Covalently Linking Oligomerization-Impaired GlpF Protomers Does Not Completely Re-establish Wild-Type Channel Activity


Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Cloning and Mutagenesis
3.2. GlpF Activity Measurements
3.3. Isolation of GlpF from Membranes and Western Blot Analysis
3.4. Purification and SDS-PAGE Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AQP | aquaporin |
| WT | wild-type |
| T | tetramer |
| NC | negative control |
| IPTG | isopropyl β-D-1-thiogalactopyranoside |
| M | molecular mass |
| TM | transmembrane |
| E. coli | Escherichia coli |
| GlpF | glycerol facilitator |
| D | Dimer |
| Tr | Trimer |
References
- Ishibashi, K. New members of mammalian aquaporins: AQP10-AQP12. Handb. Exp. Pharmacol. 2009, 251–262. [Google Scholar]
- Quigley, F.; Rosenberg, J.M.; Shachar-Hill, Y.; Bohnert, H.J. From genome to function: The Arabidopsis aquaporins. Genome Biol. 2002, 3, research0001.1–research0001.17. [Google Scholar]
- Wu, B.; Beitz, E. Aquaporins with selectivity for unconventional permeants. Cell. Mol. Life Sci. CMLS 2007, 64, 2413–2421. [Google Scholar] [CrossRef] [PubMed]
- Yasui, M.; Hazama, A.; Kwon, T.H.; Nielsen, S.; Guggino, W.B.; Agre, P. Rapid gating and anion permeability of an intracellular aquaporin. Nature 1999, 402, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Moller, A.L.; Kristiansen, K.A.; Schulz, A.; Moller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Cymer, F.; Schneider, D. Oligomerization of polytopic alpha-helical membrane proteins: Causes and consequences. Biol. Chem. 2012, 393, 1215–1230. [Google Scholar] [CrossRef] [PubMed]
