The Proliferative and Apoptotic Landscape of Basal-like Breast Cancer

 , and
, and
Abstract
:1. Basal-Like Breast Cancers Are a Clinical Challenge
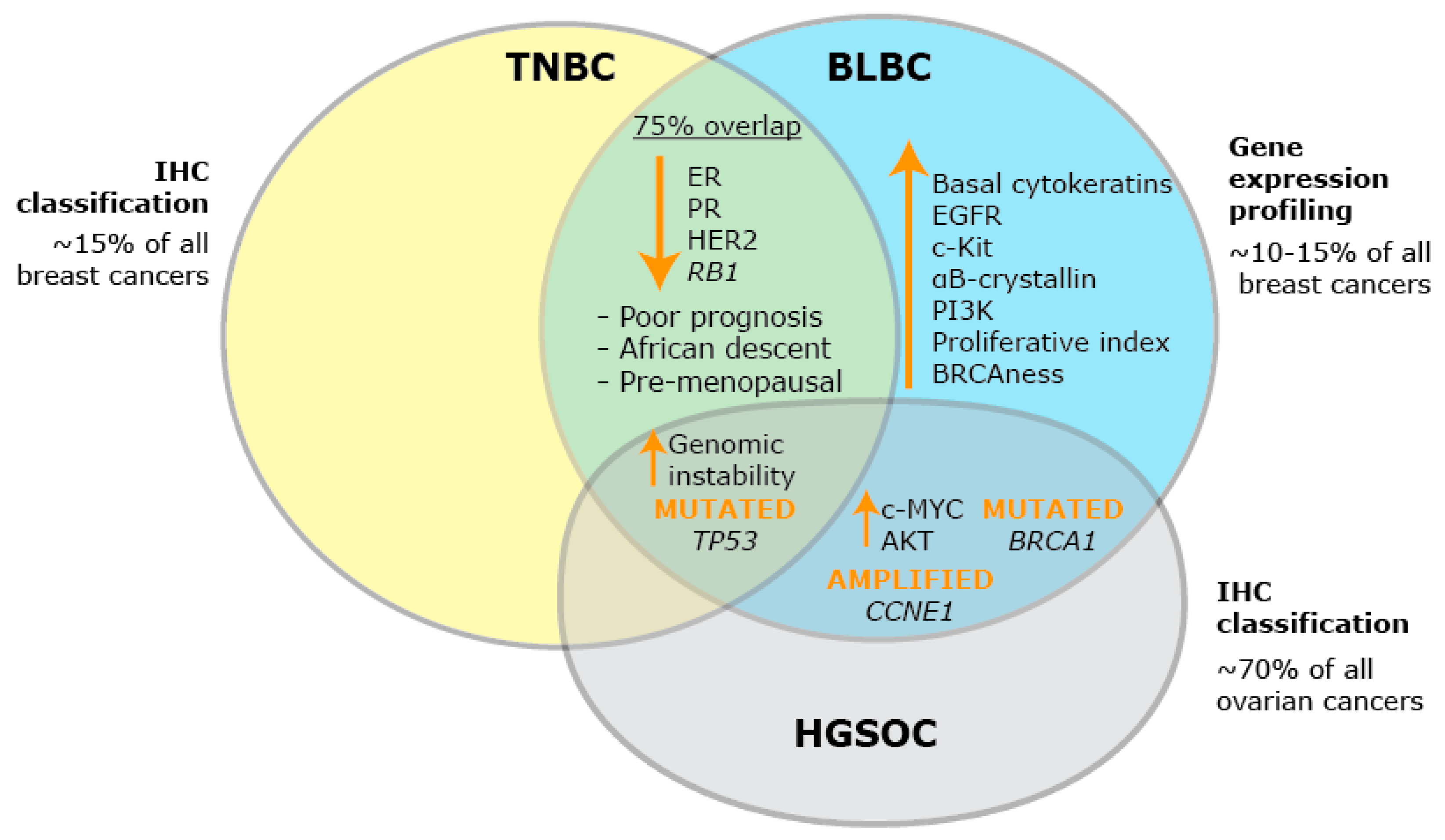
2. BLBC: A Heterogeneous Group of Breast Cancers
3. Identifying Key Hallmarks of BLBC that May Yield New Targets for Therapy
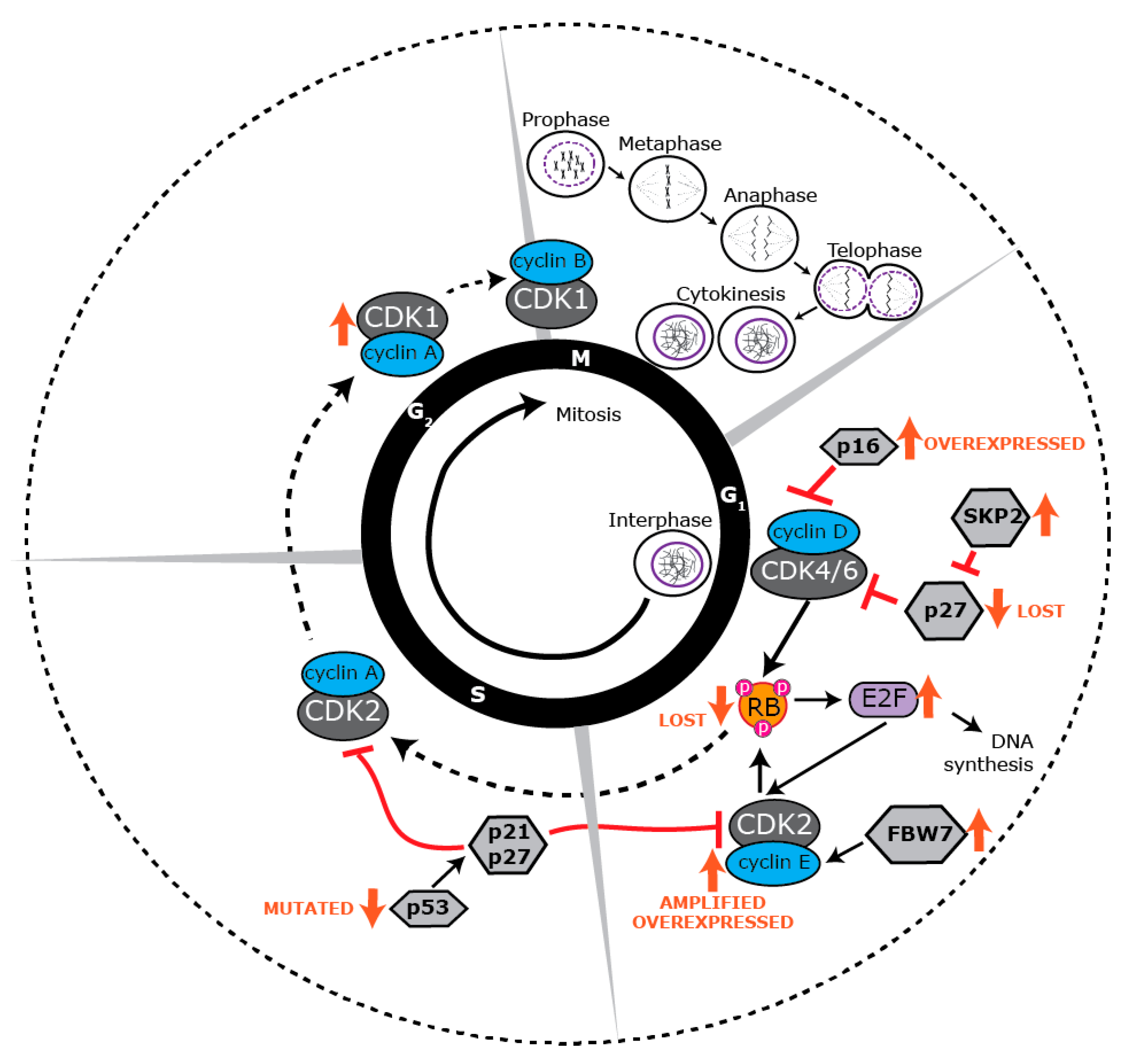
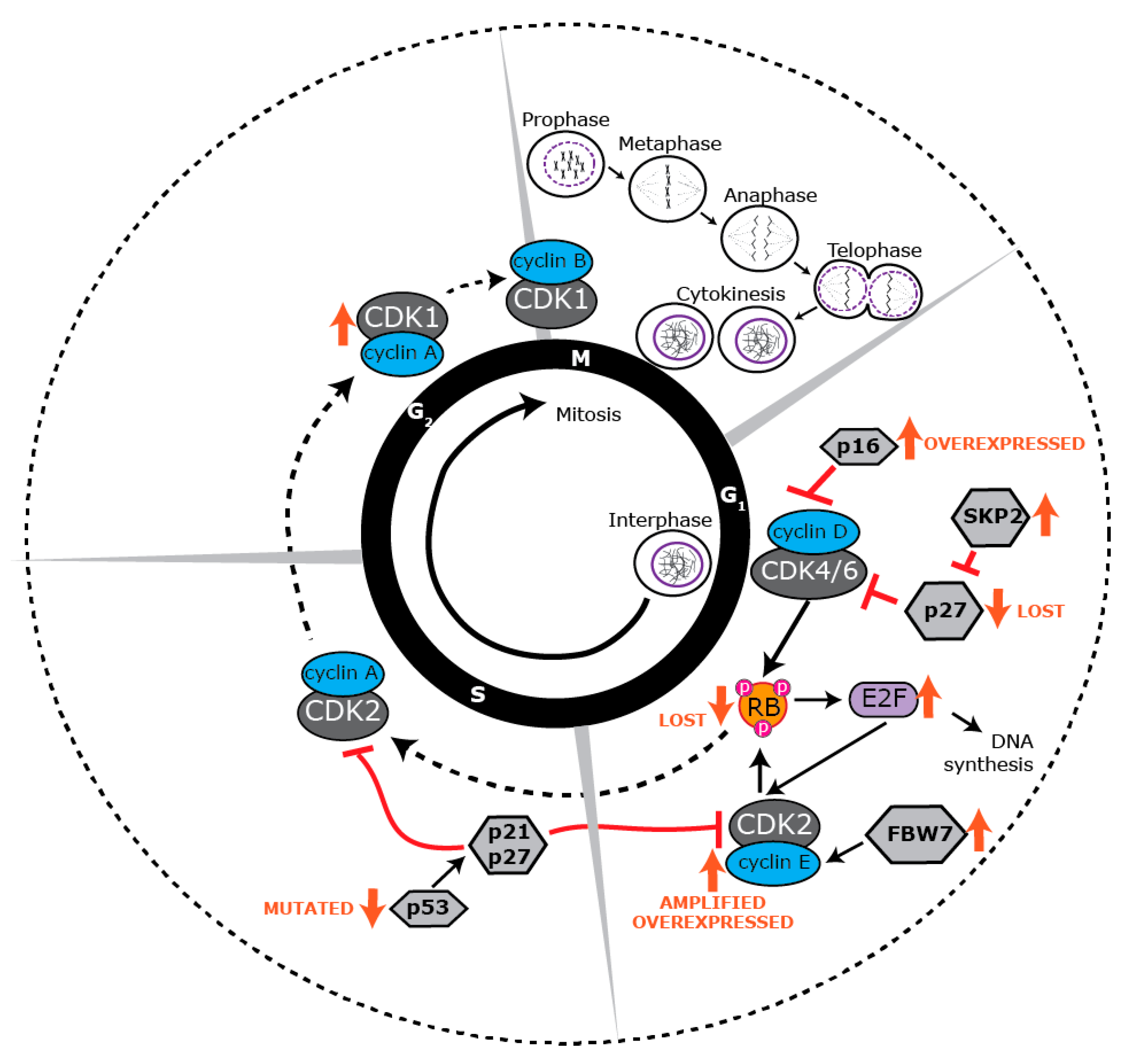
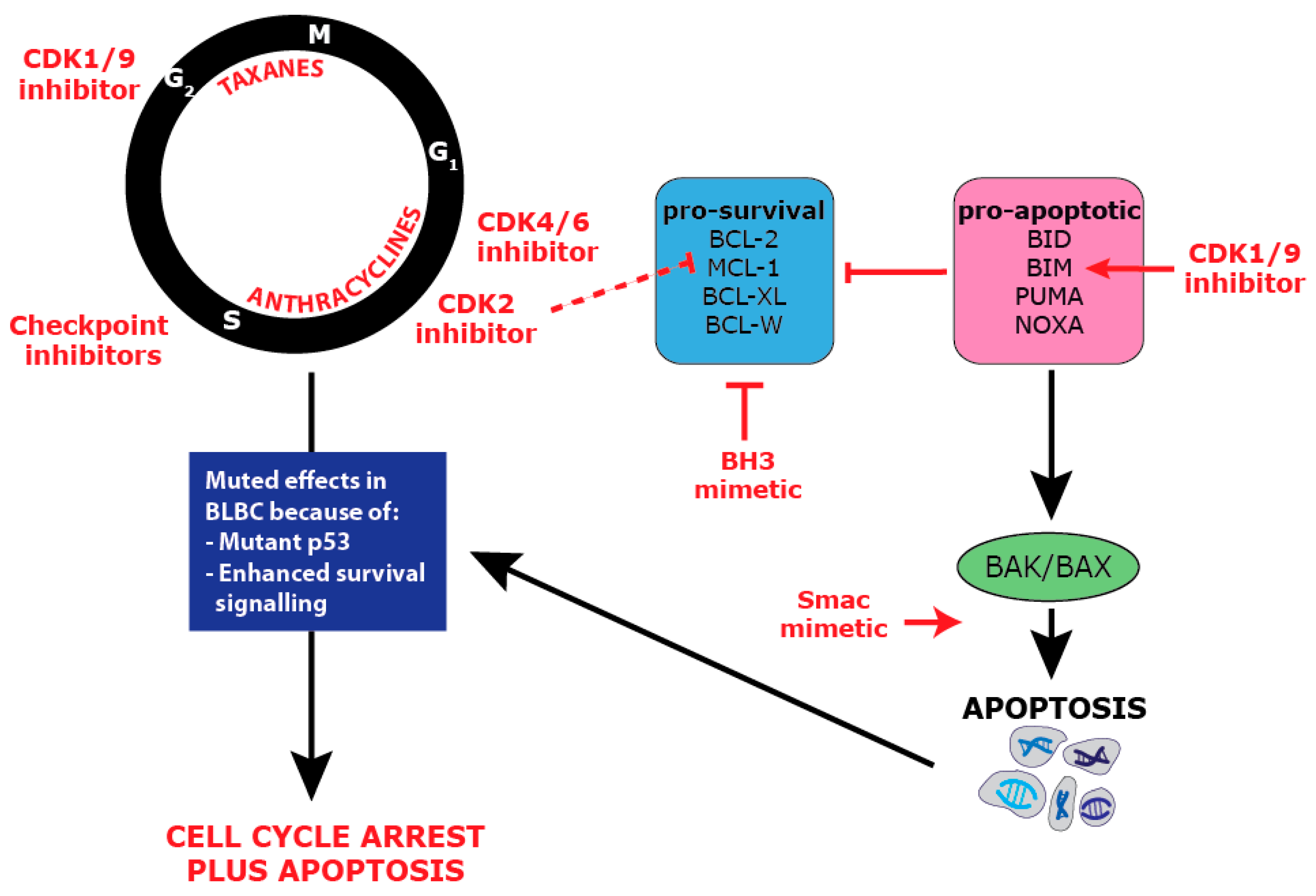
4. Proliferative Landscape
5. Deregulated Cell Cycle Control in BLBC
6. Targeting BLBC via the Cell Cycle
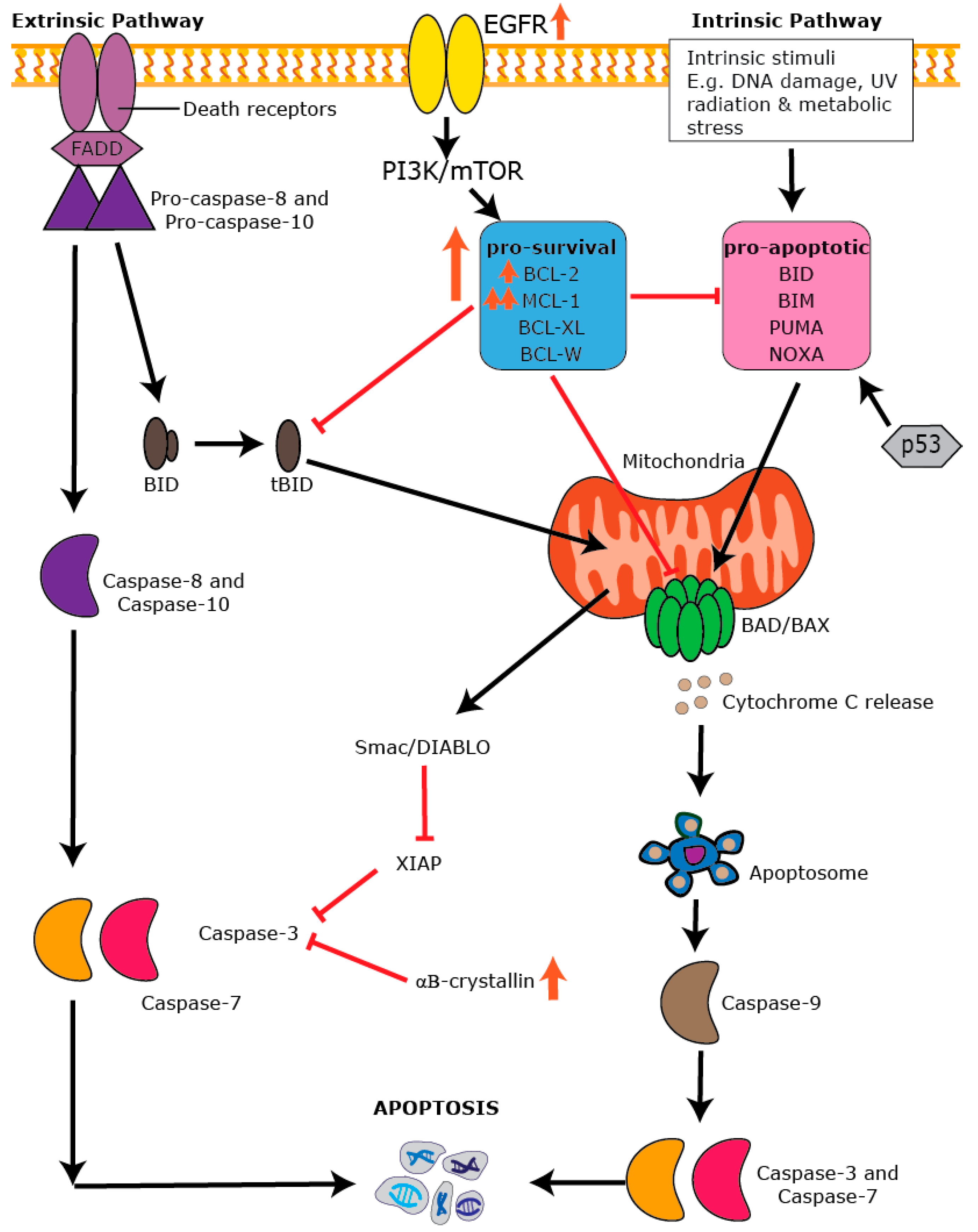
7. Apoptosis: An Essential Process in Healthy Tissues
8. The BCL-2 Family and the Intrinsic Apoptotic Pathway
9. Dysregulation of Apoptosis in BLBC
10. Targeting Survival in BLBC
11. Combination Targeting of Proliferation and Survival
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, D.G.; Short, S.M.; Prescott, N.L.; Tarr, S.M.; Coleman, K.A.; Yoder, B.J.; Crowe, J.P.; Choueiri, T.K.; Dawson, A.E.; Budd, G.T.; et al. Breast cancers with brain metastases are more likely to be estrogen receptor negative, express the basal cytokeratin CK5/6, and overexpress HER2 or EGFR. Am. J. Surg. Pathol. 2006, 30, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.; Dabbs, D.J.; Schnitt, S.J.; Baehner, F.L.; Decker, T.; Eusebi, V.; Fox, S.B.; Ichihara, S.; Jacquemier, J.; Lakhani, S.R.; et al. Basal-like and triple-negative breast cancers: A critical review with an emphasis on the implications for pathologists and oncologists. Mod. Pathol. 2011, 24, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Mustacchi, G.; De Laurentiis, M. The role of taxanes in triple-negative breast cancer: Literature review. Drug Des. Dev. Ther. 2015, 9, 4303–4318. [Google Scholar] [CrossRef]
- O’Toole, S.A.; Beith, J.M.; Millar, E.K.; West, R.; McLean, A.; Cazet, A.; Swarbrick, A.; Oakes, S.R. Therapeutic targets in triple negative breast cancer. J. Clin. Pathol. 2013, 66, 530–542. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61. [Google Scholar]
- Begg, C.B.; Rice, M.S.; Zabor, E.C.; Tworoger, S.S. Examining the common aetiology of serous ovarian cancers and basal-like breast cancers using double primaries. Br. J. Cancer 2017, 116, 1088–1091. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- De Summa, S.; Pinto, R.; Sambiasi, D.; Petriella, D.; Paradiso, V.; Paradiso, A.; Tommasi, S. BRCAness: A deeper insight into basal-like breast tumors. Ann. Oncol. 2013, 24, viii13–viii21. [Google Scholar] [CrossRef] [PubMed]
- Schuyer, M.; Berns, E.M. Is TP53 dysfunction required for BRCA1-associated carcinogenesis? Mol. Cell. Endocrinol. 1999, 155, 143–152. [Google Scholar] [CrossRef]
- Liu, X.; Holstege, H.; van der Gulden, H.; Treur-Mulder, M.; Zevenhoven, J.; Velds, A.; Kerkhoven, R.M.; van Vliet, M.H.; Wessels, L.F.; Peterse, J.L.; et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12111–12116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Laurentiis, M.; Cianniello, D.; Caputo, R.; Stanzione, B.; Arpino, G.; Cinieri, S.; Lorusso, V.; De Placido, S. Treatment of triple negative breast cancer (TNBC): Current options and future perspectives. Cancer Treat. Rev. 2010, 36 (Suppl. 3), S80–S86. [Google Scholar] [CrossRef]
- Alluri, P.; Newman, L.A. Basal-like and triple-negative breast cancers: Searching for positives among many negatives. Surg. Oncol. Clin. N. Am. 2014, 23, 567–577. [Google Scholar] [CrossRef]
- Prat, A.; Karginova, O.; Parker, J.S.; Fan, C.; He, X.; Bixby, L.; Harrell, J.C.; Roman, E.; Adamo, B.; Troester, M.; et al. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res. Treat. 2013, 142, 237–255. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef]
- Badowska-Kozakiewicz, A.M.; Budzik, M.P. Immunohistochemical characteristics of basal-like breast cancer. Contemp. Oncol. (Pozn) 2016, 20, 436–443. [Google Scholar] [CrossRef]
- Choo, J.R.; Nielsen, T.O. Biomarkers for basal-like breast cancer. Cancers 2010, 2, 1040–1065. [Google Scholar] [CrossRef] [PubMed]
- Haupt, B.; Ro, J.Y.; Schwartz, M.R. Basal-like breast carcinoma: A phenotypically distinct entity. Arch. Pathol. Lab. Med. 2010, 134, 130–133. [Google Scholar] [PubMed]
- Toft, D.J.; Cryns, V.L. Minireview: Basal-like breast cancer: From molecular profiles to targeted therapies. Mol. Endocrinol. 2011, 25, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, S.; Yashiro, M.; Takashima, T.; Aomatsu, N.; Kawajiri, H.; Ogawa, Y.; Onoda, N.; Ishikawa, T.; Wakasa, K.; Hirakawa, K. c-Kit expression as a prognostic molecular marker in patients with basal-like breast cancer. Br. J. Surg. 2013, 100, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Moyano, J.V.; Evans, J.R.; Chen, F.; Lu, M.; Werner, M.E.; Yehiely, F.; Diaz, L.K.; Turbin, D.; Karaca, G.; Wiley, E.; et al. αB-Crystallin is a novel oncoprotein that predicts poor clinical outcome in breast cancer. J. Clin. Investig. 2006, 116, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef]
- Ali, H.R.; Glont, S.E.; Blows, F.M.; Provenzano, E.; Dawson, S.J.; Liu, B.; Hiller, L.; Dunn, J.; Poole, C.J.; Bowden, S.; et al. PD-L1 protein expression in breast cancer is rare, enriched in basal-like tumours and associated with infiltrating lymphocytes. Ann. Oncol. 2015, 26, 1488–1493. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [Green Version]
- Healey, M.A.; Hirko, K.A.; Beck, A.H.; Collins, L.C.; Schnitt, S.J.; Eliassen, A.H.; Holmes, M.D.; Tamimi, R.M.; Hazra, A. Assessment of Ki67 expression for breast cancer subtype classification and prognosis in the Nurses’ Health Study. Breast Cancer Res. Treat. 2017, 166, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.K.; Lee, Y.; Lee, J.B.; Kim, H.K.; Lee, J.B.; Cho, S.J.; Kim, A. Breast carcinomas expressing basal markers have poor clinical outcome regardless of estrogen receptor status. Oncol. Rep. 2008, 19, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef]
