeileis, A. kernlab-an S4 package for kernel methods in R. J. Stat. Softw. 2004, 11, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Calle, M.L.; Urrea, V. Letter to the editor: Stability of random forest importance measures. Brief. Bioinform. 2010, 12, 86–89. [Google Scholar] [CrossRef] [Green Version]
| Method | Classifier a | Sequence Feature b | Stand-Alone Program | Webserver |
|---|---|---|---|---|
| AVPpred [41] | SVM | AAindex | − | ✓ |
| Chang et al.’s method [42] | RF | AAC, aggregation | − | − |
| AntiVPP 1.0 [44] | RF | PCP | ✓ | − |
| Meta-iAVP (This study) | Meta-predictor | AAC, Am-PseAAC | − | ✓ |
| Amino Acid | AVP (%) | Non-AVP (%) | Difference | p-Value | MDGI |
|---|---|---|---|---|---|
| K-Lys | 0.092 | 0.078 | 0.014 | <0.05 | 49.27(1) |
| T-Thr | 0.032 | 0.055 | −0.023 | <0.05 | 46.27(2) |
| L-Leu | 0.119 | 0.09 | 0.029 | <0.05 | 35.06(3) |
| I-Ile | 0.068 | 0.046 | 0.022 | <0.05 | 34.52(4) |
| S-Ser | 0.054 | 0.057 | −0.003 | 0.464 | 30.95(5) |
| W-Trp | 0.049 | 0.024 | 0.025 | <0.05 | 30.93(6) |
| N-Asn | 0.04 | 0.049 | −0.009 | <0.05 | 30.19(7) |
| R-Arg | 0.079 | 0.082 | −0.003 | 0.685 | 28.52(8) |
| C-Cys | 0.038 | 0.035 | 0.003 | 0.499 | 26.33(9) |
| E-Glu | 0.062 | 0.051 | 0.011 | <0.05 | 24.87(10) |
| D-Asp | 0.038 | 0.042 | −0.004 | 0.204 | 22.93(11) |
| A-Ala | 0.074 | 0.079 | −0.005 | 0.384 | 21.85(12) |
| V-Val | 0.049 | 0.062 | −0.013 | <0.05 | 21.1(13) |
| P-Pro | 0.033 | 0.054 | −0.021 | <0.05 | 19.73(14) |
| Q-Gln | 0.036 | 0.036 | 0 | 0.916 | 17.84(15) |
| G-Gly | 0.047 | 0.059 | −0.012 | <0.05 | 17.25(16) |
| H-His | 0.016 | 0.022 | −0.006 | <0.05 | 14.9(17) |
| F-Phe | 0.041 | 0.038 | 0.003 | 0.358 | 14.49(18) |
| Y-Tyr | 0.021 | 0.03 | −0.009 | <0.05 | 12.09(19) |
| M-Met | 0.011 | 0.014 | −0.003 | 0.085 | 6.27(20) |
| Amino Acid | AVP (%) | Non-AVP (%) | Difference | p-Value | MDGI |
|---|---|---|---|---|---|
| K-Lys | 0.092 | 0.046 | 0.045 | <0.05 | 77.11(1) |
| P-Pro | 0.033 | 0.068 | −0.035 | <0.05 | 68.87(2) |
| C-Cys | 0.038 | 0.022 | 0.015 | <0.05 | 57.68(3) |
| T-Thr | 0.032 | 0.053 | −0.021 | <0.05 | 46.84(4) |
| S-Ser | 0.054 | 0.083 | −0.029 | <0.05 | 39.57(5) |
| W-Trp | 0.049 | 0.015 | 0.033 | <0.05 | 36.83(6) |
| V-Val | 0.049 | 0.069 | −0.02 | <0.05 | 25.69(7) |
| A-Ala | 0.074 | 0.087 | −0.013 | <0.05 | 24.40(8) |
| G-Gly | 0.047 | 0.072 | −0.025 | <0.05 | 24.25(9) |
| L-Leu | 0.119 | 0.117 | 0.002 | 0.728 | 23.80(10) |
| I-Ile | 0.068 | 0.042 | 0.026 | <0.05 | 23.42(11) |
| H-His | 0.016 | 0.021 | −0.005 | <0.05 | 23.13(12) |
| E-Glu | 0.062 | 0.056 | 0.006 | 0.108 | 20.13(13) |
| Q-Gln | 0.036 | 0.04 | −0.004 | 0.18 | 18.50(14) |
| N-Asn | 0.04 | 0.03 | 0.01 | <0.05 | 18.48(15) |
| R-Arg | 0.079 | 0.061 | 0.018 | <0.05 | 17.67(16) |
| F-Phe | 0.041 | 0.038 | 0.003 | 0.321 | 16.57(17) |
| D-Asp | 0.038 | 0.038 | 0 | 0.982 | 15.75(18) |
| Y-Tyr | 0.021 | 0.023 | −0.001 | 0.537 | 10.57(19) |
| M-Met | 0.011 | 0.017 | −0.006 | <0.05 | 10.33(20) |
| Dataset | Method a | Ac (%) | Sn (%) | Sp (%) | MCC |
|---|---|---|---|---|---|