- Lagree, V.; Froger, A.; Deschamps, S.; Pellerin, I.; Delamarche, C.; Bonnec, G.; Gouranton, J.; Thomas, D.; Hubert, J.F. Oligomerization state of water channels and glycerol facilitators. Involvement of loop E. J. Biol. Chem. 1998, 273, 33949–33953. [Google Scholar] [CrossRef]
- Smith, B.L.; Agre, P. Erythrocyte Mr 28,000 transmembrane protein exists as a multisubunit oligomer similar to channel proteins. J. Biol. Chem. 1991, 266, 6407–6415. [Google Scholar]
- Fu, D.; Libson, A.; Miercke, L.J.; Weitzman, C.; Nollert, P.; Krucinski, J.; Stroud, R.M. Structure of a glycerol-conducting channel and the basis for its selectivity. Science 2000, 290, 481–486. [Google Scholar] [CrossRef]
- Galka, J.J.; Baturin, S.J.; Manley, D.M.; Kehler, A.J.; O’Neil, J.D. Stability of the glycerol facilitator in detergent solutions. Biochemistry 2008, 47, 3513–3524. [Google Scholar] [CrossRef]
- Baturin, S.; Galka, J.J.; Piyadasa, H.; Gajjeraman, S.; O’Neil, J.D. The effects of a protein osmolyte on the stability of the integral membrane protein glycerol facilitator. Biochem. Cell Biol. 2014, 92, 564–575. [Google Scholar] [CrossRef]
- Klein, N.; Neumann, J.; O’Neil, J.D.; Schneider, D. Folding and stability of the aquaglyceroporin GlpF: Implications for human aqua(glycero)porin diseases. Biochim. Biophys. Acta 2015, 1848, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Uehlein, N.; Lovisolo, C.; Siefritz, F.; Kaldenhoff, R. The tobacco aquaporin NtAQP1 is a membrane CO2 pore with physiological functions. Nature 2003, 425, 734–737. [Google Scholar] [CrossRef]