- Leidy, J.; Khan, A.; Kandil, D. Basal-like breast cancer: Update on clinicopathologic, immunohistochemical, and molecular features. Arch. Pathol. Lab. Med. 2014, 138, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007, 8, 235–244. [Google Scholar] [CrossRef]
- Lerma, E.; Peiro, G.; Ramón, T.; Fernandez, S.; Martinez, D.; Pons, C.; Munoz, F.; Sabate, J.M.; Alonso, C.; Ojeda, B. Immunohistochemical heterogeneity of breast carcinomas negative for estrogen receptors, progesterone receptors and Her2/neu (basal-like breast carcinomas). Mod. Pathol. 2007, 20, 1200. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Bai, J.; Li, Y.; Zhang, G. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Peyressatre, M.; Prevel, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From small molecules to Peptide inhibitors. Cancers (Basel) 2015, 7, 179–237. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.S.; Weinberg, R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 1998, 18, 753–761. [Google Scholar] [CrossRef]
- Meyerson, M.; Harlow, E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol. Cell. Biol. 1994, 14, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Kato, J.; Matsushime, H.; Hiebert, S.W.; Ewen, M.E.; Sherr, C.J. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993, 7, 331–342. [Google Scholar]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef]
- Cakir, A.; Gonul, I.I.; Uluoglu, O. A comprehensive morphological study for basal-like breast carcinomas with comparison to nonbasal-like carcinomas. Diagn. Pathol. 2012, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Ertel, A.; Dean, J.L.; Rui, H.; Liu, C.; Witkiewicz, A.K.; Knudsen, K.E.; Knudsen, E.S. RB-pathway disruption in breast cancer: Differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 2010, 9, 4153–4163. [Google Scholar] [CrossRef]
- Trere, D.; Brighenti, E.; Donati, G.; Ceccarelli, C.; Santini, D.; Taffurelli, M.; Montanaro, L.; Derenzini, M. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann. Oncol. 2009, 20, 1818–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herschkowitz, J.I.; He, X.; Fan, C.; Perou, C.M. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008, 10, R75. [Google Scholar] [CrossRef] [PubMed]
- Branham, M.T.; Marzese, D.M.; Laurito, S.R.; Gago, F.E.; Orozco, J.I.; Tello, O.M.; Vargas-Roig, L.M.; Roque, M. Methylation profile of triple-negative breast carcinomas. Oncogenesis 2012, 1, e17. [Google Scholar] [CrossRef]
- Gauthier, M.L.; Berman, H.K.; Miller, C.; Kozakeiwicz, K.; Chew, K.; Moore, D.; Rabban, J.; Chen, Y.Y.; Kerlikowske, K.; Tlsty, T.D. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell 2007, 12, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.; Green, A.R.; Aleskandarany, M.A.; Grainge, M.; Paish, C.E.; Lambros, M.B.; Reis-Filho, J.S.; Ellis, I.O. CCND1 amplification and cyclin D1 expression in breast cancer and their relation with proteomic subgroups and patient outcome. Breast Cancer Res. Treat. 2008, 109, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef]
- Dai, M.; Zhang, C.; Ali, A.; Hong, X.; Tian, J.; Lo, C.; Fils-Aimé, N.; Burgos, S.A.; Ali, S.; Lebrun, J.-J. CDK4 regulates cancer stemness and is a novel therapeutic target for triple-negative breast cancer. Sci. Rep. 2016, 6, 35383. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-H.; Yao, J.; Chan, L.-C.; Wu, T.-J.; Hsu, J.L.; Fang, Y.-F.; Wei, Y.; Wu, Y.; Huang, W.-C.; Liu, C.-L. Definition of PKC-α, CDK6, and MET as therapeutic targets in triple-negative breast cancer. Cancer Res. 2014, 74, 4822–4835. [Google Scholar] [CrossRef]
- Cretella, D.; Ravelli, A.; Fumarola, C.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Alfieri, R.; Biondi, A.; Generali, D.; Bonelli, M. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J. Exp. Clin. Cancer Res. 2018, 37, 72. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Yu, J.; Deng, M.; Yin, Y.; Zhang, H.; Luo, K.; Qin, B.; Li, Y.; Wu, C.; Ren, T. CDK4/6-dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat. Commun. 2017, 8, 13923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, S.; Shirane, M.; Takekoshi, S.; Kusakabe, T.; Itoh, J.; Egashira, N.; Tokuda, Y.; Mori, K.; Osamura, Y.R. Overexpression of E2F-5 correlates with a pathological basal phenotype and a worse clinical outcome. Br. J. Cancer 2009, 100, 764–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulkes, W.D.; Brunet, J.S.; Stefansson, I.M.; Straume, O.; Chappuis, P.O.; Begin, L.R.; Hamel, N.; Goffin, J.R.; Wong, N.; Trudel, M.; et al. The prognostic implication of the basal-like (cyclin E high/p27 low/p53+/glomeruloid-microvascular-proliferation+) phenotype of BRCA1-related breast cancer. Cancer Res. 2004, 64, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Caldon, C.E.; Sergio, C.M.; Kang, J.; Muthukaruppan, A.; Boersma, M.N.; Stone, A.; Barraclough, J.; Lee, C.S.; Black, M.A.; Miller, L.D.; et al. Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Mol. Cancer Ther. 2012, 11, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, R.; Mackay, A.; Wilkerson, P.M.; Lambros, M.B.; Wetterskog, D.; Arnedos, M.; Shiu, K.K.; Geyer, F.C.; Langerod, A.; Kreike, B.; et al. Functional characterization of the 19q12 amplicon in grade III breast cancers. Breast Cancer Res. 2012, 14, R53. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voduc, D.; Nielsen, T.O.; Cheang, M.C.; Foulkes, W.D. The combination of high cyclin E and Skp2 expression in breast cancer is associated with a poor prognosis and the basal phenotype. Hum. Pathol. 2008, 39, 1431–1437. [Google Scholar] [CrossRef]
- Corsino, P.E.; Davis, B.J.; Norgaard, P.H.; Parker, N.N.; Law, M.; Dunn, W.; Law, B.K. Mammary tumors initiated by constitutive Cdk2 activation contain an invasive basal-like component. Neoplasia 2008, 10, 1240–1252. [Google Scholar] [CrossRef]
- Liu, J.C.; Granieri, L.; Shrestha, M.; Wang, D.-Y.; Vorobieva, I.; Rubie, E.A.; Jones, R.; Ju, Y.; Pellecchia, G.; Jiang, Z. Identification of CDC25 as a Common Therapeutic Target for Triple-Negative Breast Cancer. Cell Rep. 2018, 23, 112–126. [Google Scholar] [CrossRef]
