Folate Repletion after Deficiency Induces Irreversible Genomic and Transcriptional Changes in Human Papillomavirus Type 16 (HPV16)-Immortalized Human Keratinocytes

 ,
,
Abstract
:1. Introduction
2. Results
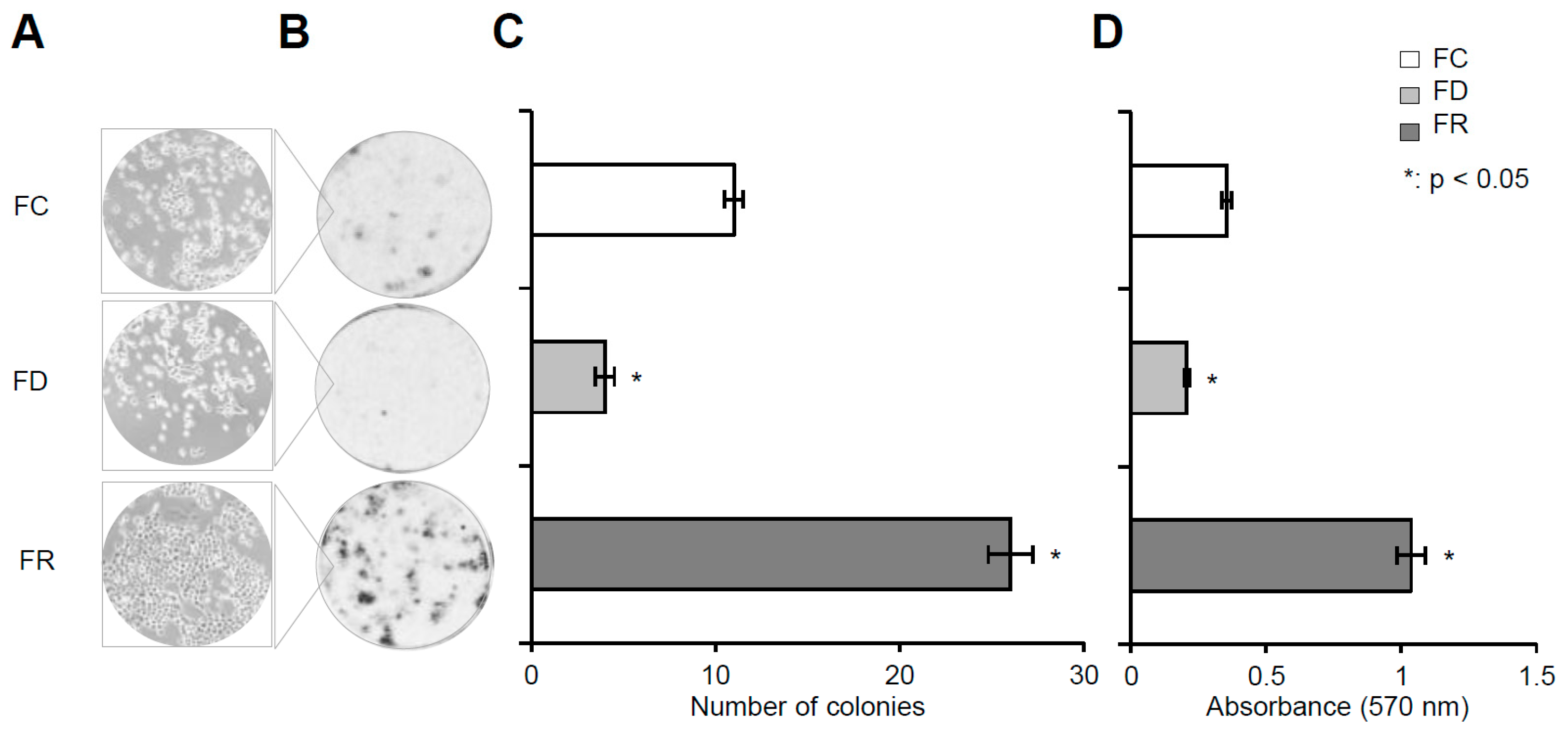
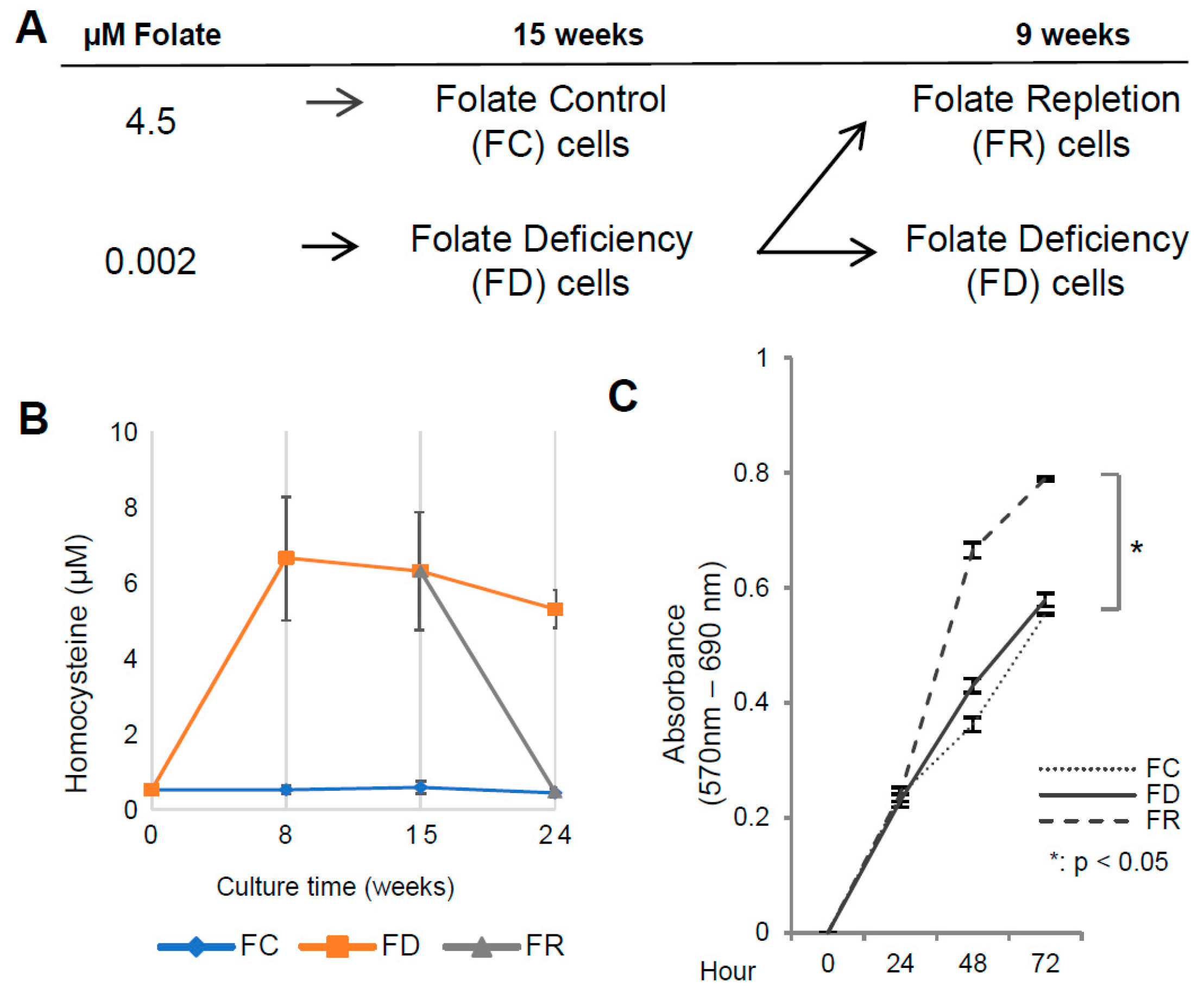
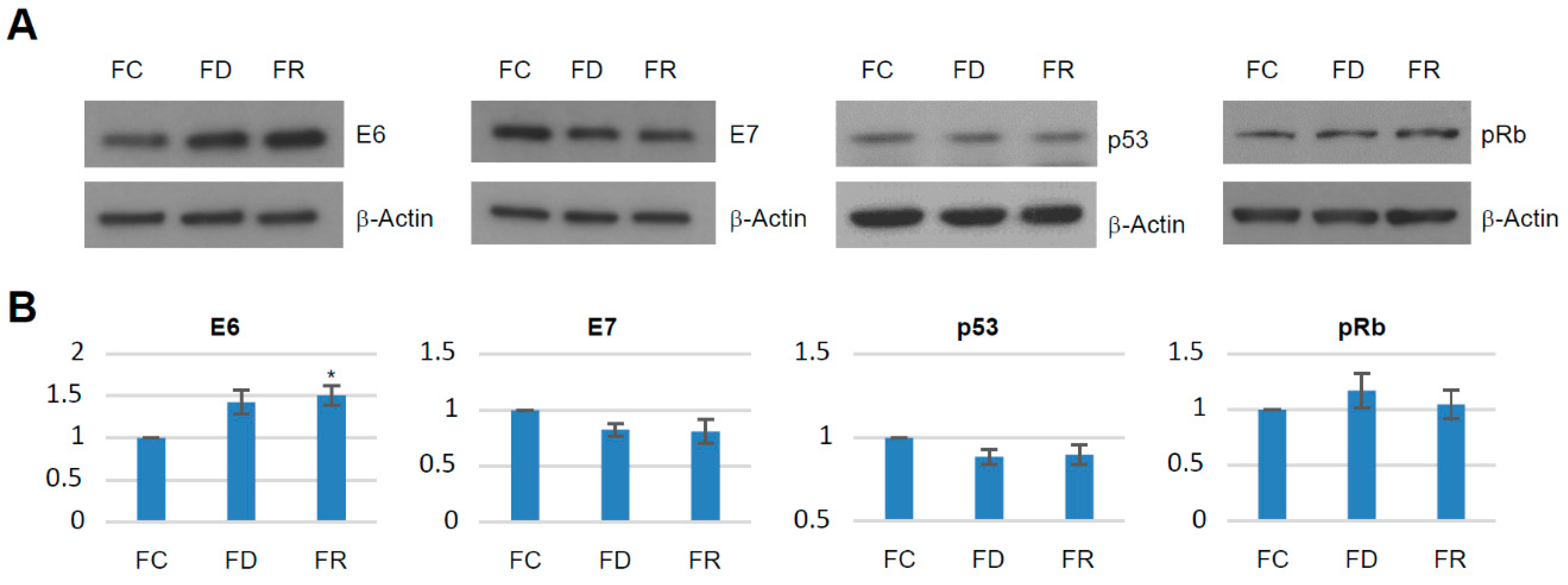
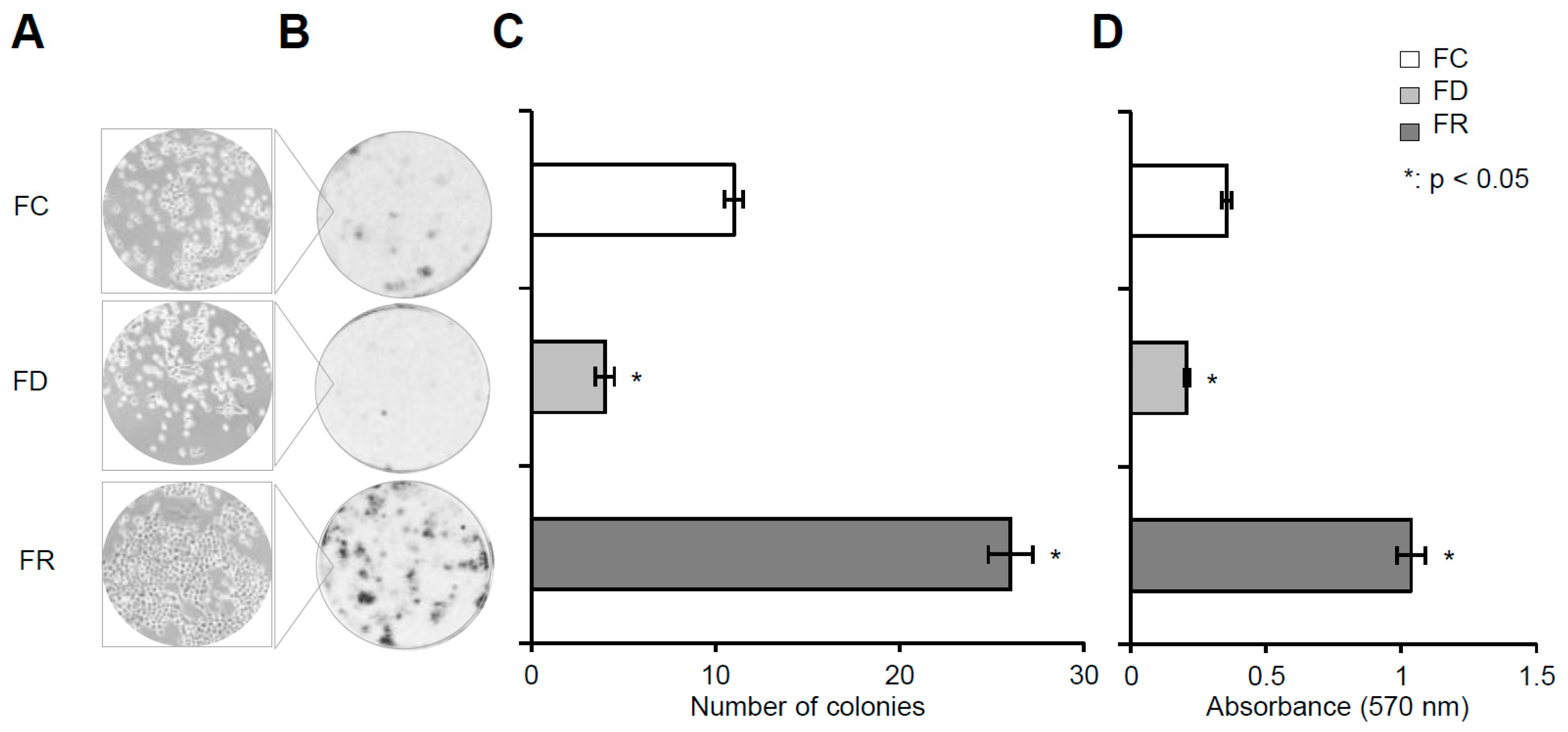
2.1. Folate Deficiency and Repletion Generate Phenotypes with Altered Metabolism and Proliferation
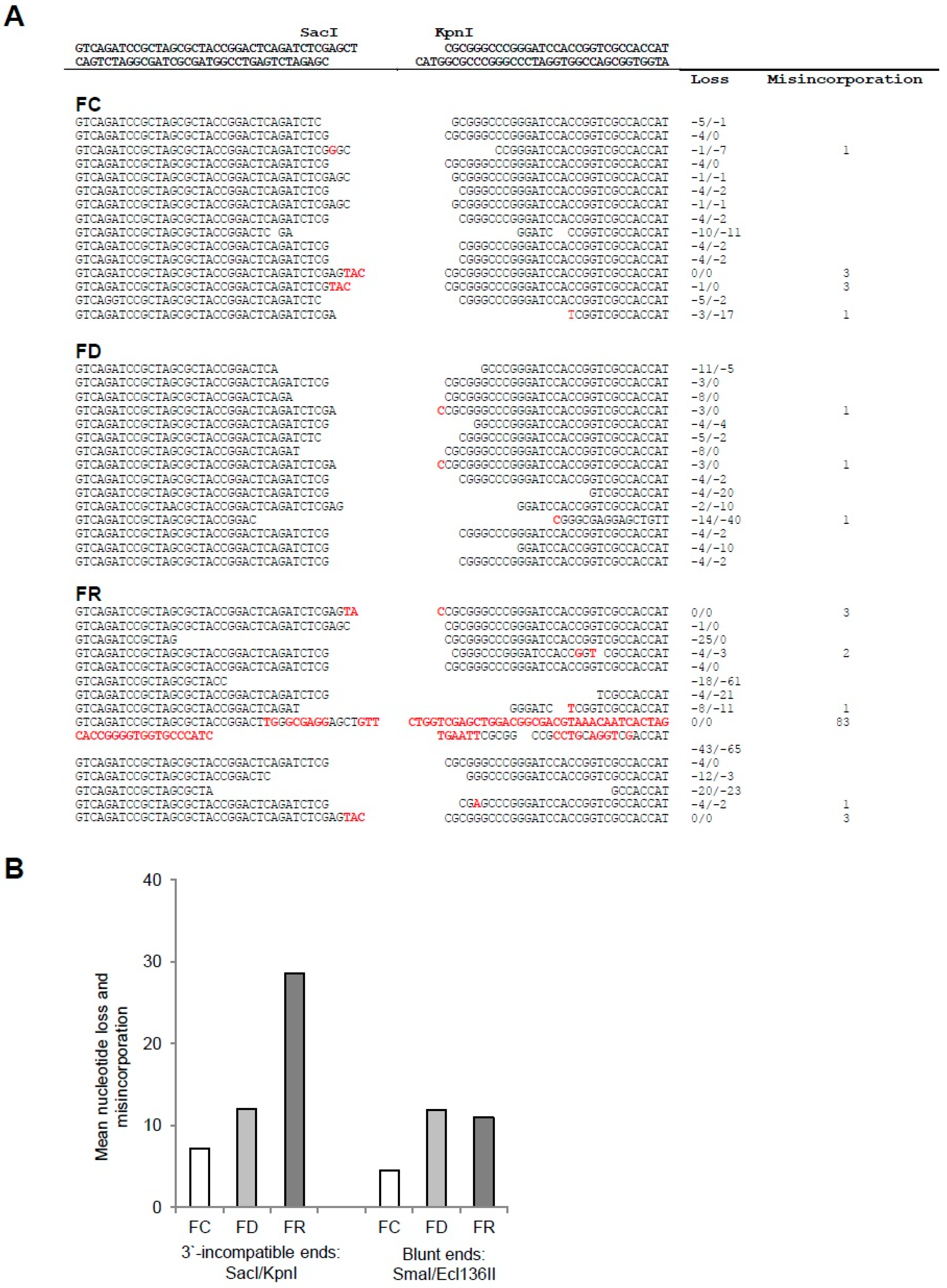
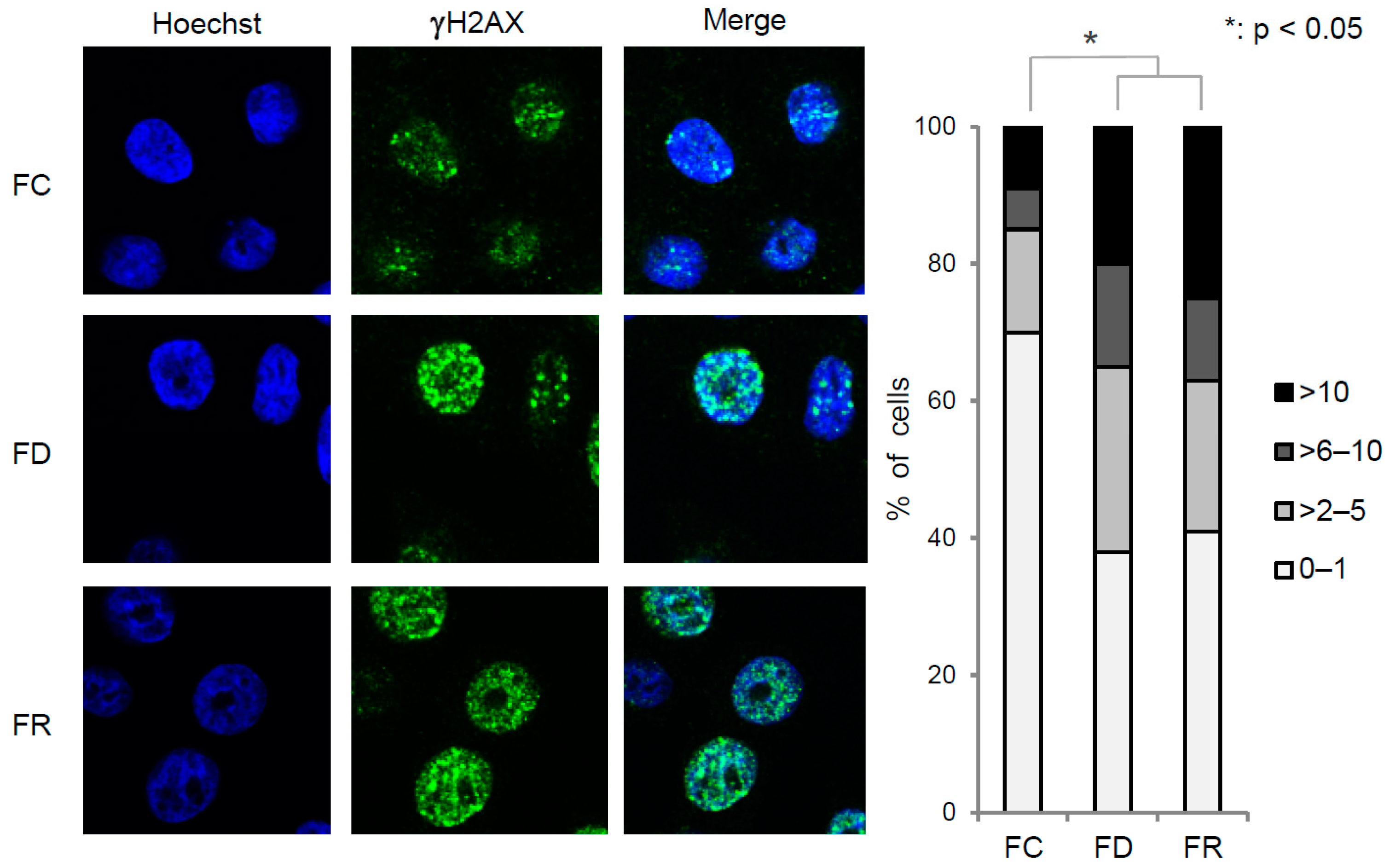
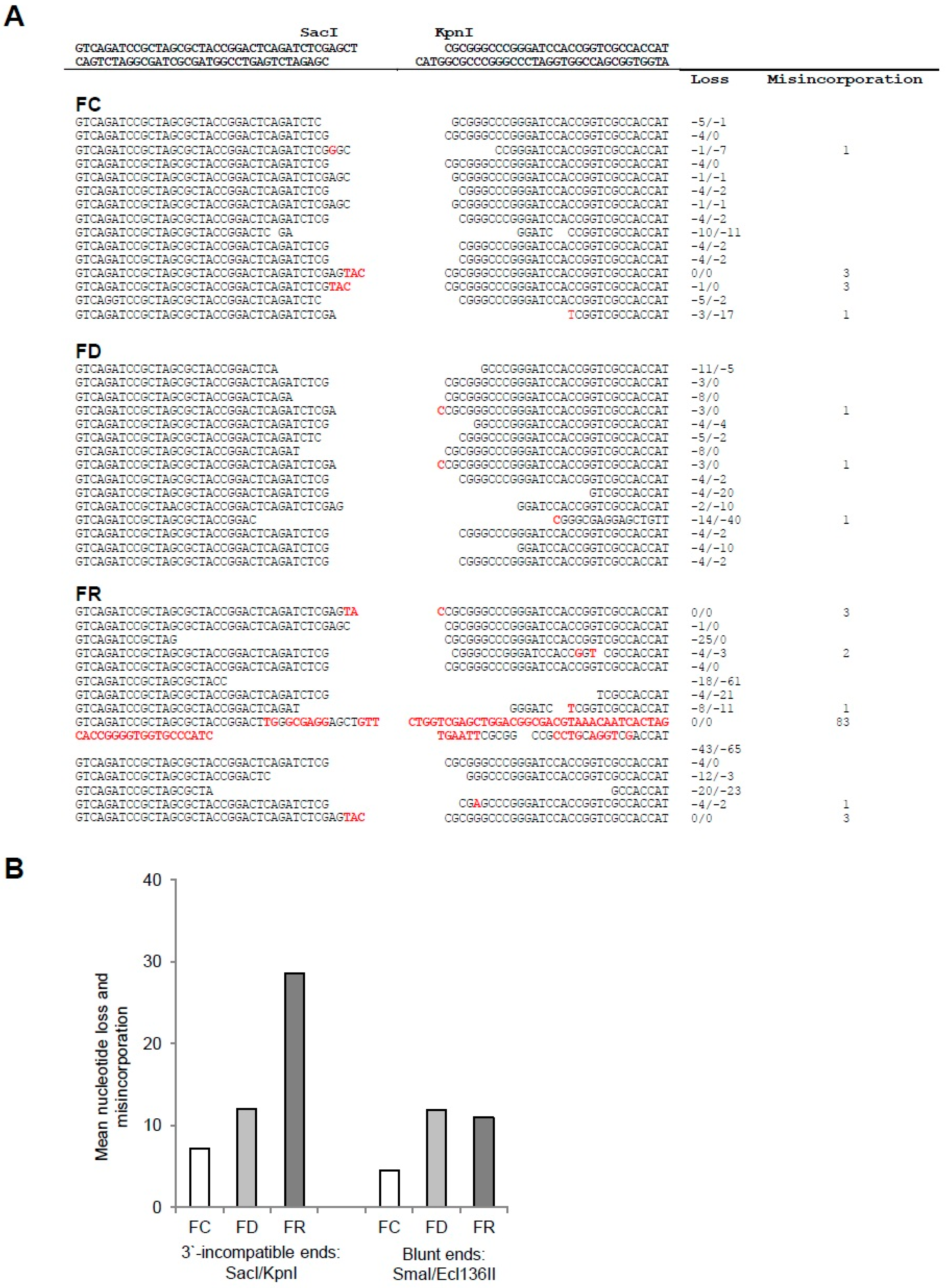
2.2. Folate Deficiency Results in Irreversible DNA Damage, Nucleotide Loss, and Misincorporation
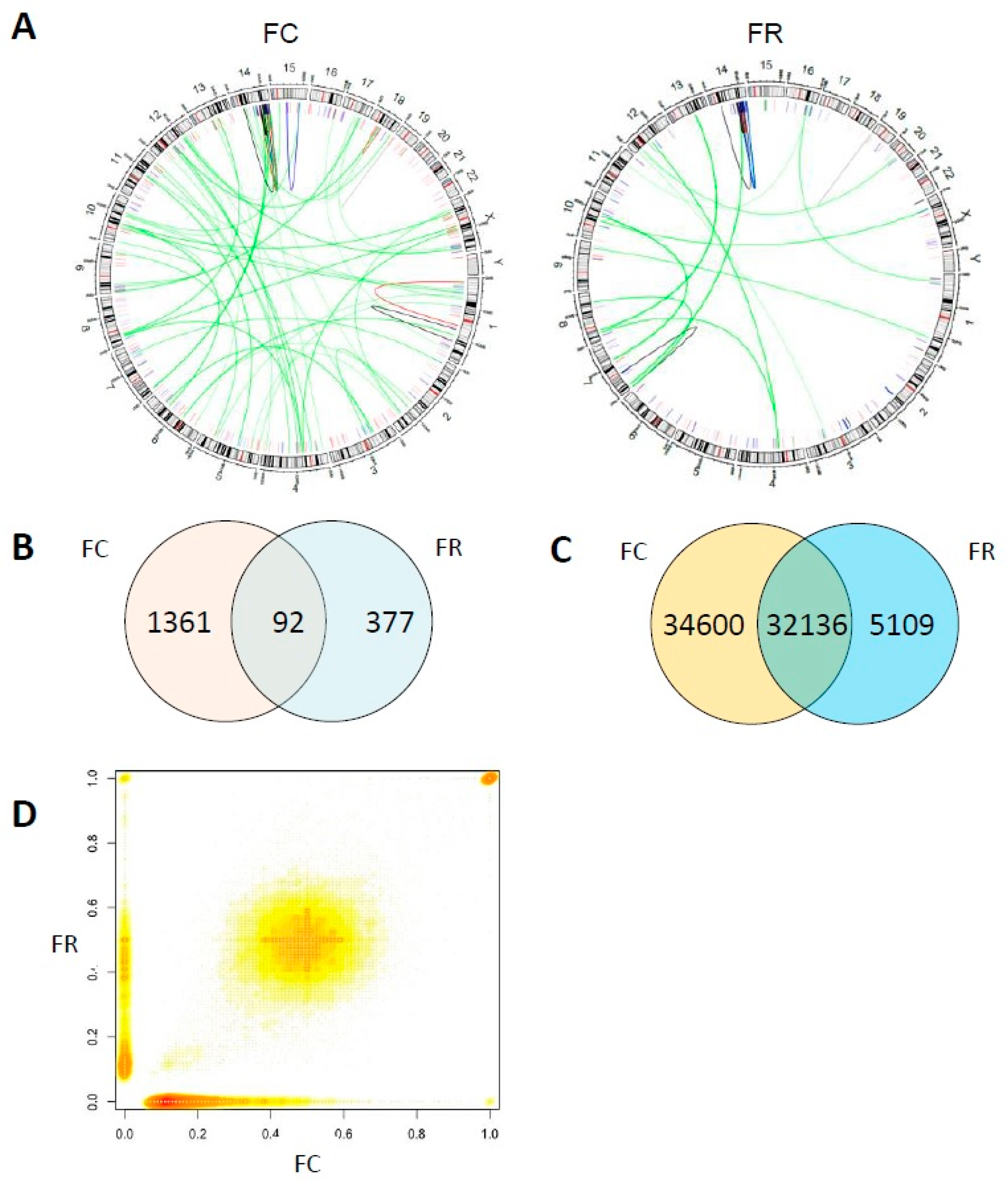
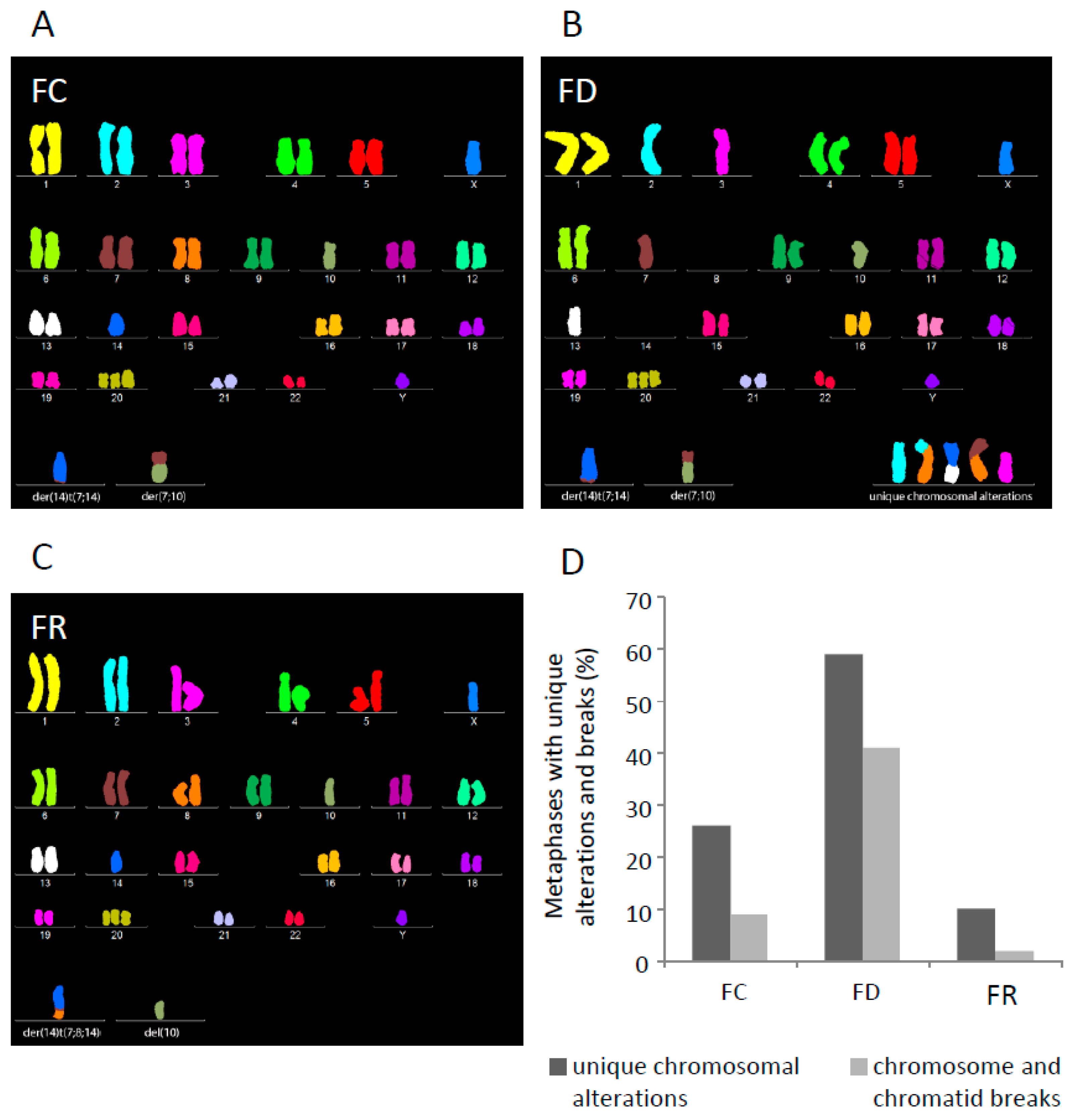
2.3. Folate Deficiency Cells Harbor Chromosomal Rearrangements and Folate Repletion Results in a Clonal Selection
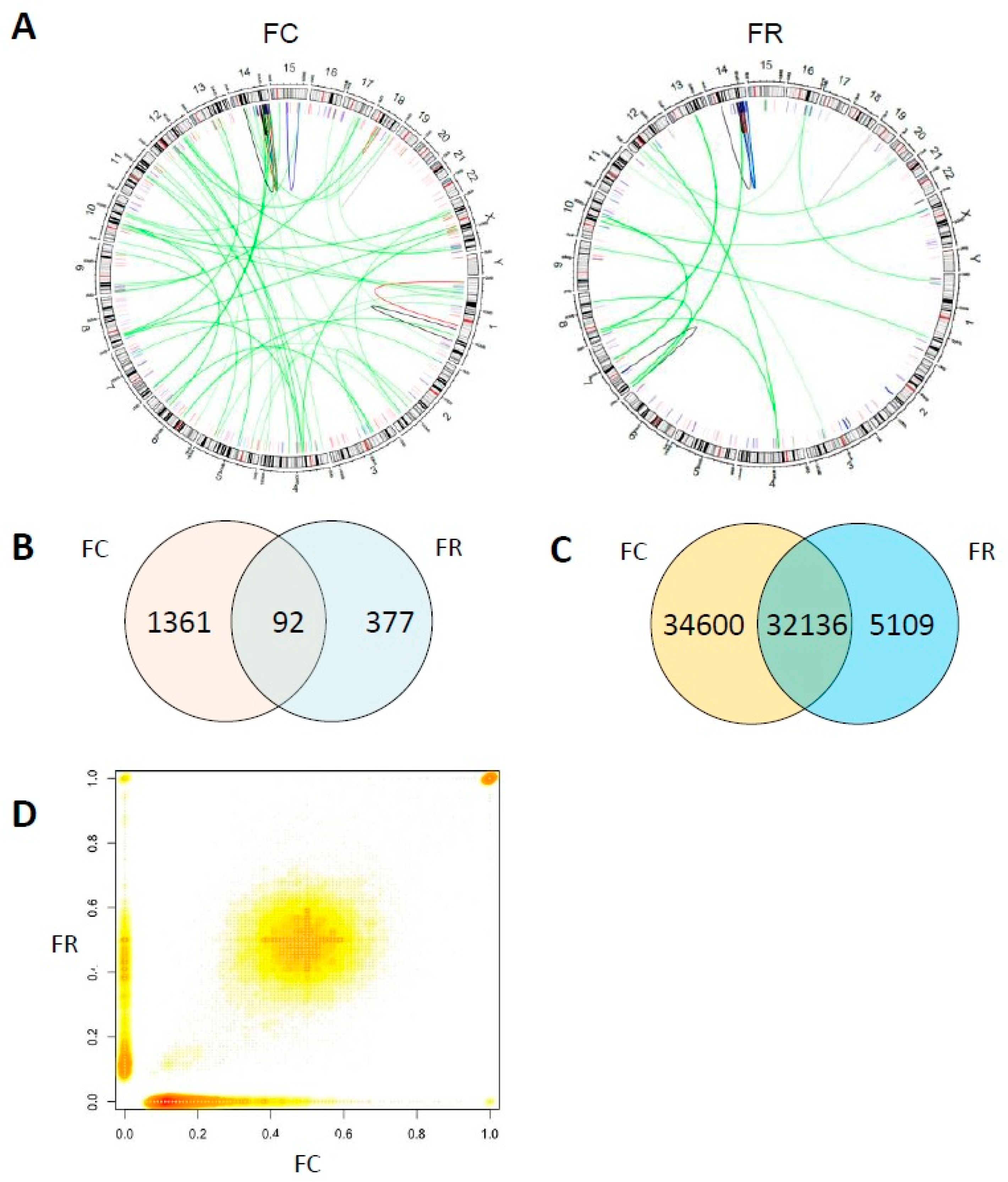
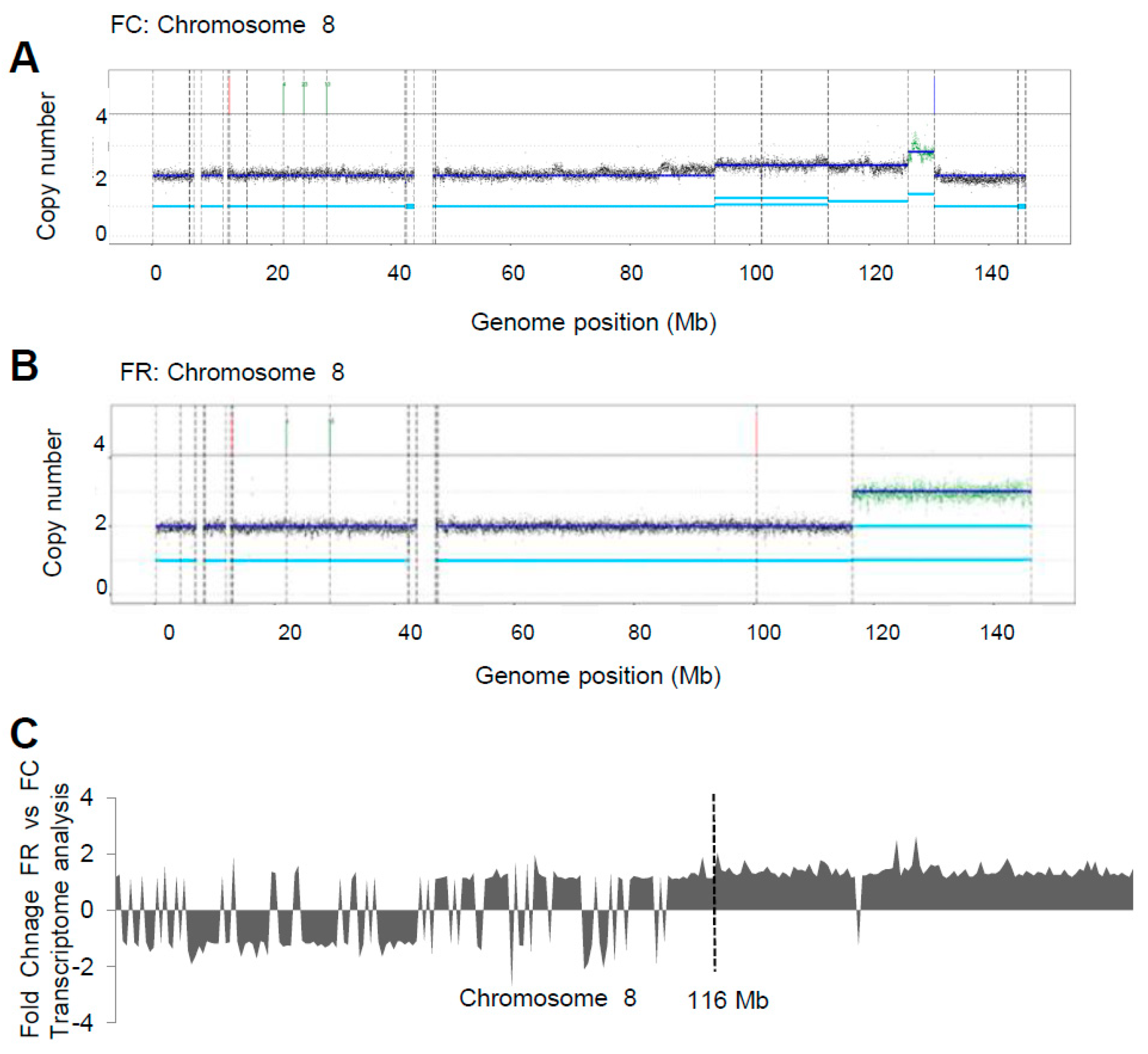
2.4. Whole Genome Sequencing Reveals Distinct Genomic Alterations in FC Cells
2.5. DNA Alterations in Folate Repleted Cells Can Disrupt Gene Structure
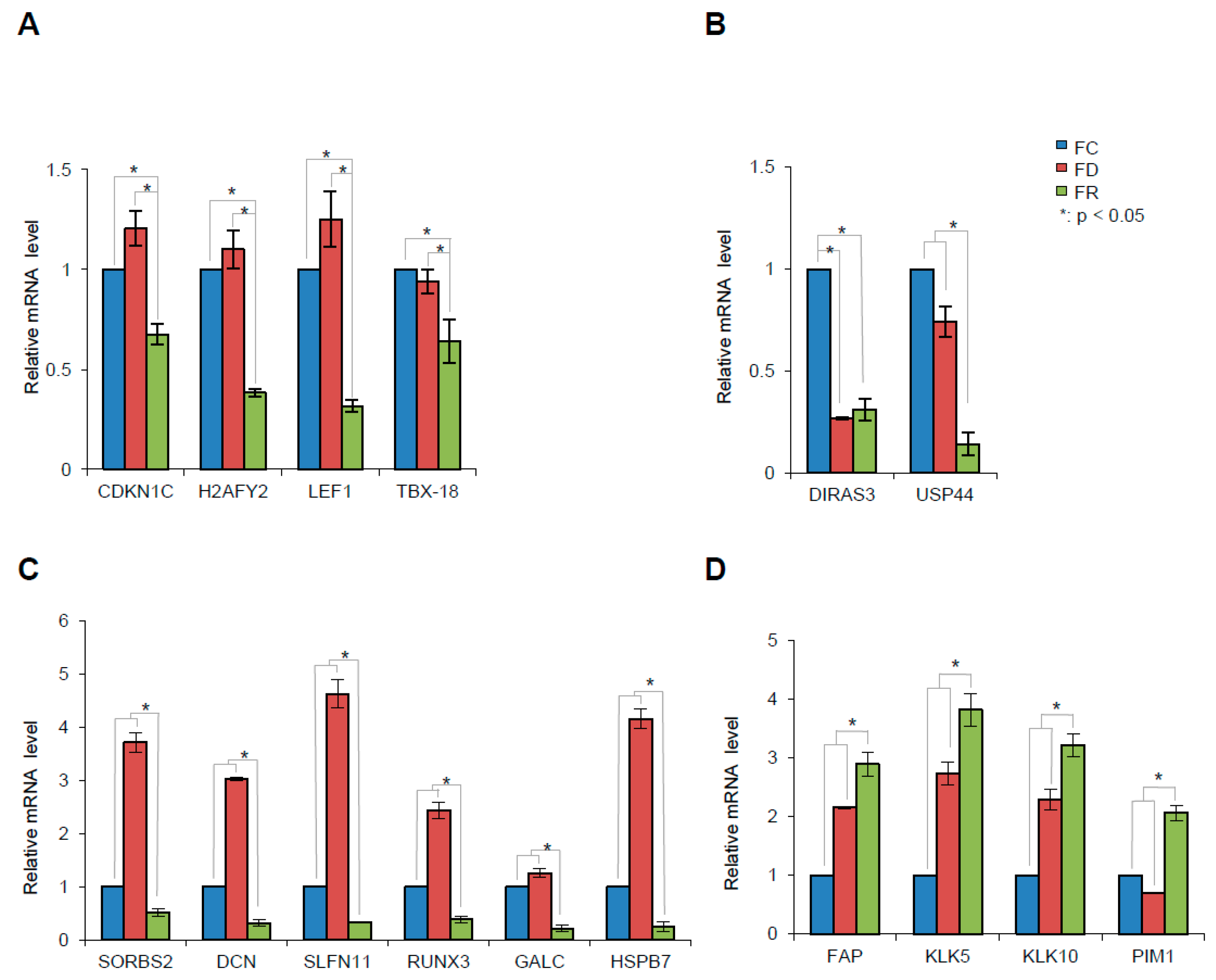
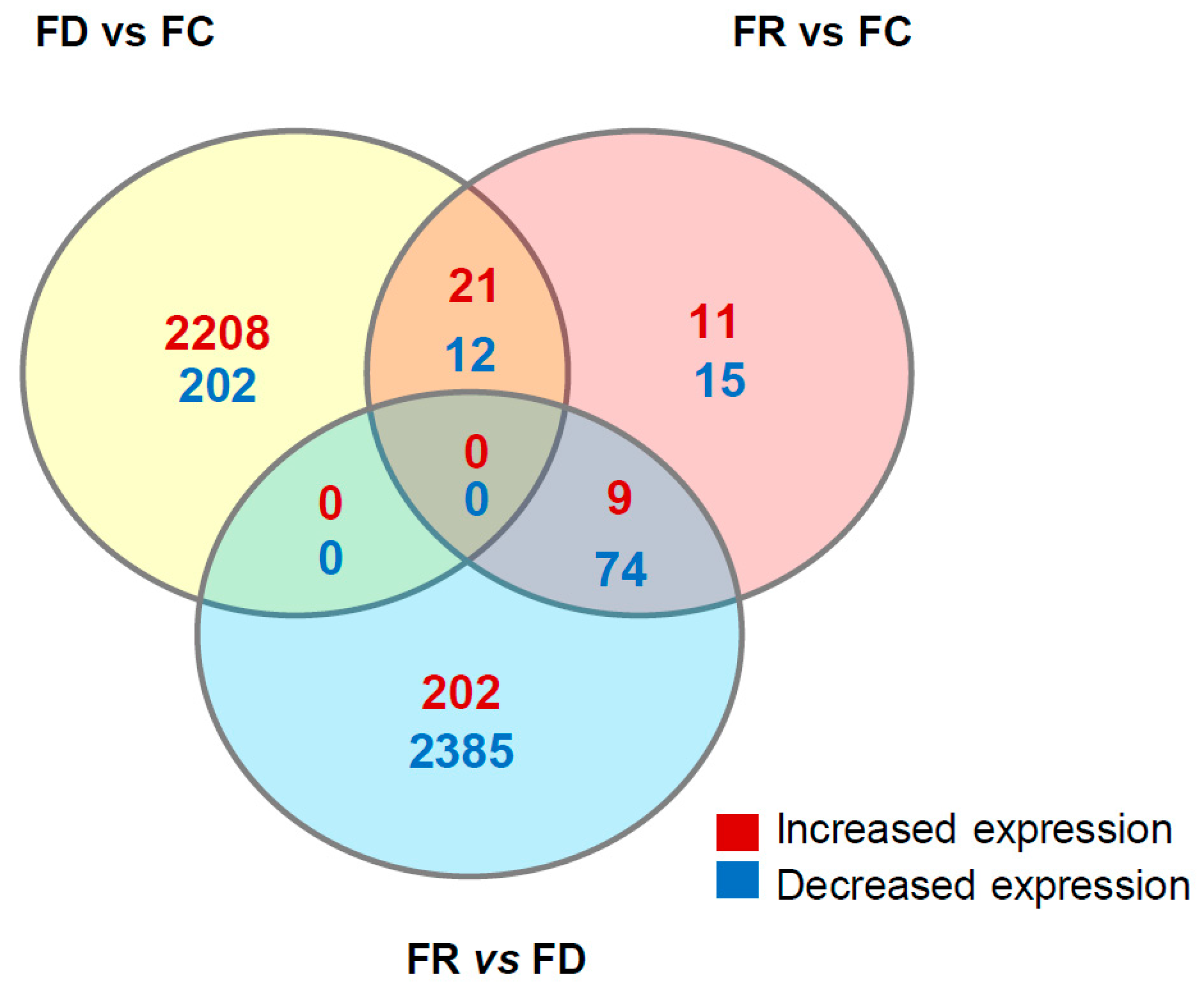
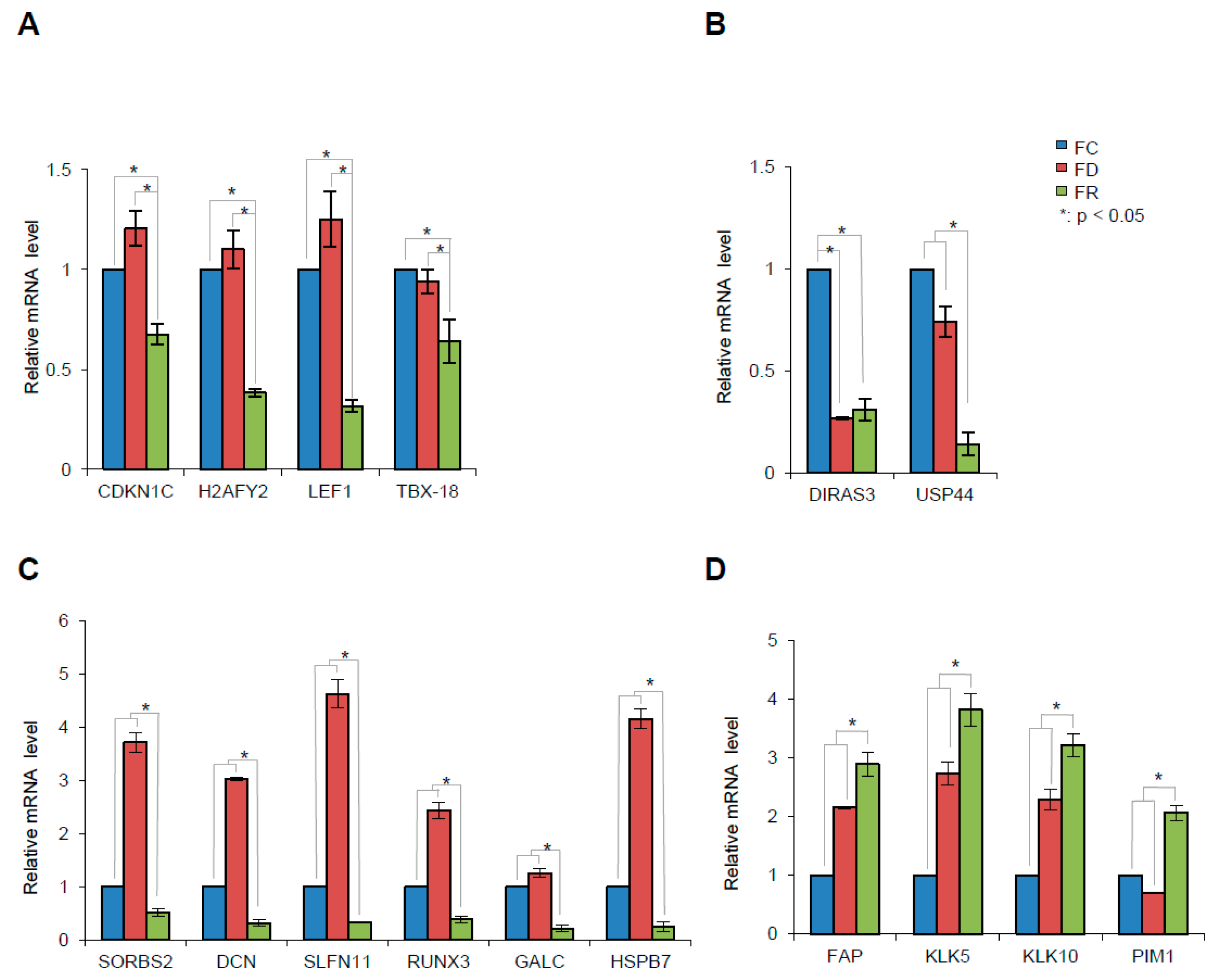
2.6. Folate Deficiency and Repletion Show Gene Expression Variations
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Homocysteine Level Determination
4.3. MTT Assay
4.4. Colony Formation Assay
4.5. Immunofluorescence
4.6. In Vivo DNA-Ligation Assay
4.7. Karyotype Analysis by Multicolour FISH (M-FISH)
4.8. Immunoblotting
4.9. Real-Time qPCR
4.10. Whole Genome Sequencing
4.11. Bioinformatic Analysis of Whole Genome Sequencing
4.12. RNA Sequencing
4.13. Bioinformatics Analysis of RNA Sequencing Data
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FC | Control cells grown in medium with standard folate content |
| FD | Cells adapted to growing in medium with low folate level |
| FR | FD cells reconstituted with standard folate medium |
| HPV | Human papillomavirus |
| M-FISH | Multiplex fluorescence in situ hybridization |
| HPLC | High-pressure liquid chromatography |
| NHEJ | Non-homologous end joining |
| WGS | Whole genome sequencing |
| SV | Structural variations |
| SNV | Single nucleotide variants |
| MAF | Minor allele frequency |
| FS | Fragile sites |
| MoMuLV | Moloney murine leukemia virus |
References