- Herrera, M.; Garvin, J.L. Aquaporins as gas channels. Pflug. Arch. 2011, 462, 623. [Google Scholar] [CrossRef]
- Fang, X.; Yang, B.; Matthay, M.A.; Verkman, A.S. Evidence against aquaporin-1-dependent CO2 permeability in lung and kidney. J. Physiol. 2002, 542, 63–69. [Google Scholar] [CrossRef]
- Cymer, F.; Schneider, D. A single glutamate residue controls the oligomerization, function, and stability of the aquaglyceroporin GlpF. Biochemistry 2010, 49, 279–286. [Google Scholar] [CrossRef]
- Buck, T.M.; Wagner, J.; Grund, S.; Skach, W.R. A novel tripartite motif involved in aquaporin topogenesis, monomer folding and tetramerization. Nat. Struct. Mol. Biol. 2007, 14, 762–769. [Google Scholar] [CrossRef]
- Gratkowski, H.; Lear, J.D.; DeGrado, W.F. Polar side chains drive the association of model transmembrane peptides. Proc. Natl. Acad. Sci. USA 2001, 98, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.X.; Merianos, H.J.; Brunger, A.T.; Engelman, D.M. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. USA 2001, 98, 2250–2255. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Park, S.; Jimenez, R.H.; Rinehart, D.; Tamm, L.K. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. Proc. Natl. Acad. Sci. USA 2007, 129, 8320–8327. [Google Scholar] [CrossRef]
- Sal-Man, N.; Gerber, D.; Shai, Y. The composition rather than position of polar residues (QxxS) drives aspartate receptor transmembrane domain dimerization in vivo. Biochemistry 2004, 43, 2309–2313. [Google Scholar] [CrossRef]