- Nogi, H.; Uchida, K.; Kamio, M.; Kato, K.; Toriumi, Y.; Akiba, T.; Morikawa, T.; Suzuki, M.; Kobayashi, T.; Takeyama, H. Triple-negative breast cancer exhibits a favorable response to neoadjuvant chemotherapy independent of the expression of topoisomerase IIα. Mol. Clin. Oncol. 2016, 4, 383–389. [Google Scholar] [CrossRef]
- Mrklic, I.; Pogorelic, Z.; Capkun, V.; Tomic, S. Expression of topoisomerase II-α in triple negative breast cancer. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Chandriani, S.; Frengen, E.; Cowling, V.H.; Pendergrass, S.A.; Perou, C.M.; Whitfield, M.L.; Cole, M.D. A Core MYC Gene Expression Signature Is Prominent in Basal-Like Breast Cancer but Only Partially Overlaps the Core Serum Response. PLoS ONE 2009, 4, e6693. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grushko, T.A.; Nwachukwu, C.; Charoenthammaraksa, S.; Huo, D.; Khramtsov, A.; Mashek, H.; Zhang, C.; Xu, J.; Perou, C.M.; Olopade, O.I. Evaluation of BRCA1 inactivation by promoter methylation as a marker of triple-negative and basal-like breast cancers. J. Clin. Oncol. 2010, 28, 10510–10510. [Google Scholar] [CrossRef]
- Verlinden, L.; Bempt, I.V.; Eelen, G.; Drijkoningen, M.; Verlinden, I.; Marchal, K.; De Wolf-Peeters, C.; Christiaens, M.-R.; Michiels, L.; Bouillon, R. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor−/progesterone receptor−/HER-2− breast carcinomas. Cancer Res. 2007, 67, 6574–6581. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Osmanbeyoglu, H.U.; Pelossof, R.; Bromberg, J.; Leslie, C.S. Linking signaling pathways to transcriptional programs in breast cancer. Genome Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Bartek, J. Cell cycle–targeted cancer therapies. Ann. Rev. Cancer Biol. 2017, 1, 41–57. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Ocaña, A.; Pérez-Peña, J.; Díez-González, L.; Sánchez-Corrales, V.; Templeton, A.; Seruga, B.; Amir, E.; Pandiella, A. Transcriptomic analyses identify association between mitotic kinases, PDZ-binding kinase and BUB1, and clinical outcome in breast cancer. Breast Cancer Res. Treat. 2016, 156, 1–8. [Google Scholar] [CrossRef]
- Rogers, S.; McCloy, R.A.; Parker, B.L.; Gallego-Ortega, D.; Law, A.M.; Chin, V.T.; Conway, J.R.; Fey, D.; Millar, E.K.; O’Toole, S. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene 2018, 37, 4518–4533. [Google Scholar] [CrossRef] [PubMed]
- Marzec, K.A.; Burgess, A. Oncogenic functions of MASTL kinase. Front. Cell Dev. Biol. 2018, 6, 162. [Google Scholar] [CrossRef] [PubMed]
- Thorner, A.R.; Hoadley, K.A.; Parker, J.S.; Winkel, S.; Millikan, R.C.; Perou, C.M. In vitro and in vivo analysis of B-Myb in basal-like breast cancer. Oncogene 2008, 28, 742. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908. [Google Scholar] [CrossRef] [Green Version]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Ganguly, A.; Yang, H.; Cabral, F. Paclitaxel-dependent cell lines reveal a novel drug activity. Mol. Cancer Ther. 2010, 9, 2914–2923. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules and actin filaments: Dynamic targets for cancer chemotherapy. Curr. Opin. Cell Biol. 1998, 10, 123–130. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER positive breast cancer. Endocr.-Relat. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, N.; Conklin, D.; Beckmann, R.; Luo, T.; Chau, K.; Thomas, J.; Mc Nulty, A.; Marchal, C.; Kalous, O.; von Euw, E.; et al. Preclinical Activity of Abemaciclib Alone or in Combination with Antimitotic and Targeted Therapies in Breast Cancer. Mol. Cancer Ther. 2018, 17, 897–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.S.; Barr, A.R.; Cutts, R.; Beaney, M.; Babina, I.; Sampath, D.; Giltnane, J.; Lacap, J.A.; Crocker, L.; Young, A. Single-cell dynamics determines response to CDK4/6 inhibition in triple-negative breast cancer. Clin. Cancer Res. 2017, 23, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.; McClendon, A.K.; Hickey, T.E.; Butler, L.M.; Tilley, W.D.; Witkiewicz, A.K.; Knudsen, E.S. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 2012, 11, 2756–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alba, E.; Chacon, J.I.; Lluch, A.; Anton, A.; Estevez, L.; Cirauqui, B.; Carrasco, E.; Calvo, L.; Segui, M.A.; Ribelles, N.; et al. A randomized phase II trial of platinum salts in basal-like breast cancer patients in the neoadjuvant setting. Results from the GEICAM/2006-03, multicenter study. Breast Cancer Res. Treat. 2012, 136, 487–493. [Google Scholar] [CrossRef] [PubMed]
- McClendon, A.K.; Dean, J.L.; Rivadeneira, D.B.; Yu, J.E.; Reed, C.A.; Gao, E.; Farber, J.L.; Force, T.; Koch, W.J.; Knudsen, E.S. CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle 2012, 11, 2747–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajput, S.; Khera, N.; Guo, Z.; Hoog, J.; Li, S.; Ma, C.X. Inhibition of cyclin dependent kinase 9 by dinaciclib suppresses cyclin B1 expression and tumor growth in triple negative breast cancer. Oncotarget 2016, 7, 56864–56875. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E.; et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, S.; Caldon, E.; Tilley, W.; Wang, S. Cyclin Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Low, K.H.; Alexander, A.; Jiang, Y.; Karakas, C.; Hess, K.R.; Carey, J.P.; Bui, T.; Vijayaraghavan, S.; Evans, K.W.; et al. Cyclin E overexpression sensitizes triple negative breast cancer to Wee1 kinase Inhibition. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Jimenez, C.; Alcaraz-Sanabria, A.; Perez-Pena, J.; Corrales-Sanchez, V.; Serrano-Heras, G.; Galan-Moya, E.M.; Serrano-Oviedo, L.; Montero, J.C.; Burgos, M.; Llopis, J.; et al. Targeting basal-like breast tumors with bromodomain and extraterminal domain (BET) and polo-like kinase inhibitors. Oncotarget 2017, 8, 19478–19490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Braud, F.G.; Cascinu, S.; Spitaleri, G.; Pilz, K.; Clementi, L.; Liu, D.; Glomb, P.; De Pas, T. A phase I dose-escalation study of volasertib (BI 6727) combined with nintedanib (BIBF 1120) in advanced solid tumors. J. Clin. Oncol. 2013, 31, 2556–2556. [Google Scholar]