- Bailey, L.B.; Gregory, J.F., 3rd. Folate metabolism and requirements. J. Nutr. 1999, 129, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Mayne, S.T.; Playdon, M.C.; Rock, C.L. Diet, nutrition, and cancer: Past, present and future. Nat. Rev. Clin. Oncol. 2016, 13, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Campos, N.G.; Tsu, V.; Jeronimo, J.; Mvundura, M.; Kim, J.J. Estimating the value of point-of-care HPV testing in three low- and middle-income countries: A modeling study. BMC Cancer 2017, 17, 791. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hao, M.; Wang, Y.; Feng, N.; Wang, Z.; Wang, W.; Wang, J.; Ding, L. Association between folate status and cervical intraepithelial neoplasia. Eur. J. Clin. Nutr. 2016, 70, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.P.; Scarpini, C.G.; Groves, I.J.; Odle, R.I.; Coleman, N. Identification of host transcriptional networks showing concentration-dependent regulation by HPV16 E6 and E7 proteins in basal cervical squamous epithelial cells. Sci. Rep. 2016, 6, 29832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moazzen, S.; Dolatkhah, R.; Tabrizi, J.S.; Shaarbafi, J.; Alizadeh, B.Z.; de Bock, G.H.; Dastgiri, S. Folic acid intake and folate status and colorectal cancer risk: A systematic review and meta-analysis. Clin. Nutr. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Yu, S.; Zhang, S.; Ding, X.; Su, J.; Cheng, Z. Association between folate intake and risk of head and neck squamous cell carcinoma: An overall and dose-response PRISMA meta-analysis. Medicine (Baltimore) 2017, 96, e8182. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, H.; Hossain, M.B.; Lera, L.; Hirsch, S.; Albala, C.; Uauy, R.; Broberg, K.; Ronco, A.M. High levels of circulating folate concentrations are associated with DNA methylation of tumor suppressor and repair genes p16, MLH1, and MGMT in elderly Chileans. Clin. Epigenet. 2017, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, C.D.; Ragusa, D.; Giammanco, M.; Tosi, S. Folate deficiency as predisposing factor for childhood leukaemia: A review of the literature. Genes Nutr. 2017, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Ebbing, M.; Bonaa, K.H.; Nygard, O.; Arnesen, E.; Ueland, P.M.; Nordrehaug, J.E.; Rasmussen, K.; Njolstad, I.; Refsum, H.; Nilsen, D.W.; et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. Jama 2009, 302, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Weisbeck, A.; Jansen, R.J. Nutrients and the pancreas: An epigenetic perspective. Nutrients 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Wien, T.N.; Pike, E.; Wisloff, T.; Staff, A.; Smeland, S.; Klemp, M. Cancer risk with folic acid supplements: A systematic review and meta-analysis. BMJ Open 2012, 2, e000653. [Google Scholar] [CrossRef] [PubMed]