- Nemeth-Cahalan, K.L.; Clemens, D.M.; Hall, J.E. Regulation of AQP0 water permeability is enhanced by cooperativity. J. Gen. Physiol. 2013, 141, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Nemeth-Cahalan, K.L.; Kalman, K.; Froger, A.; Hall, J.E. Zinc modulation of water permeability reveals that aquaporin 0 functions as a cooperative tetramer. J. Gen. Physiol 2007, 130, 457–464. [Google Scholar] [CrossRef]
- Reichow, S.L.; Gonen, T. Noncanonical binding of calmodulin to aquaporin-0: Implications for channel regulation. Structure 2008, 16, 1389–1398. [Google Scholar] [CrossRef]
- Otto, B.; Uehlein, N.; Sdorra, S.; Fischer, M.; Ayaz, M.; Belastegui-Macadam, X.; Heckwolf, M.; Lachnit, M.; Pede, N.; Priem, N.; et al. Aquaporin tetramer composition modifies the function of tobacco aquaporins. J. Biol. Chem. 2010, 285, 31253–31260. [Google Scholar] [CrossRef]
- Neumann, J.; Klein, N.; Otzen, D.E.; Schneider, D. Folding energetics and oligomerization of polytopic alpha-helical transmembrane proteins. Arch. Biochem. Biophys. 2014, 564, 281–296. [Google Scholar] [CrossRef]
- Tamas, M.J.; Karlgren, S.; Bill, R.M.; Hedfalk, K.; Allegri, L.; Ferreira, M.; Thevelein, J.M.; Rydstrom, J.; Mullins, J.G.; Hohmann, S. A short regulatory domain restricts glycerol transport through yeast Fps1p. J. Biol. Chem. 2003, 278, 6337–6345. [Google Scholar] [CrossRef]
- Trefz, M.; Keller, R.; Vogt, M.; Schneider, D. The GlpF residue Trp219 is part of an amino-acid cluster crucial for aquaglyceroporin oligomerization and function. Biochim. Biophys. Acta Biomembr. 2018, 1860, 887–894. [Google Scholar] [CrossRef]