| T544p+407n | k-NN | 78.79 | 88.24 | 66.13 | 0.56 |
| rpart | 74.09 | 81.03 | 64.82 | 0.47 | |
| glm | 70.15 | 82.87 | 53.27 | 0.38 | |
| RF | 84.22 | 85.70 | 82.34 | 0.68 | |
| XGBoost | 84.33 | 86.69 | 80.97 | 0.68 | |
| SVM | 79.53 | 83.81 | 73.86 | 0.58 | |
| Meta-predictor | 88.17 | 89.23 | 86.94 | 0.76 | |
| T544p+544n | k-NN | 84.15 | 82.53 | 86.07 | 0.68 |
| rpart | 80.63 | 82.37 | 79.73 | 0.62 | |
| glm | 77.11 | 77.78 | 76.78 | 0.54 | |
| RF | 89.44 | 84.18 | 94.68 | 0.79 | |
| XGBoost | 89.16 | 87.48 | 90.90 | 0.78 | |
| SVM | 88.79 | 87.13 | 90.71 | 0.78 | |
| Meta-predictor | 92.31 | 88.44 | 96.16 | 0.85 | |
| V60p+45n | k-NN | 80.77 | 95.00 | 61.36 | 0.61 |
| rpart | 75.96 | 86.67 | 61.36 | 0.50 | |
| glm | 68.27 | 86.67 | 43.18 | 0.34 | |
| RF | 86.54 | 86.67 | 86.36 | 0.73 | |
| XGBoost | 83.65 | 85.00 | 81.82 | 0.67 | |
| SVM | 86.54 | 93.33 | 77.27 | 0.72 | |
| Meta-predictor | 95.19 | 96.67 | 93.18 | 0.90 | |
| V60p+60n | k-NN | 89.83 | 85.00 | 94.83 | 0.80 |
| rpart | 83.05 | 88.33 | 77.59 | 0.66 | |
| glm | 73.73 | 78.33 | 68.97 | 0.48 | |
| RF | 91.53 | 90.00 | 93.10 | 0.83 | |
| XGBoost | 90.68 | 90.00 | 91.38 | 0.81 | |
| SVM | 89.83 | 88.33 | 91.38 | 0.80 | |
| Meta-predictor | 94.92 | 93.33 | 96.55 | 0.90 |
| Dataset | Method a | Ac (%) | Sn (%) | Sp (%) | MCC |
|---|---|---|---|---|---|
| T544p+407n | AVPpred | 85.00 | 82.20 | 88.20 | 0.70 |
| Chang et al.’s method | 85.10 | 86.60 | 83.00 | 0.70 | |
| AntiVPP 1.0 | - | - | - | - | |
| Meta-iAVP | 88.20 | 89.20 | 86.90 | 0.76 | |
| T544p+544n | AVPpred | 90.00 | 89.70 | 90.30 | 0.80 |
| Chang et al.’s method | 91.50 | 89.00 | 94.10 | 0.83 | |
| AntiVPP 1.0 | - | - | - | - | |
| Meta-iAVP | 93.20 | 89.00 | 97.40 | 0.87 | |
| V60p+45n | AVPpred | 85.70 | 88.30 | 82.20 | 0.71 |
| Chang et al.’s method | 89.50 | 91.70 | 86.70 | 0.79 | |
| AntiVPP 1.0 | - | - | - | - | |
| Meta-iAVP | 95.20 | 96.70 | 93.20 | 0.90 | |
| V60p+60n | AVPpred | 92.50 | 93.30 | 91.70 | 0.85 |
| Chang et al.’s method | 93.30 | 91.70 | 95.00 | 0.87 | |
| AntiVPP 1.0 | 93.00 | 87.00 | 97.00 | 0.87 | |
| Meta-iAVP | 94.90 | 91.70 | 98.30 | 0.90 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. Meta-iAVP: A Sequence-Based Meta-Predictor for Improving the Prediction of Antiviral Peptides Using Effective Feature Representation. Int. J. Mol. Sci. 2019, 20, 5743. https://doi.org/10.3390/ijms20225743
Schaduangrat N, Nantasenamat C, Prachayasittikul V, Shoombuatong W. Meta-iAVP: A Sequence-Based Meta-Predictor for Improving the Prediction of Antiviral Peptides Using Effective Feature Representation. International Journal of Molecular Sciences. 2019; 20(22):5743. https://doi.org/10.3390/ijms20225743
Chicago/Turabian StyleSchaduangrat, Nalini, Chanin Nantasenamat, Virapong Prachayasittikul, and Watshara Shoombuatong. 2019. "Meta-iAVP: A Sequence-Based Meta-Predictor for Improving the Prediction of Antiviral Peptides Using Effective Feature Representation" International Journal of Molecular Sciences 20, no. 22: 5743. https://doi.org/10.3390/ijms20225743