- Liew, S.C.; Gupta, E.D. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: Epidemiology, metabolism and the associated diseases. Eur. J. Med. Genet. 2015, 58, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Protiva, P.; Mason, J.B.; Liu, Z.; Hopkins, M.E.; Nelson, C.; Marshall, J.R.; Lambrecht, R.W.; Pendyala, S.; Kopelovich, L.; Kim, M.; et al. Altered folate availability modifies the molecular environment of the human colorectum: Implications for colorectal carcinogenesis. Cancer Prev. Res. 2011, 4, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Lemarroy, A.; Gariglio, P.; Whitaker, N.J.; Eichhorst, S.T.; zur Hausen, H.; Krammer, P.H.; Rösl, F. Restoration of p53 expression sensitizes human papillomavirus type 16 immortalized human keratinocytes to CD95-mediated apoptosis. Oncogene 2002, 21, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, M.R.; Chacko, P.; Kesari, L.A.; Jayaprakash, P.G.; Jayaram, H.N.; Antony, A.C. Expression of folate receptors and heterogeneous nuclear ribonucleoprotein E1 in women with human papillomavirus mediated transformation of cervical tissue to cancer. J. Clin. Pathol. 2003, 56, 569–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwaśniewska, A.; Tukendorf, A.; Goździcka-Józefiak, A.; Semczuk-Sikora, A.; Korobowicz, E. Content of folic acid and free homocysteine in blood serum of human papillomavirus-infected women with cervical dysplasia. Eur. J. Gynaecol. Oncol. 2002, 23, 311–316. [Google Scholar] [PubMed]
- Antony, A.; Tang, Y.S.; Khan, R.A.; Biju, M.P.; Xiao, X.; Li, Q.J.; Sun, X.L.; Jayaram, H.N.; Stabler, S.P. Translational upregulation of folate receptors is mediated by homocysteine via RNA-heterogeneous nuclear ribonucleoprotein E1 interactions. J. Clin. Invest. 2004, 113, 285–301. [Google Scholar] [CrossRef] [Green Version]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends. Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Rotem, A.; Janzer, A.; Izar, B.; Ji, Z.; Doench, J.G.; Garraway, L.A.; Struhl, K. Alternative to the soft-agar assay that permits high-throughput drug and genetic screens for cellular transformation. Proc. Natl. Acad. Sci. USA 2015, 112, 5708–5713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafehi, H.; Orlowski, C.; Georgiadis, G.T.; Ververis, K.; El-Osta, A.; Karagiannis, T.C. Clonogenic assay: Adherent cells. J. Vis. Exp. JoVE 2011. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Maoz, K.; Bester, A.C.; Im, M.M.; Shewach, D.S.; Karni, R.; Kerem, B. Folate levels modulate oncogene-induced replication stress and tumorigenicity. EMBO Mol. Med. 2015, 7, 1138–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Pastwa, E.; Somiari, R.I.; Malinowski, M.; Somiari, S.B.; Winters, T.A. In vitro non-homologous DNA end joining assays--the 20th anniversary. Int. J. Biochem. Cell Biol. 2009, 41, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, R.N.; Chudoba, I. The use of M-FISH and M-BAND to define chromosome abnormalities. Methods Mol. Biol. 2011, 730, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Kleinheinz, K.; Bludau, I.; Huebschmann, D.; Heinold, M.; Kensche, P.; Gu, Z.; Lopez, C.; Hummel, M.; Klapper, W.; Moeller, P.; et al. ACEseq—Allele specific copy number estimation from whole genome sequencing. bioRxiv 2017. [Google Scholar] [CrossRef]
- Sutherland, G.R. Heritable fragile sites on human chromosomes I. Factors affecting expression in lymphocyte culture. Am. J. Hum. Genet. 1979, 31, 125–135. [Google Scholar] [PubMed]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, Y.; Liu, Y.; Yu, S.; Xie, H.; Shi, X.; Qin, S.; Ma, F.; Tan, T.Z.; Thiery, J.P.; et al. A central role for TRPS1 in the control of cell cycle and cancer development. Oncotarget 2014, 5, 7677–7690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, M. Nothing in cancer makes sense except. BMC Biol. 2018, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Tang, Y.S.; Khan, R.A.; Zhang, Y.; Kusumanchi, P.; Stabler, S.P.; Jayaram, H.N.; Antony, A.C. Influence of physiologic folate deficiency on human papillomavirus type 16 (HPV16)-harboring human keratinocytes in vitro and in vivo. J. Biol. Chem. 2012, 287, 12559–12577. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Coleman, N. Human papillomavirus genome integration in squamous carcinogenesis: What have next-generation sequencing studies taught us? J. Pathol. 2018, 245, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Meng, Y. Association between serum folate level and cervical cancer: A meta-analysis. Arch. Gynecol. Obstet. 2016, 293, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Tang, Y.S.; Kusumanchi, P.; Stabler, S.P.; Zhang, Y.; Antony, A.C. Folate deficiency facilitates genomic integration of human papillomavirus type 16 dna in vivo in a novel mouse model for rapid oncogenic transformation of human keratinocytes. J. Nutr. 2018, 148, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Duthie, S.J.; Narayanan, S.; Blum, S.; Pirie, L.; Brand, G.M. Folate deficiency in vitro induces uracil misincorporation and DNA hypomethylation and inhibits DNA excision repair in immortalized normal human colon epithelial cells. Nutr. Cancer 2000, 37, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Ivashkevich, A.; Redon, C.E.; Nakamura, A.J.; Martin, R.F.; Martin, O.A. Use of the gamma-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Betermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.; Savini, C.; Krufczik, M.; Cremer, C.; Roesl, F.; Hausmann, M. Super-resolution localization microscopy of γ-H2AX and heterochromatin after folate deficiency. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.M.; Cimini, D. Cancer karyotypes: Survival of the fittest. Front. Oncol. 2013, 3, 148. [Google Scholar] [CrossRef] [PubMed]
- Duensing, A.; Chin, A.; Wang, L.; Kuan, S.F.; Duensing, S. Analysis of centrosome overduplication in correlation to cell division errors in high-risk human papillomavirus (HPV)-associated anal neoplasms. Virology 2008, 372, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schutze, D.M.; Krijgsman, O.; Snijders, P.J.; Ylstra, B.; Weischenfeldt, J.; Mardin, B.R.; Stutz, A.M.; Korbel, J.O.; de Winter, J.P.; Meijer, C.J.; et al. Immortalization capacity of HPV types is inversely related to chromosomal instability. Oncotarget 2016, 7, 37608–37621. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Ni, J.; Zhu, Y.; Zhou, T.; Ma, X.; Xue, J.; Wang, X. Folate deficiency induces mitotic aberrations and chromosomal instability by compromising the spindle assembly checkpoint in cultured human colon cells. Mutagenesis 2017, 32, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Bofin, A.M.; Ytterhus, B.; Fjosne, H.E.; Hagmar, B.M. Abnormal chromosome 8 copy number in cytological smears from breast carcinomas detected by means of fluorescence in situ hybridization (FISH). Cytopathol. Off. J. Br. Soc. Clin. Cytol. 2003, 14, 5–11. [Google Scholar] [CrossRef]
- Mark, H.F.; Afify, A.M.; Werness, B.A.; Das, S.; Mark, S.; Samy, M. Trisomy 8 in stage I and stage III ovarian cancer detected by fluorescence in situ hybridization. Exp. Mol. Pathol. 1999, 66, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Maserati, E.; Aprili, F.; Vinante, F.; Locatelli, F.; Amendola, G.; Zatterale, A.; Milone, G.; Minelli, A.; Bernardi, F.; Lo Curto, F.; et al. Trisomy 8 in myelodysplasia and acute leukemia is constitutional in 15–20% of cases. Genes Chromosom. Cancer 2002, 33, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Policht, F.A.; Song, M.; Sitailo, S.; O’Hare, A.; Ashfaq, R.; Muller, C.Y.; Morrison, L.E.; King, W.; Sokolova, I.A. Analysis of genetic copy number changes in cervical disease progression. BMC Cancer 2010, 10, 432. [Google Scholar] [CrossRef] [PubMed]