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Fraiser, L.H.; Kanekal, S.; Kehrer, J.P. Cyclophosphamide toxicity. Characterising and avoiding the problem. Drugs 1991, 42, 781–795. [Google Scholar] [CrossRef]
- Hall, A.G.; Tilby, M.L. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood Rev. 1992, 6, 163–173. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs--phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [PubMed]
- Shah, M.; Nunes, M.R.; Stearns, V. CDK4/6 Inhibitors: Game Changers in the Management of Hormone Receptor-Positive Advanced Breast Cancer? Oncology (Williston Park) 2018, 32, 216–222. [Google Scholar] [PubMed]
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shohdy, K.S.; Lasheen, S.; Kassem, L.; Abdel-Rahman, O. Gastrointestinal adverse effects of cyclin-dependent kinase 4 and 6 inhibitors in breast cancer patients: A systematic review and meta-analysis. Ther. Adv. Drug Saf. 2017, 8, 337–347. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Gregory, G.P.; Hogg, S.J.; Kats, L.M.; Vidacs, E.; Baker, A.J.; Gilan, O.; Lefebure, M.; Martin, B.P.; Dawson, M.A.; Johnstone, R.W.; et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 2015, 29, 1437–1441. [Google Scholar] [CrossRef]
- Fabre, C.; Gobbi, M.; Ezzili, C.; Zoubir, M.; Sablin, M.P.; Small, K.; Im, E.; Shinwari, N.; Zhang, D.; Zhou, H.; et al. Clinical study of the novel cyclin-dependent kinase inhibitor dinaciclib in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia patients. Cancer Chemother. Pharmacol. 2014, 74, 1057–1064. [Google Scholar] [CrossRef]
- Blachly, J.S.; Byrd, J.C. Emerging drug profile: Cyclin-dependent kinase inhibitors. Leuk. Lymphoma 2013, 54, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Karp, J.E. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk. Res. 2015, 39, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.I.; Bible, K.; Martell, R.E.; Adelman, D.C.; LoRusso, P.M. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Investig. New Drugs 2008, 26, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Saladino, C.; Davis, S.; Blake, D.; Zheleva, D. CYC065, potential therapeutic agent for AML and MLL leukaemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, S183. [Google Scholar] [CrossRef]
- Vriend, L.E.; De Witt Hamer, P.C.; Van Noorden, C.J.; Wurdinger, T. WEE1 inhibition and genomic instability in cancer. Biochim. Biophys. Acta 2013, 1836, 227–235. [Google Scholar] [CrossRef]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.M.; Kaufmann, S.H. Prospects for the use of ATR inhibitors to treat cancer. Pharmaceuticals 2010, 3, 1311–1334. [Google Scholar] [CrossRef]
- Schoffski, P. Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology. Oncologist 2009, 14, 559–570. [Google Scholar] [CrossRef]
- de Braud, F.; Cascinu, S.; Spitaleri, G.; Pilz, K.; Clementi, L.; Liu, D.; Sikken, P.; De Pas, T. A phase I, dose-escalation study of volasertib combined with nintedanib in advanced solid tumors. Ann. Oncol. 2015, 26, 2341–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Torchinsky, A.; Carp, H.; Toder, V. The role of apoptosis in normal and abnormal embryonic development. J. Assist. Reprod. Genet. 1999, 16, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Marsden, V.S.; Strasser, A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu. Rev. Immunol. 2003, 21, 71–105. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Callus, B.A.; Vaux, D.L. Caspase inhibitors: Viral, cellular and chemical. Cell Death Differ. 2007, 14, 73–78. [Google Scholar] [CrossRef]
- Mahoney, D.; Cheung, H.; Mrad, R.L.; Plenchette, S.; Simard, C.; Enwere, E.; Arora, V.; Mak, T.; Lacasse, E.; Waring, J. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc. Natl. Acad. Sci. USA 2008, 105, 11778–11783. [Google Scholar] [CrossRef] [PubMed]
- Adrain, C.; Creagh, E.M.; Martin, S.J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001, 20, 6627–6636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Young, A.I.; Law, A.M.; Castillo, L.; Chong, S.; Cullen, H.D.; Koehler, M.; Herzog, S.; Brummer, T.; Lee, E.F.; Fairlie, W.D.; et al. MCL-1 inhibition provides a new way to suppress breast cancer metastasis and increase sensitivity to dasatinib. Breast Cancer Res. 2016, 18, 125. [Google Scholar] [CrossRef]
- Bouchalova, K.; Svoboda, M.; Kharaishvili, G.; Vrbkova, J.; Bouchal, J.; Trojanec, R.; Koudelakova, V.; Radova, L.; Cwiertka, K.; Hajduch, M.; et al. BCL2 is an independent predictor of outcome in basal-like triple-negative breast cancers treated with adjuvant anthracycline-based chemotherapy. Tumor Biol. 2015, 36, 4243–4252. [Google Scholar] [CrossRef]
- Bhargava, V.; Kell, D.L.; van de Rijn, M.; Warnke, R.A. Bcl-2 immunoreactivity in breast carcinoma correlates with hormone receptor positivity. Am. J. Pathol. 1994, 145, 535–540. [Google Scholar] [PubMed]
- Oakes, S.R.; Vaillant, F.; Lim, E.; Lee, L.; Breslin, K.; Feleppa, F.; Deb, S.; Ritchie, M.E.; Takano, E.; Ward, T.; et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl. Acad. Sci. USA 2012, 109, 2766–2771. [Google Scholar] [CrossRef] [PubMed]
- Petrocca, F.; Altschuler, G.; Tan, S.M.; Mendillo, M.L.; Yan, H.; Jerry, D.J.; Kung, A.L.; Hide, W.; Ince, T.A.; Lieberman, J. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 2013, 24, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.M.; Rossanese, O.W.; Olejniczak, E.T.; Fesik, S.W. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ. 2015, 22, 2098–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Nimmer, P.; Sheppard, G.S.; Bruncko, M.; Hessler, P.; Lu, X.; Roberts-Rapp, L.; Pappano, W.N.; Elmore, S.W.; Souers, A.J.; et al. MCL-1 Is a Key Determinant of Breast Cancer Cell Survival: Validation of MCL-1 Dependency Utilizing a Highly Selective Small Molecule Inhibitor. Mol. Cancer Ther. 2015, 14, 1837–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inuzuka, H.; Shaik, S.; Onoyama, I.; Gao, D.; Tseng, A.; Maser, R.S.; Zhai, B.; Wan, L.; Gutierrez, A.; Lau, A.W.; et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011, 471, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Wang, Y.; Zhang, P.; Lu, J.; Mao, J.H. Evaluating the prognostic significance of FBXW7 expression level in human breast cancer by a meta-analysis of transcriptional profiles. J. Cancer Sci. Ther. 2012, 4, 299–305. [Google Scholar] [CrossRef]