- Finger, C.; Volkmer, T.; Prodohl, A.; Otzen, D.E.; Engelman, D.M.; Schneider, D. The stability of transmembrane helix interactions measured in a biological membrane. J. Mol. Biol. 2006, 358, 1221–1228. [Google Scholar] [CrossRef]
- Bearer, E.L. Overview of image analysis, image importing, and image processing using freeware. Curr. Protoc. Mol. Biol. 2003. [CrossRef]
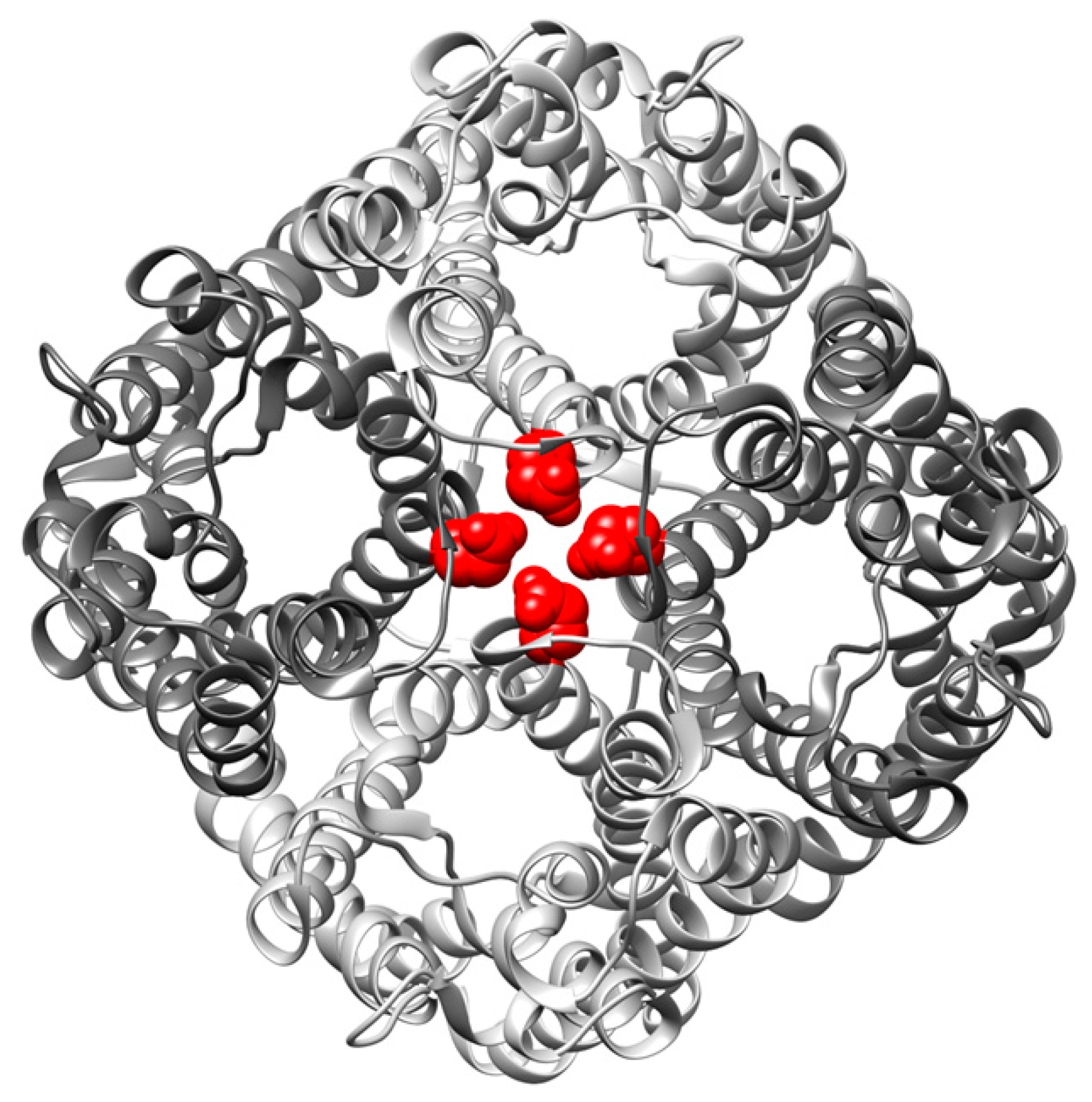
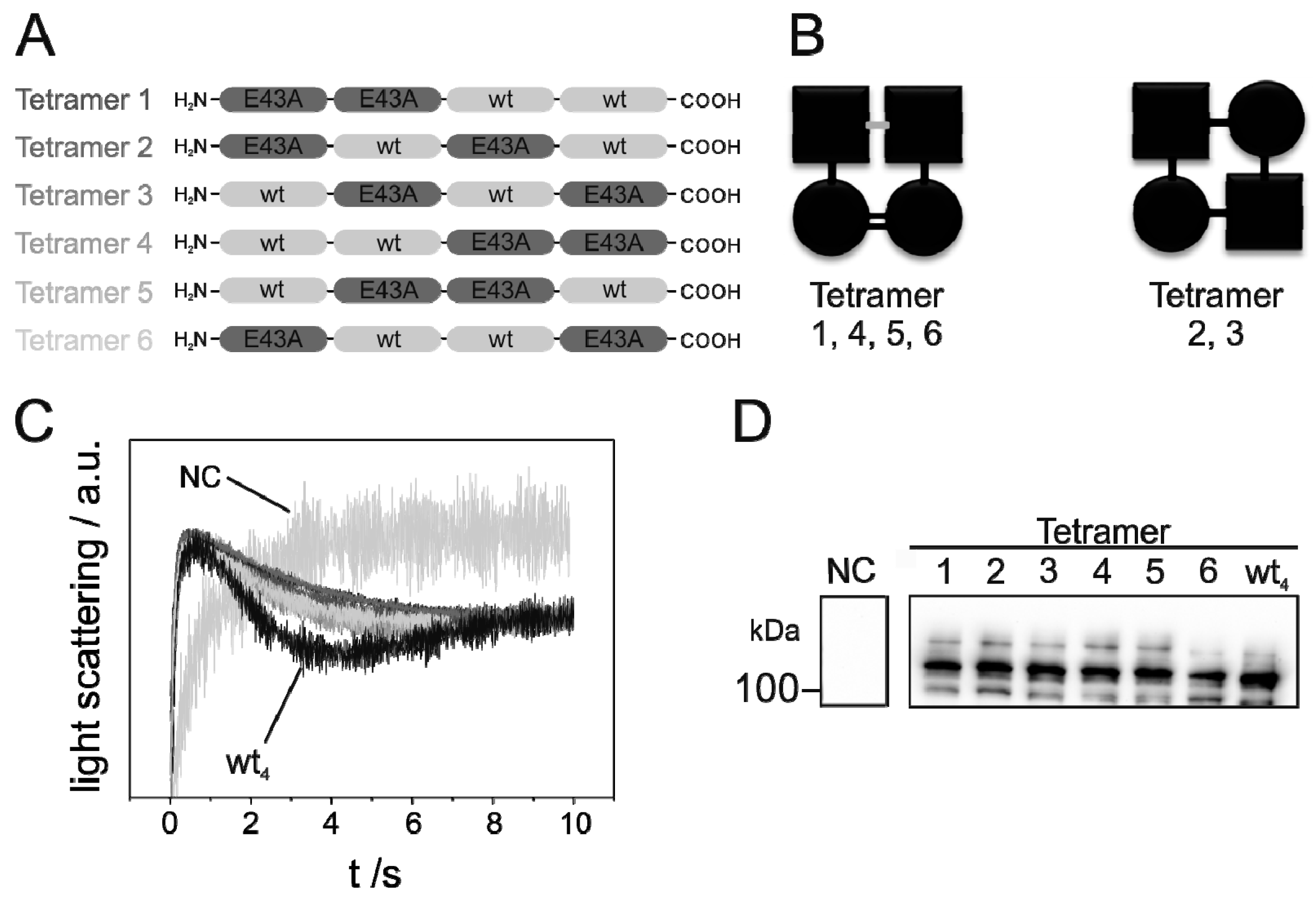
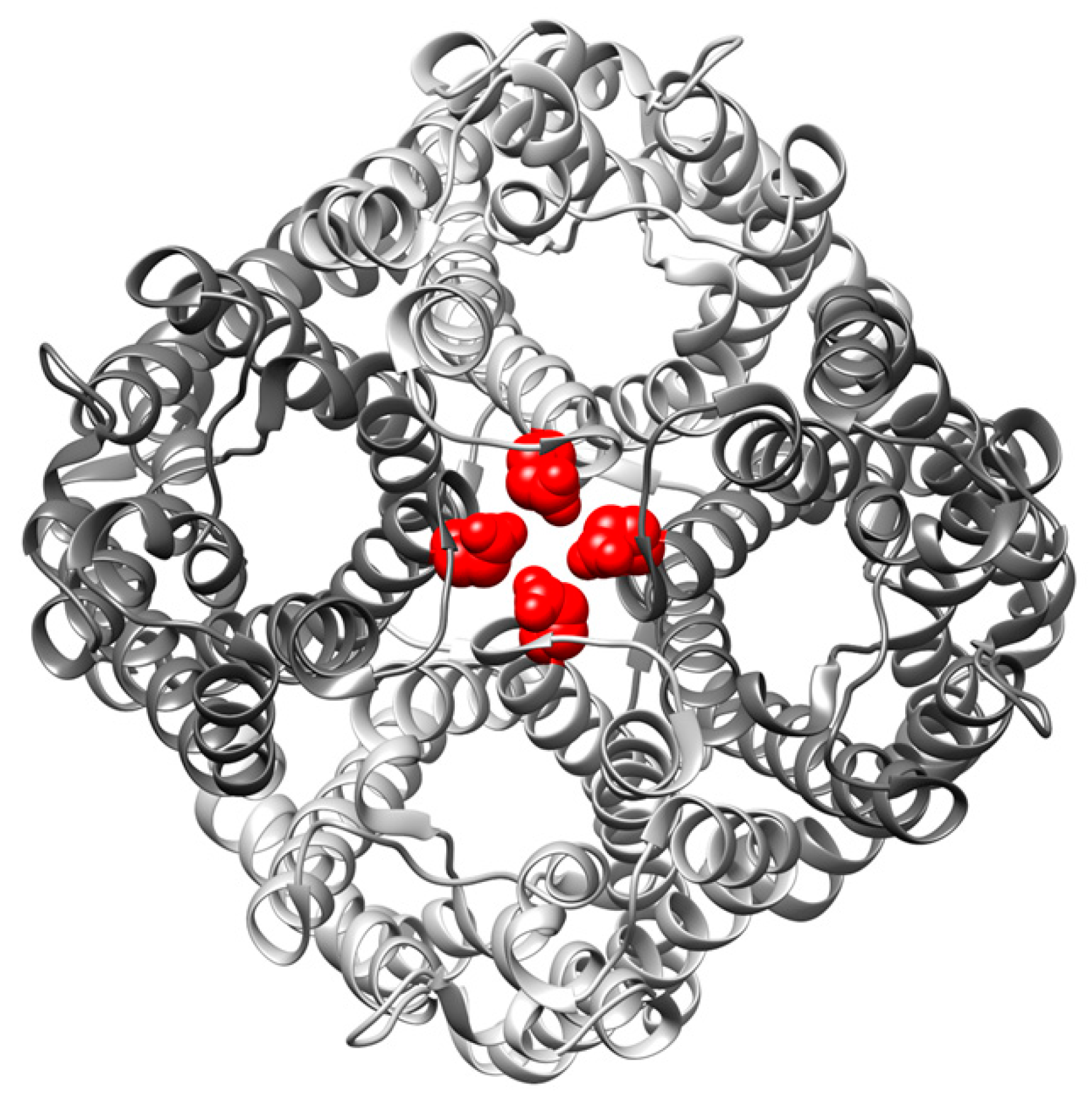
 ), were determined by approximation of the light-scattering curves using a single exponential decay function (n = 3 ± SD) (see also Figure S1). (D) Western blot analysis, determining the expression level of the studied GlpF homooligomer at increasing isopropyl β-D-1-thiogalactopyranoside (IPTG) concentrations. For the Western blot analysis, an antibody recognizing the GlpF C-terminus was used.
), were determined by approximation of the light-scattering curves using a single exponential decay function (n = 3 ± SD) (see also Figure S1). (D) Western blot analysis, determining the expression level of the studied GlpF homooligomer at increasing isopropyl β-D-1-thiogalactopyranoside (IPTG) concentrations. For the Western blot analysis, an antibody recognizing the GlpF C-terminus was used.
), were determined by approximation of the light-scattering curves using a single exponential decay function (n = 3 ± SD) (see also Figure S1). (D) Western blot analysis, determining the expression level of the studied GlpF homooligomer at increasing isopropyl β-D-1-thiogalactopyranoside (IPTG) concentrations. For the Western blot analysis, an antibody recognizing the GlpF C-terminus was used.