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, F.; Fisher, D.; Fort, P.; Marie, J.P.; Daoust, S.; Roche, B.; Grunau, C.; Cosseau, C.; Mitta, G.; Baghdiguian, S.; et al. Applying ecological and evolutionary theory to cancer: A long and winding road. Evol. Appl. 2013, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Laughney, A.M.; Elizalde, S.; Genovese, G.; Bakhoum, S.F. Dynamics of tumor heterogeneity derived from clonal karyotypic evolution. Cell Rep. 2015, 12, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Plutynski, A. Cancer and the goals of integration. Stud. Hist. Philos. Biol. Biomed. Sci. 2013, 44, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Villacorta-Martin, C.; Craig, A.J.; Villanueva, A. Divergent evolutionary trajectories in transplanted tumor models. Nat. Genet. 2017, 49, 1565–1566. [Google Scholar] [CrossRef] [PubMed]
- Demlova, R.; Zdrazilova-Dubska, L.; Sterba, J.; Stanta, G.; Valik, D. Host-dependent variables: The missing link to personalized medicine. Eur. J. Surg. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.B.; Stover, P.J.; McNulty, H.; Fenech, M.F.; Gregory, J.F., 3rd; Mills, J.L.; Pfeiffer, C.M.; Fazili, Z.; Zhang, M.; Ueland, P.M.; et al. Biomarkers of nutrition for development-folate review. J. Nutr. 2015, 145, 1636S–1680S. [Google Scholar] [CrossRef] [PubMed]
- Cordero, A.M.; Crider, K.S.; Rogers, L.M.; Cannon, M.J.; Berry, R.J. Optimal serum and red blood cell folate concentrations in women of reproductive age for prevention of neural tube defects: World Health Organization guidelines. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 421–423. [Google Scholar] [PubMed]
- Selhub, J.; Rosenberg, I.H. Excessive folic acid intake and relation to adverse health outcome. Biochimie 2016, 126, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, C.; Thomas-Virnig, C.; Allen-Hoffmann, B.L. Classical human epidermal keratinocyte cell culture. Methods Mol. Biol. 2013, 945, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, M.; Droux, M.; Hell, R. O-acetylserine (thiol) lyase: An enigmatic enzyme of plant cysteine biosynthesis revisited in Arabidopsis thaliana. J. Exp. Bot. 2004, 55, 1785–1798. [Google Scholar] [CrossRef] [PubMed]
- Buck, D.; Malivert, L.; de Chasseval, R.; Barraud, A.; Fondanèche, M.C.; Sanal, O.; Plebani, A.; Stéphan, J.L.; Hufnagel, M.; le Deist, F.; et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 2006, 124, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Hirt, B. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 1967, 26, 365–369. [Google Scholar] [CrossRef]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabs, J.; Zickgraf, F.M.; Park, J.; Wagner, S.; Jiang, X.; Jechow, K.; Kleinheinz, K.; Toprak, U.H.; Schneider, M.A.; Meister, M.; et al. Screening drug effects in patient-derived cancer cells links organoid responses to genome alterations. Mol. Syst. Biol. 2017, 13, 955. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold Change | |||||