- Pervin, S.; Tran, A.; Tran, L.; Urman, R.; Braga, M.; Chaudhuri, G.; Singh, R. Reduced association of anti-apoptotic protein Mcl-1 with E3 ligase Mule increases the stability of Mcl-1 in breast cancer cells. Br. J. Cancer 2011, 105, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Campbell, K.J.; Dhayade, S.; Ferrari, N.; Sims, A.H.; Johnson, E.; Mason, S.M.; Dickson, A.; Ryan, K.M.; Kalna, G.; Edwards, J.; et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 2018, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, B.; Park, S.; Pak, J.H.; Kim, I. Downregulation of Mcl-1 by daunorubicin pretreatment reverses resistance of breast cancer cells to TNF-related apoptosis-inducing ligand. Biochem. Biophys. Res. Commun. 2012, 422, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Min, K.J.; Seo, B.R.; Nam, J.O.; Choi, K.S.; Yoo, Y.H.; Kwon, T.K. Cafestol overcomes ABT-737 resistance in Mcl-1-overexpressed renal carcinoma Caki cells through downregulation of Mcl-1 expression and upregulation of Bim expression. Cell Death Dis. 2014, 5, e1514. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Sitterding, S.M.; Wiseman, W.R.; Schiller, C.L.; Luan, C.; Chen, F.; Moyano, J.V.; Watkin, W.G.; Wiley, E.L.; Cryns, V.L.; Diaz, L.K. AlphaB-crystallin: A novel marker of invasive basal-like and metaplastic breast carcinomas. Ann. Diagn. Pathol. 2008, 12, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, O.; Chen, F.; Wiley, E.L.; Keswani, A.; Diaz, L.K.; Memmel, H.C.; Rademaker, A.; Gradishar, W.J.; Morrow, M.; Khan, S.A.; et al. alphaB-crystallin is a novel predictor of resistance to neoadjuvant chemotherapy in breast cancer. Breast Cancer Res. Treat. 2008, 111, 411–417. [Google Scholar] [CrossRef]
- Voduc, K.D.; Nielsen, T.O.; Perou, C.M.; Harrell, J.C.; Fan, C.; Kennecke, H.; Minn, A.J.; Cryns, V.L.; Cheang, M.C.U. αB-crystallin expression in breast cancer is associated with brain metastasis. NPJ Breast Cancer 2015, 1, 15014. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ruan, Q.; Han, S.; Xi, L.; Jiang, W.; Jiang, H.; Ostrov, D.A.; Cai, J. Discovery of structure-based small molecular inhibitor of alphaB-crystallin against basal-like/triple-negative breast cancer development in vitro and in vivo. Breast Cancer Res Treat. 2014, 145, 45–59. [Google Scholar] [CrossRef]
- Launay, N.; Tarze, A.; Vicart, P.; Lilienbaum, A. Serine 59 phosphorylation of {alpha}B-crystallin down-regulates its anti-apoptotic function by binding and sequestering Bcl-2 in breast cancer cells. J. Biol. Chem. 2010, 285, 37324–37332. [Google Scholar] [CrossRef]
- Xu, F.; Yu, H.; Liu, J.; Cheng, L. alphaB-crystallin regulates oxidative stress-induced apoptosis in cardiac H9c2 cells via the PI3K/AKT pathway. Mol. Biol. Rep. 2013, 40, 2517–2526. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef]
- Fojo, T. p53 as a therapeutic target: Unresolved issues on the road to cancer therapy targeting mutant p53. Drug Resist. Updat 2002, 5, 209–216. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Vachhani, P.; Bose, P.; Rahmani, M.; Grant, S. Rational combination of dual PI3K/mTOR blockade and Bcl-2/-xL inhibition in AML. Physiol. Genom. 2014, 46, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.W.; Hong, W.; Kang, H.J.; Kim, H.J.; Zhao, W.; Wang, A.; Seong, Y.S.; Bae, I. Inhibition of the PI3K/AKT pathway potentiates cytotoxicity of EGFR kinase inhibitors in triple-negative breast cancer cells. J. Cell. Mol. Med. 2013, 17, 648–656. [Google Scholar] [CrossRef]
- Fulda, S. Smac Mimetics to Therapeutically Target IAP Proteins in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Allensworth, J.L.; Sauer, S.J.; Lyerly, H.K.; Morse, M.A.; Devi, G.R. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Res. Treat. 2013, 137, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Masoumi, K.C.; Cornmark, L.; Lonne, G.K.; Hellman, U.; Larsson, C. Identification of a novel protein kinase Cdelta-Smac complex that dissociates during paclitaxel-induced cell death. FEBS Lett. 2012, 586, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Cornmark, L.; Holmgren, C.; Masoumi, K.; Larsson, C. PKC activation sensitizes basal-like breast cancer cell lines to Smac mimetics. Cell Death Discov. 2016, 2, 16002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dine, J.L.; O’Sullivan, C.C.; Voeller, D.; Greer, Y.E.; Chavez, K.J.; Conway, C.M.; Sinclair, S.; Stone, B.; Amiri-Kordestani, L.; Merchant, A.S.; et al. The TRAIL receptor agonist drozitumab targets basal B triple-negative breast cancer cells that express vimentin and Axl. Breast Cancer Res. Treat. 2016, 155, 235–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baell, J.B.; Huang, D.C. Prospects for targeting the Bcl-2 family of proteins to develop novel cytotoxic drugs. Biochem. Pharmacol. 2002, 64, 851–863. [Google Scholar] [CrossRef]
- Cragg, M.S.; Harris, C.; Strasser, A.; Scott, C.L. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat. Rev. Cancer 2009, 9, 321–326. [Google Scholar] [CrossRef]
- Lessene, G.; Czabotar, P.E.; Colman, P.M. BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 989–1000. [Google Scholar] [CrossRef]
- Fujise, K.; Zhang, D.; Liu, J.; Yeh, E.T. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J. Biol. Chem. 2000, 275, 39458–39465. [Google Scholar] [CrossRef]
- Dai, H.; Meng, X.W.; Kaufmann, S.H. Mitochondrial apoptosis and BH3 mimetics. F1000Res 2016, 5, 2804. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, G.J.; Lok, S.W.; Bergin, A.R.; Whittle, J.R.; Shackleton, K.; Sherman, P.; Dawson, S.-J.; Desai, J.; Liew, D.; Mann, B.; et al. Safety and efficacy of the BCL2 inhibitor venetoclax in estrogen receptor (ER) and BCL2-positive metastatic breast cancer: The mBEP study. J. Clin. Oncol. 2017, 35, 1044–1044. [Google Scholar] [CrossRef]