), were determined by approximation of the light-scattering curves using a single exponential decay function (n = 3 ± SD) (see also Figure S1). (D) Western blot analysis, determining the expression level of the studied GlpF homooligomer at increasing isopropyl β-D-1-thiogalactopyranoside (IPTG) concentrations. For the Western blot analysis, an antibody recognizing the GlpF C-terminus was used.
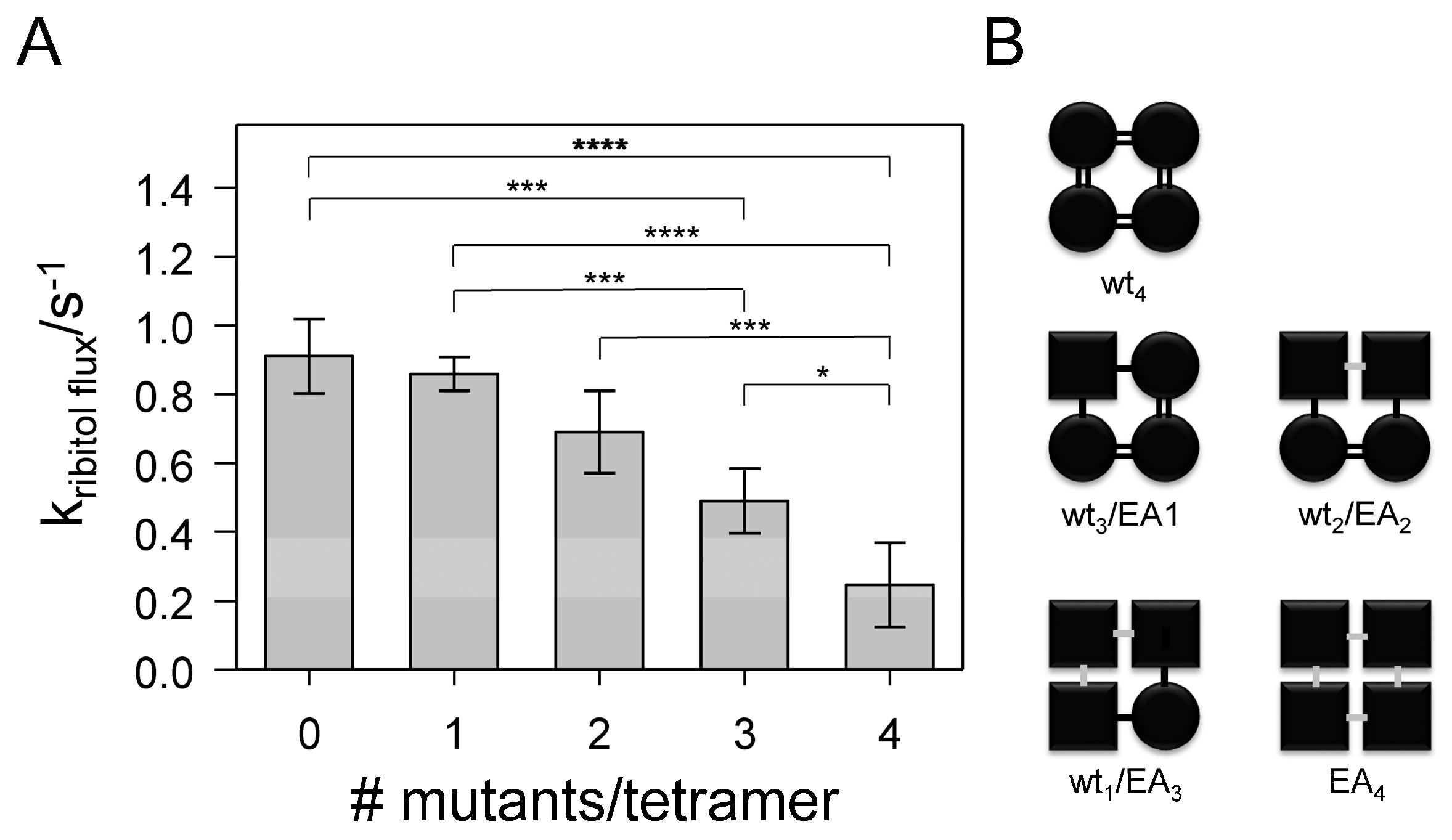
 ), WT2/EA2 (
), WT2/EA2 (  ), WT1/EA3 (
), WT1/EA3 (  ) and EA4 (◀) were determined by approximation of the light- scattering curves using a single exponential decay function (n = 3 ± SD). For comparison, the rate constants of the ribitol flux facilitated by the fused tetramer WT4 (
) and EA4 (◀) were determined by approximation of the light- scattering curves using a single exponential decay function (n = 3 ± SD). For comparison, the rate constants of the ribitol flux facilitated by the fused tetramer WT4 (  ) are also depicted. (E) Western blot analysis, determining the expression level of the various fused GlpF tetramers. For the Western blot analysis, an antibody recognizing the GlpF C-terminus was used.