|---|---|---|---|---|---|
| RNA-Seq | |||||
| Gene Name | FD vs. FC | FR vs. FC | FR vs. FD | Comments | |
| SORBS2 | Sorbin And SH3 Domain Containing 2 | 3.5 | –2.1 | –7.6 | Putative tumor suppressor gene involved in cervical carcinogenesis, PMID: 21602178. |
| DCN | Decorin | 2.6 | –2.8 | –8.2 | Growth suppressor of various tumor cell lines. Reduced expression or a total disappearance of DCN takes place during tumor progression, PMID: 2669749. |
| SLFN11 | Schlafen Family Member 11 | 3.3 | –4 | –15.1 | Putative DNA/RNA helicase, can sensitize cancer cells to DNA-damaging agents, PMID: 22927417. |
| GALC | Galactosylceramidase | N.S. | –3.4 | –3.4 | Lysosomal enzyme responsible for glycosphingolipids degradation by products which are important for synthesis of apoptosis. Mediator ceramide. DNA promoter hypermethylation contributes to GALC down-regulation in lung, head, and neck cancers, PMID: 26617822. |
| HSPB7 | Heat Shock 27kDa Protein Family, Member 7 | –2.9 | –2 | –9.8 | Tumor suppressor gene involved in p53 signaling and down-regulated in renal cell carcinoma by hypermethylation, PMID: 24585183. |
| DIRAS3 | Family, GTP-Binding RAS-Like3 | –4.7 | –4.6 | N.S. | Tumor suppressor gene whose expression is lost in ovarian and breast cancers, PMID 16757345. |
| RUNX3 | Runt-Related Transcription Factor 3 | 1.9 | –2.6 | –5.4 | RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer, PMID: 16818622. Association between methylation of the RUNX3 promoter and gastric cancer was reported, confirming the role of RUNX3 as a tumor suppressor gene, PMID: 21867527. RUNX3 does play important functions in immunity and inflammation and may thereby indirectly influence epithelial tumor development, PMID: 25641675. |
| USP44 | Ubiquitin Specific Peptidase 44 | –1.6 | –3.1 | –2.9 | Tumor suppressor, which is epigenetically inactivated in colorectal neoplasia, PMID: 24837038. It protects against chromosome missegregation and it regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis, PMID: 23187131, PMID: 23187126. |
| CDKN1C | Cyclin-dependent kinase inhibitor 1C | N.S. | –2.1 | –2.1 | Negative regulator of cell proliferation. It regulates several hallmarks of cancer, including apoptosis, cell invasion and metastasis, tumor differentiation and angiogenesis, PMID: 21447370. |
| H2AFY2 | H2A Histone Family, Member Y2 | N.S. | –3.9 | –4.2 | Tumor suppressor function in lung cancer and melanoma, PMID: 26658220. |