- Inao, T.; Iida, Y.; Moritani, T.; Okimoto, T.; Tanino, R.; Kotani, H.; Harada, M. Bcl-2 inhibition sensitizes triple-negative human breast cancer cells to doxorubicin. Oncotarget 2018, 9, 25545–25556. [Google Scholar] [CrossRef]
- Williams, M.M.; Lee, L.; Hicks, D.J.; Joly, M.M.; Elion, D.; Rahman, B.; McKernan, C.; Sanchez, V.; Balko, J.M.; Stricker, T.; et al. Key Survival Factor, Mcl-1, Correlates with Sensitivity to Combined Bcl-2/Bcl-xL Blockade. Mol. Cancer Res. 2017, 15, 259–268. [Google Scholar] [CrossRef]
- Perciavalle, R.M.; Opferman, J.T. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013, 23, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, J.; Nimmer, P.; Jin, S.; Smith, M.; Xiao, Y.; et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.L.; Roberts, D.J.; Kubli, D.A.; Lee, Y.; Quinsay, M.N.; Owens, J.B.; Fischer, K.M.; Sussman, M.A.; Miyamoto, S.; Gustafsson, A.B. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013, 27, 1365–1377. [Google Scholar] [PubMed] [Green Version]
- Wang, X.; Bathina, M.; Lynch, J.; Koss, B.; Calabrese, C.; Frase, S.; Schuetz, J.D.; Rehg, J.E.; Opferman, J.T. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013, 27, 1351–1364. [Google Scholar] [PubMed] [Green Version]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.; Whittle, J.R.; Vaillant, F.; Serrano, A.; Gong, J.-N.; Giner, G.; Maragno, A.L.; Chanrion, M.; Schneider, E.; Pal, B.; et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Wuilleme-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.L.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, Y. Targeting p53 for Novel Anticancer Therapy. Transl. Oncol. 2010, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G. The role of gefitinib in lung cancer treatment. Clin. Cancer Res. 2004, 10, 4233s–4237s. [Google Scholar] [CrossRef] [PubMed]
- Segovia-Mendoza, M.; Gonzalez-Gonzalez, M.E.; Barrera, D.; Diaz, L.; Garcia-Becerra, R. Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of HER2-positive breast cancer: Preclinical and clinical evidence. Am. J. Cancer Res. 2015, 5, 2531–2561. [Google Scholar] [PubMed]
- Fulda, S. Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin. Cancer Res. 2015, 21, 5030–5036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrucci, E.; Pasquini, L.; Bernabei, M.; Saulle, E.; Biffoni, M.; Accarpio, F.; Sibio, S.; Di Giorgio, A.; Di Donato, V.; Casorelli, A.; et al. A small molecule SMAC mimic LBW242 potentiates TRAIL- and anticancer drug-mediated cell death of ovarian cancer cells. PLoS ONE 2012, 7, e35073. [Google Scholar] [CrossRef] [PubMed]
- Parton, M.; Bardia, A.; Kummel, S.; Estevez, L.G.; Huang, C.-S.; Castan, J.C.; Ruiz Borrego, M.; Telli, M.L.; Lluch, A.; Lopez, R. A phase II, open-label, neoadjuvant, randomized study of LCL161 with paclitaxel in patients with triple-negative breast cancer (TNBC). Am. Soc. Clin. Oncol. 2015, 33, 1014–1014. [Google Scholar]
- Rocha Lima, C.M.; Bayraktar, S.; Flores, A.M.; MacIntyre, J.; Montero, A.; Baranda, J.C.; Wallmark, J.; Portera, C.; Raja, R.; Stern, H.; et al. Phase Ib study of drozitumab combined with first-line mFOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Cancer Investig. 2012, 30, 727–731. [Google Scholar] [CrossRef]
- Xiang, H.; Reyes, A.E., 2nd; Eppler, S.; Kelley, S.; Damico-Beyer, L.A. Death receptor 5 agonistic antibody PRO95780: Preclinical pharmacokinetics and concentration-effect relationship support clinical dose and regimen selection. Cancer Chemother. Pharmacol. 2013, 72, 405–415. [Google Scholar] [CrossRef]
- Kang, Z.; Chen, J.J.; Yu, Y.; Li, B.; Sun, S.Y.; Zhang, B.; Cao, L. Drozitumab, a human antibody to death receptor 5, has potent antitumor activity against rhabdomyosarcoma with the expression of caspase-8 predictive of response. Clin. Cancer Res. 2011, 17, 3181–3192. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Lessene, G.; Czabotar, P.E.; Sleebs, B.E.; Zobel, K.; Lowes, K.N.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Shapiro, G.I. Cyclin-dependent kinases (cdks) and the DNA damage response: Rationale for cdk inhibitor-chemotherapy combinations as an anticancer strategy for solid tumors. Expert Opin. Ther. Targets 2010, 14, 1199–1212. [Google Scholar] [CrossRef]
- Panayotopoulou, E.G.; Müller, A.-K.; Börries, M.; Busch, H.; Hu, G.; Lev, S. Targeting of apoptotic pathways by SMAC or BH3 mimetics distinctly sensitizes paclitaxel-resistant triple negative breast cancer cells. Oncotarget 2017, 8, 45088–45104. [Google Scholar] [CrossRef]
- Chen, J.; Jin, S.; Abraham, V.; Huang, X.; Liu, B.; Mitten, M.J.; Nimmer, P.; Lin, X.; Smith, M.; Shen, Y.; et al. The Bcl-2/Bcl-XL/Bcl-w Inhibitor, Navitoclax, Enhances the Activity of Chemotherapeutic Agents In Vitro and In Vivo. Mol. Cancer Ther. 2011, 10, 2340–2349. [Google Scholar] [CrossRef] [Green Version]
- Inoue-Yamauchi, A.; Jeng, P.S.; Kim, K.; Chen, H.-C.; Han, S.; Ganesan, Y.T.; Ishizawa, K.; Jebiwott, S.; Dong, Y.; Pietanza, M.C.; et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat. Commun. 2017, 8, 16078. [Google Scholar] [CrossRef] [Green Version]
- Paoluzzi, L.; Gonen, M.; Gardner, J.R.; Mastrella, J.; Yang, D.; Holmlund, J.; Sorensen, M.; Leopold, L.; Manova, K.; Marcucci, G.; et al. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood 2008, 111, 5350–5358. [Google Scholar] [CrossRef] [Green Version]