), WT2/EA2 ( ), WT1/EA3 ( ) and EA4 (◀) were determined by approximation of the light- scattering curves using a single exponential decay function (n = 3 ± SD). For comparison, the rate constants of the ribitol flux facilitated by the fused tetramer WT4 ( ) are also depicted. (E) Western blot analysis, determining the expression level of the various fused GlpF tetramers. For the Western blot analysis, an antibody recognizing the GlpF C-terminus was used.
) are also depicted. (E) Western blot analysis, determining the expression level of the various fused GlpF tetramers. For the Western blot analysis, an antibody recognizing the GlpF C-terminus was used.
), WT2/EA2 ( ), WT1/EA3 ( ) and EA4 (◀) were determined by approximation of the light- scattering curves using a single exponential decay function (n = 3 ± SD). For comparison, the rate constants of the ribitol flux facilitated by the fused tetramer WT4 ( ) are also depicted. (E) Western blot analysis, determining the expression level of the various fused GlpF tetramers. For the Western blot analysis, an antibody recognizing the GlpF C-terminus was used.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Reference |
|---|---|
| pGlpF | [16] |
| pMalp2 | [16] |
| p4xGlpF | This study |
| p1xGlpF-E43A | This study |
| p3xGlpF-1xGlpF-E43A | This study |
| p2xGlpF-2xGlpF-E43A | This study |
| p1xGlpF-3xGlpF-E43A | This study |
| p4xGlpF-E43A | This study |
| p2xGlpF-2xGlpF-E43A tetramer 2 | This study |
| p2xGlpF-2xGlpF-E43A tetramer 3 | This study |
| p2xGlpF-2xGlpF-E43A tetramer 4 | This study |
| p2xGlpF-2xGlpF-E43A tetramer 5 | This study |
| p2xGlpF-2xGlpF-E43A tetramer 6 | This study |
| pRSET-His-GlpF | [16] |
| Primer | 5′-Sequence-3′ |
|---|---|
| GlpF NdeI XhoI for | GCGCGCCATATGGGCAGCGGCCTCGAGATGAGTCAAACATCAACC |
| GlpF SalI rev | GCGCGCGGATCCGTCGACCAGCGAAGCTTTTTG |
| QC GlpF Stop SalI for | CAAAAAGCTTCGCTGTAAGTCGACGGATCCGGC |
| QC GlpF Stop SalI rev | GCCGGATCCGTCGACTTACAGCGAAGCTTTTTG |
| QC GlpF-E43A for | CGTCTTTTGGTCAGTGGGCAATCAGTGTCATTTGGG |
| QC GlpF-E43A rev | CCCCAAATGACACTGATTGCCCACTGACAAAAGAC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, N.; Trefz, M.; Schneider, D. Covalently Linking Oligomerization-Impaired GlpF Protomers Does Not Completely Re-establish Wild-Type Channel Activity. Int. J. Mol. Sci. 2019, 20, 927. https://doi.org/10.3390/ijms20040927
Klein N, Trefz M, Schneider D. Covalently Linking Oligomerization-Impaired GlpF Protomers Does Not Completely Re-establish Wild-Type Channel Activity. International Journal of Molecular Sciences. 2019; 20(4):927. https://doi.org/10.3390/ijms20040927
Chicago/Turabian StyleKlein, Noreen, Margareta Trefz, and Dirk Schneider. 2019. "Covalently Linking Oligomerization-Impaired GlpF Protomers Does Not Completely Re-establish Wild-Type Channel Activity" International Journal of Molecular Sciences 20, no. 4: 927. https://doi.org/10.3390/ijms20040927