| LEF1 | Lymphoid Enhancer–Binding Factor 1 | N.S. | –3.7 | –4.4 | Implication with AKT pathway in repairing DSB, PMID: 20956948. |
| TBX18 | T-Box 18 | N.S. | –2 | –2.2 | Tumor suppressive properties, hypermethylated in colon cancer. TBX18 inhibits growth and clonogenic survival of colon cancer cells in vitro, PMID: 20454457. |
| FAP | Fibroblast Activation Protein, Alpha | 1.6 | 2.1 | 1.2 | Over-expression of FAP increases tumor growth in xenografts of ovarian cancer cells, PMID: 24028972. Involved in the control of human breast cancer cell line growth and motility via the FAK pathway, PMID: 24885257. |
| KLK5 | Kallikrein-Related Peptidase 5 | 1.7 | 2.5 | 1.3 | Kallikreins are a subgroup of serine proteases having diverse physiological functions. Growing evidence suggests that many kallikreins are implicated in carcinogenesis and some have potential as novel cancer and other disease biomarkers, PMID: 19921697. |
| KLK10 | Kallikrein-Related Peptidase 10 | 1.9 | 2.2 | 1.2 | |
| PIM1 | Pim-1 Proto-Oncogene, Serine/Threonine Kinase | 1.8 | 1.2 | 2.2 | PIM genes encode serine/threonine kinases that have been shown to counteract the increased sensitivity to apoptosis induction that is associated with MYC-driven tumorigenesis, PMID: 21150935. The PIM1 kinase promotes prostate cancer cell migration and adhesion via multiple signaling pathways, PMID: 26934497. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savini, C.; Yang, R.; Savelyeva, L.; Göckel-Krzikalla, E.; Hotz-Wagenblatt, A.; Westermann, F.; Rösl, F. Folate Repletion after Deficiency Induces Irreversible Genomic and Transcriptional Changes in Human Papillomavirus Type 16 (HPV16)-Immortalized Human Keratinocytes. Int. J. Mol. Sci. 2019, 20, 1100. https://doi.org/10.3390/ijms20051100
Savini C, Yang R, Savelyeva L, Göckel-Krzikalla E, Hotz-Wagenblatt A, Westermann F, Rösl F. Folate Repletion after Deficiency Induces Irreversible Genomic and Transcriptional Changes in Human Papillomavirus Type 16 (HPV16)-Immortalized Human Keratinocytes. International Journal of Molecular Sciences. 2019; 20(5):1100. https://doi.org/10.3390/ijms20051100
Chicago/Turabian StyleSavini, Claudia, Ruwen Yang, Larisa Savelyeva, Elke Göckel-Krzikalla, Agnes Hotz-Wagenblatt, Frank Westermann, and Frank Rösl. 2019. "Folate Repletion after Deficiency Induces Irreversible Genomic and Transcriptional Changes in Human Papillomavirus Type 16 (HPV16)-Immortalized Human Keratinocytes" International Journal of Molecular Sciences 20, no. 5: 1100. https://doi.org/10.3390/ijms20051100