- Thangavel, C.; Boopathi, E.; Liu, Y.; McNair, C.; Haber, A.; Perepelyuk, M.; Bhardwaj, A.; Addya, S.; Ertel, A.; Shoyele, S.; et al. Therapeutic Challenge with a CDK 4/6 Inhibitor Induces an RB-Dependent SMAC-Mediated Apoptotic Response in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.S.; Tat, T.T.; Misra, S.; Hill, B.T.; Smith, M.R.; Almasan, A.; Mazumder, S. Cyclin E/Cdk2-dependent phosphorylation of Mcl-1 determines its stability and cellular sensitivity to BH3 mimetics. Oncotarget 2015, 6, 16912–16925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Dai, Y.; Pei, X.Y.; Myers, J.; Wang, L.; Kramer, L.B.; Garnett, M.; Schwartz, D.M.; Su, F.; Simmons, G.L.; et al. CDK inhibitors upregulate BH3-only proteins to sensitize human myeloma cells to BH3 mimetic therapies. Cancer Res. 2012, 72, 4225–4237. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Lee, M.J.; Ye, A.S.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; MacBeath, G.; Yaffe, M.B. Sequential Application of Anticancer Drugs Enhances Cell Death by Rewiring Apoptotic Signaling Networks. Cell 2012, 149, 780–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadimitriou, M.; Mountzios, G.; Papadimitriou, C.A. The role of PARP inhibition in triple-negative breast cancer: Unraveling the wide spectrum of synthetic lethality. Cancer Treat. Rev. 2018, 67, 34–44. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Drug Examples | Mechanism of Action | Drug Status | Clinical Use | Mode of Administration | Adverse Events |
|---|---|---|---|---|---|---|
| Chemotherapy | ||||||
| Anthracycline [84,90,105,106,107] | Doxorubicin–Adriamycin (Bedford Laboratories) |
Effect: cytostatic and cytotoxic | FDA approved | ALL, AML, lymphoma, neuroblastoma, sarcoma, Wilms’ tumor, SCLC, breast, gastric, ovarian, thyroid, bladder cancers | IV | Alopecia, nausea, cardiotoxicity |
| Epirubicin–Ellence (Pfizer) | Breast cancer | |||||
| Alkylating Agent: Nitrogen mustards [108,109] | Cyclophosphamide–Cytoxan (Baxter Healthcare Corporation) | Covalent linkage of alkyl groups to DNA (N-7 position of guanine), preventing DNA synthesis Arrests in all cell cycle phases, particularly G1/S. Effect: cytotoxic | FDA approved | Hematological malignancies, neuroblastoma, retinoblastoma, ovarian, breast cancers | IV | Myelosuppression, nausea, alopecia, hemorrhagic cystitis |
| Alkylating Agent: Platinum [110,111,112] | Carboplatin–Paraplatin (Bristol-Myers Squibb Company) | Platinates DNA Covalent binding of platinum to DNA (N-7 position on purine residues), causing DNA damage. Induces an S and G2/M arrest. Effect: anti-mitotic and cytotoxic | FDA approved | Head and neck, ovarian, lung cancers | IV | Myelosuppression |
| Cisplatin–Platinol (Bristol-Myers Squibb Company) | FDA approved | Lung, SCCHN, ovarian, uterine, breast, bladder, testicular cancers | Nephrotoxicity, nausea, vomiting | |||
| Taxane [6,87] | Docetaxel–Taxotere (Sanofi-Aventis) | Disrupts microtubule de-polymerization Induces a G2/M arrest. Effect: anti-mitotic | FDA approved | Breast, prostate, gastric cancers, NSCLC, SCCHN | IV | Neurotoxicity, myelosuppression |
| Paclitaxel–Taxol [88] (Bristol-Myers Squibb Company) | Ovarian, breast cancers, AIDS-related Kaposi sarcoma, NSCLC | |||||
| Cell Cycle Inhibition | ||||||
| CDK4/6 inhibitor | Palbociclib-Ibrance [113] (Pfizer) | Targets CDK4/6 Induces G1 phase arrest. Effect: cytostatic and anti-proliferative | FDA approved (in combination with endocrine therapy, or as monotherapy (abemaciclib)) | ER+ HER2− breast cancer | Oral | Nausea, diarrhoea, fatigue, neutropenia [114] |
| Ribociclib–Kisqali [115] (Novartis) | ||||||
| Abemaciclib-Verzenio [116] (Eli Lilly) | Gastrointestinal toxicity [117] | |||||
| Pan-CDK inhibitor | Dinaciclib [99,118,119] (MK7965, SCH727965; Merck & Co.) | Targets CDK1, CDK2, CDK5, CDK9 Induces G1 and G2/M phase arrest, inhibits phosphorylation of RNA polymerase II and down-regulates MCL-1, and is synthetically lethal in MYC-amplified tumors. Effect: cytostatic and cytotoxic | Orphan drug designation | CLL | IV | Hematological and gastrointestinal toxicity [120] |
| Investigational compound in clinical trials: Phase III (n = 1) Phase II (n = 3) Phase I (n = 11) | Various advanced solid tumors and hematological malignancies | Toxicity due to lack of efficacy | ||||
| Flavopiridol–Alvocidib [118,121] (Sanofi-Aventis) | Targets CDK1, CDK2, CDK4, CDK6, CDK7, CDK9 Induces G1 and G2 phase arrest, inhibits phosphorylation of RNA polymerase II and down-regulates MCL-1. Effect: cytostatic, pro-apoptotic and inhibits transcription | Orphan drug designation | AML | IV | Tumor lysis syndrome, oral mucositis, gastrointestinal toxicity [122] | |
| Investigational compound in clinical trials: Phase II (n = 24) Phase I (n = 33) | Various advanced solid tumors and hematological malignancies | Considerable toxicity due to limited efficacy | ||||
| SNS032 [123] (Sunesis) | Targets CDK2, CDK7, CDK9 Inhibits phosphorylation of RNA polymerase II and down-regulates MCL-1 and XIAP. Effect: S phase arrest, pro-apoptotic and inhibits transcription | Investigational compound in clinical trials: Phase I (n = 2) | B-lymphoid malignancies [NCT00446342][124] | IV | Myelosuppression | |
| Various solid tumors [NCT00292864][125] | Fatigue, abdominal pain | |||||
| CYC065 [100,126] (Cyclacel Pharmaceuticals, Inc.) | Targets CDK2, CDK9 Inhibits phosphorylation of RNA polymerase II. Effect: S phase arrest, pro-apoptotic and inhibits transcription | Investigational compound in clinical trials: Phase I (n = 2) | CLL in combination with venetoclax [NCT03739554] Advanced solid tumors/lymphoma [NCT02552953] | IV | Not yet reported | |
| Cell Cycle Checkpoint Inhibition | ||||||
| WEE1 inhibitor [101,127] | Adavosertib (AZD1775, MK-1775; AstraZeneca) | Targets WEE1 Inhibiting WEE1 promotes G2 checkpoint escape, premature mitotic entry and DNA damage during S phase. Damaged DNA is not repaired prior to mitosis, encouraging mitotic catastrophe. Effect: pro-apoptotic | Orphan drug designation | Ovarian cancer | Oral | Not yet reported |
| Investigational compound in clinical trials: Phase II (n = 5) Phase I (n = 5) | Various advanced solid tumors and hematological malignancies | |||||
| ATR inhibitor [128,129] | AZD6738 [101](AstraZeneca) | Targets ATR activity ATR is a DNA damage response kinase, activated by DNA damage. ATR inhibition prevents activation of CHEK1 and the detection of DNA damage and replication stress at the G2/M checkpoint.Effect: pro-apoptotic | Investigational compound in clinical trials: Phase II (n = 10) Phase I (n = 8) | Various advanced solid tumors and hematological malignancies | Oral | Not yet reported |
| M6620/VX-970 (Vertex Pharmaceuticals Inc.) | Investigational compound in clinical trials: Phase II (n = 6) Phase I (n = 7) | Various advanced solid tumors | ||||
| PLK1 inhibitor | Volasertib [130,131,132] (BI 6727; Boehringer Ingekheim) | Cell cycle kinase inhibitor targeting PLK1, PLK2 and PLK3 Induces pro-metaphase arrest by interfering with mitosis (e.g., entry, spindle formation, chromosome separation and cytokinesis). Effect: anti-mitotic, pro-apoptotic and anti-proliferative | Orphan drug designation | AML | IV | Not yet reported |
| Investigational compound in clinical trials: Phase III (n = 1) Phase II (n = 4) Phase I (n = 11) | Various advanced solid tumors and hematological malignancies | Hematological toxicity | ||||
| Therapy | Drug Examples | Mechanism of Action | Drug Status | Clinical Use | Mode of Administration | Adverse Events |
|---|---|---|---|---|---|---|
| Heat Shock Protein Inhibition | ||||||
| αB-crystallin antagonist [169] | NCI-41356 | Targets the interaction between αB-crystallin and VEGF165 αB-crystallin repairs misfolded proteins (e.g., VEGF), and αB-crystallin antagonism can inhibit VEGF production in tumor cells. Effect: reduces tumor growth and invasiveness in vitro and in vivo, decreases angiogenesis | Investigational compound in pre-clinical studies | Triple negative breast cancer models | - | Not yet reported |
| p53 Inhibition | ||||||
| MDM2 inhibitor | MI series [201] e.g., MI-77301 (SAR405838; Sanofi-Aventis) | Targets MDM2 Inhibits the MDM2-p53 interaction and activates p53 transcriptional activity. Effect: anti-proliferative and induction of p53-mediated apoptosis | Investigational compound in clinical trials: Phase I (n = 2) | Solid tumors | Oral | Not yet reported |
| Nutlins [202] e.g., Idasanutlin (RG7388; Roche) | Investigational compound in clinical trials: Phase III (n = 1) Phase II (n = 1) Phase I (n = 9) | Various advanced solid tumors and hematological malignancies | ||||
| Signal Transductor Inhibition | ||||||
| Tyrosine kinase inhibitor [203,204] | Gefitinib–Iressa (AstraZeneca) | Targets EGFR at the intracellular domain Inhibits the kinase activity of EGFR to promote proliferation and survival. Effect: anti-proliferative and pro-apoptotic | FDA approved | NSCLC | Oral | Gastrointestinal toxicity |
| Second mitochondria-derived activator of caspase (Smac) mimetic | ||||||
| Smac mimetics/IAP antagonist [205] | LBW242 [184,206] (Novartis Oncology) | Targets IAPs Binds to and supresses IAPs (e.g., XIAP, c-IAP1 and c-IAP2), inducing cell death via caspase activation. Effect: pro-apoptotic | Investigational compound in pre-clinical studies | Breast, ovarian cancers, MM, AML, melanoma cell lines | Oral | Not yet reported |
| LCL161 [207] (Novartis Oncology) | Investigational compound in clinical trials: Phase II (n = 3) Phase I (n = 6) | Various solid tumors and hematological malignancies | Gastrointestinal toxicity | |||
| Pro-apoptotic Receptor Agonists | ||||||
| Apo2/TRAIL agonist [208,209,210] | Drozitumab (PRO95780; Genentech Inc.) | Targets DR5 Binding of TRAIL to DR5 initiates an apoptotic signaling cascade (extrinsic pathway) via assembly of the death induced signaling complex involving caspases-8 and 10. Effect: pro-apoptotic | Investigational compound in clinical trials: Phase II (n = 2) Phase I (n = 2) | NSCLC, non-Hodgkin’s lymphoma, colorectal cancer | IV | Hematological toxicity, diarrhoea, nausea |
| BH3-only Pro-survival Proteins (BH3 mimetics) | ||||||
| BCL-2 inhibitor [190] | ABT-737 [211] (Abbott Laboratories) | Targets BCL-2, BCL-XL and BCL-W, blocking the interaction with pro-apoptotic BCL-2 family members Effect: BAX/BAK-dependent apoptosis | Investigational compound in pre-clinical studies | SCLC, follicular lymphoma, CLL cell lines | - | Not yet reported |
| Navitoclax [212,213] (ABT-263; Abbott Laboratories) | Investigational compound in clinical trials: Phase II (n = 4) Phase 1 (n = 21) | Various advanced solid tumors and hematological malignancies | Oral | Thrombocytopenia, neutropenia | ||
| Venetoclax - Venclexta [214,215] (ABT-199; AbbVie Inc.) | Targets BCL-2, blocking the interaction with pro-apoptotic BCL-2 family members Effect: BAX/BAK-dependent apoptosis | FDA approved | CLL and SLL | Neutropenia, tumor lysis syndrome | ||
| BCL-XL inhibitor | WEHI-539 [216] | Targets BCL-XL, blocking the interaction with pro-apoptotic BCL-2 family members Effect: BAK-dependent apoptosis | Investigational compound in pre-clinical studies | Murine associated fibroblast cells | - | Not yet reported |
| MCL-1 inhibitor | S63845 [198] (Servier) | Targets MCL-1 and disrupts the binding of BAX/BAK to MCL-1 Effect: BAX/BAK-dependent apoptosis | Investigational compound in pre-clinical studies | MM, AML, lymphoma cell lines | - | Not yet reported |
| A-1210477 [195] (AbbVie Inc.) | Targets MCL-1 and disrupts the MCL-1/BIM protein complex Effect: BAX/BAK-dependent apoptosis | NSCLC, MM cell lines | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandrou, S.; George, S.M.; Ormandy, C.J.; Lim, E.; Oakes, S.R.; Caldon, C.E. The Proliferative and Apoptotic Landscape of Basal-like Breast Cancer. Int. J. Mol. Sci. 2019, 20, 667. https://doi.org/10.3390/ijms20030667
Alexandrou S, George SM, Ormandy CJ, Lim E, Oakes SR, Caldon CE. The Proliferative and Apoptotic Landscape of Basal-like Breast Cancer. International Journal of Molecular Sciences. 2019; 20(3):667. https://doi.org/10.3390/ijms20030667
Chicago/Turabian StyleAlexandrou, Sarah, Sandra Marie George, Christopher John Ormandy, Elgene Lim, Samantha Richelle Oakes, and C. Elizabeth Caldon. 2019. "The Proliferative and Apoptotic Landscape of Basal-like Breast Cancer" International Journal of Molecular Sciences 20, no. 3: 667. https://doi.org/10.3390/ijms20030667