HPV-Mediated Resistance to TNF and TRAIL Is Characterized by Global Alterations in Apoptosis Regulatory Factors, Dysregulation of Death Receptors, and Induction of ROS/RNS

 , , and
, , and
Abstract
:1. Introduction
2. Results
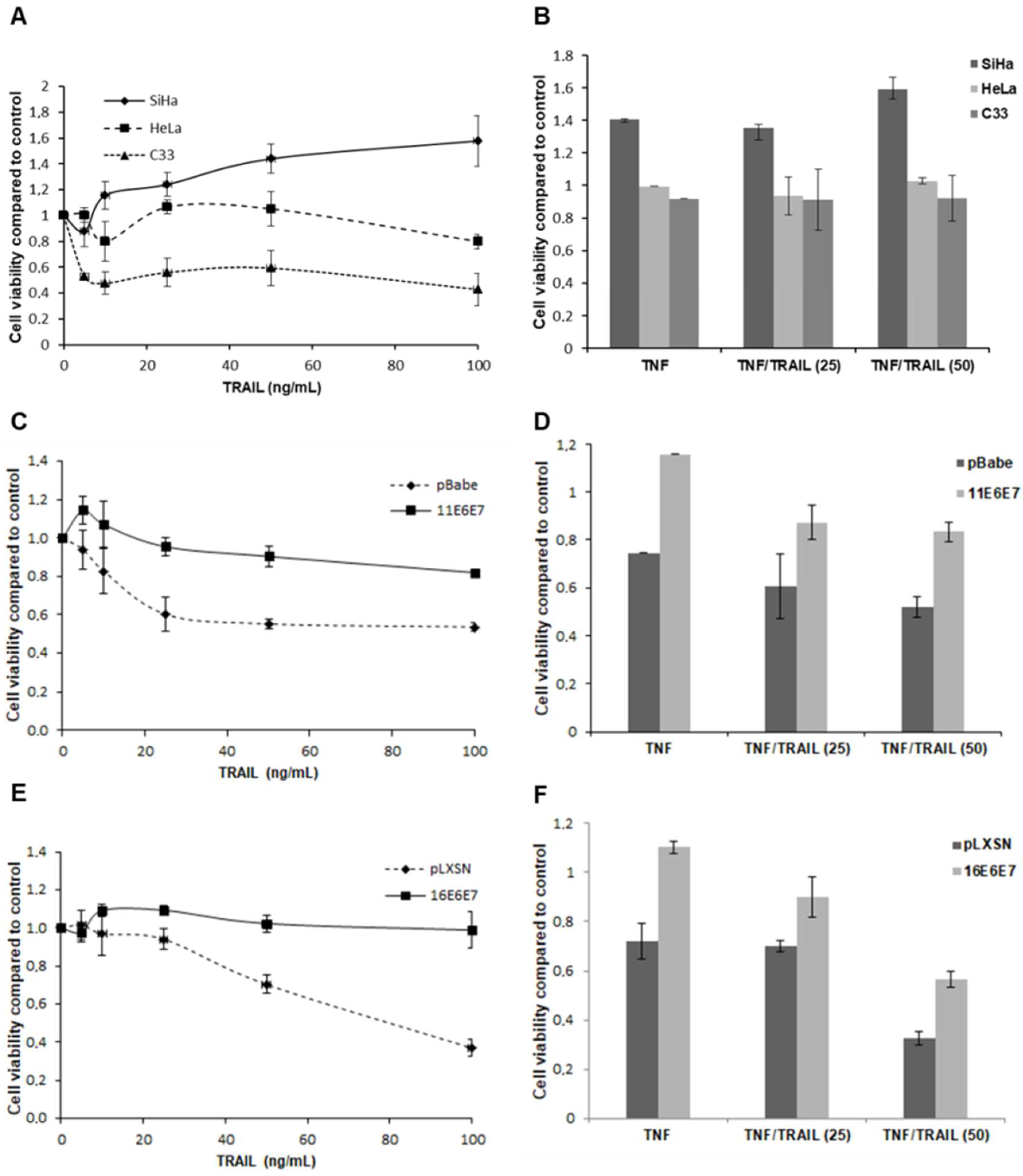
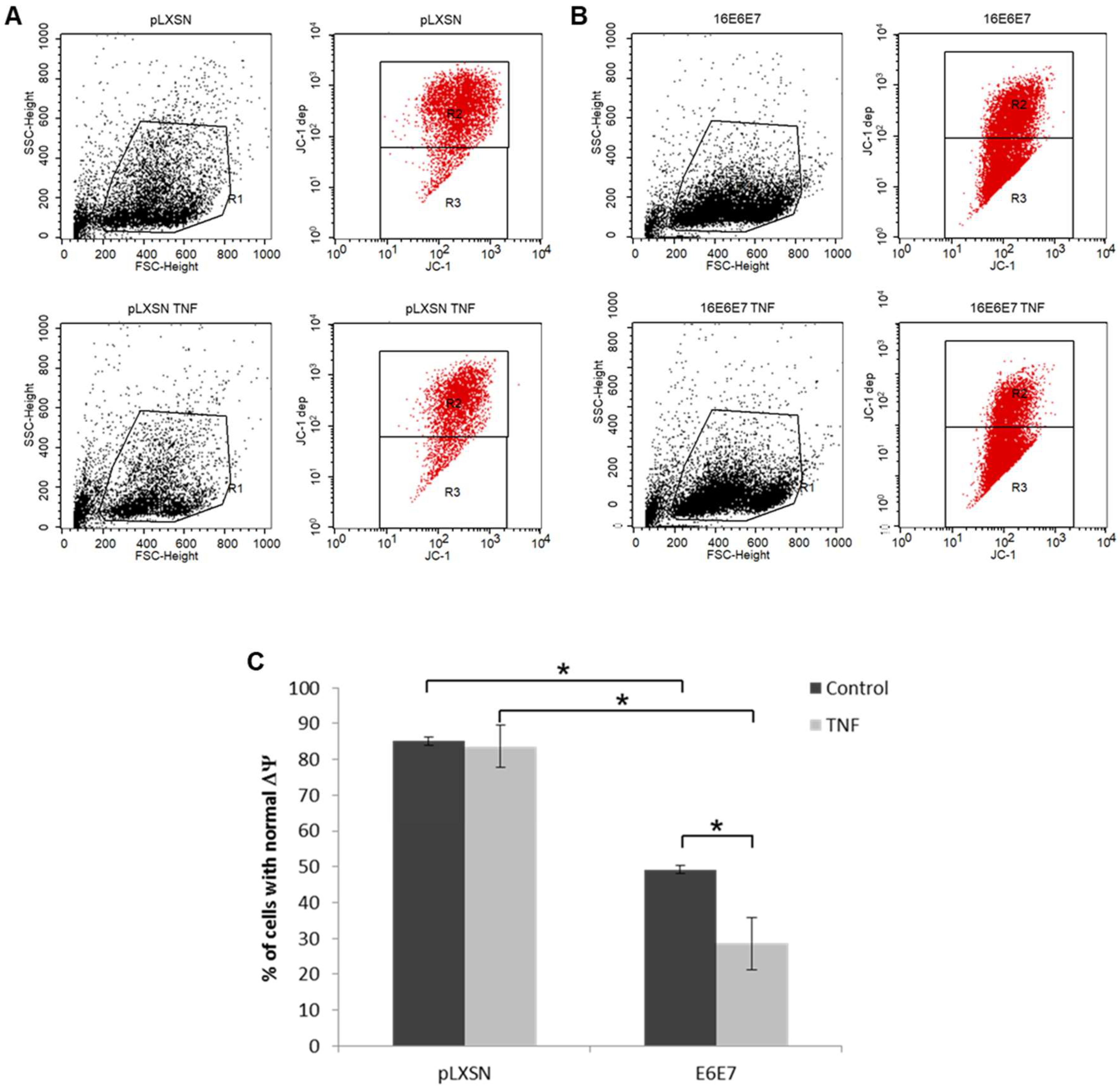
2.1. HPV E6 and E7 Confer Resistance to both TNF and TRAIL
2.2. HPV16 E6 and E7 Differentially Alter the Expression of Genes Involved in the NFκB Pathway
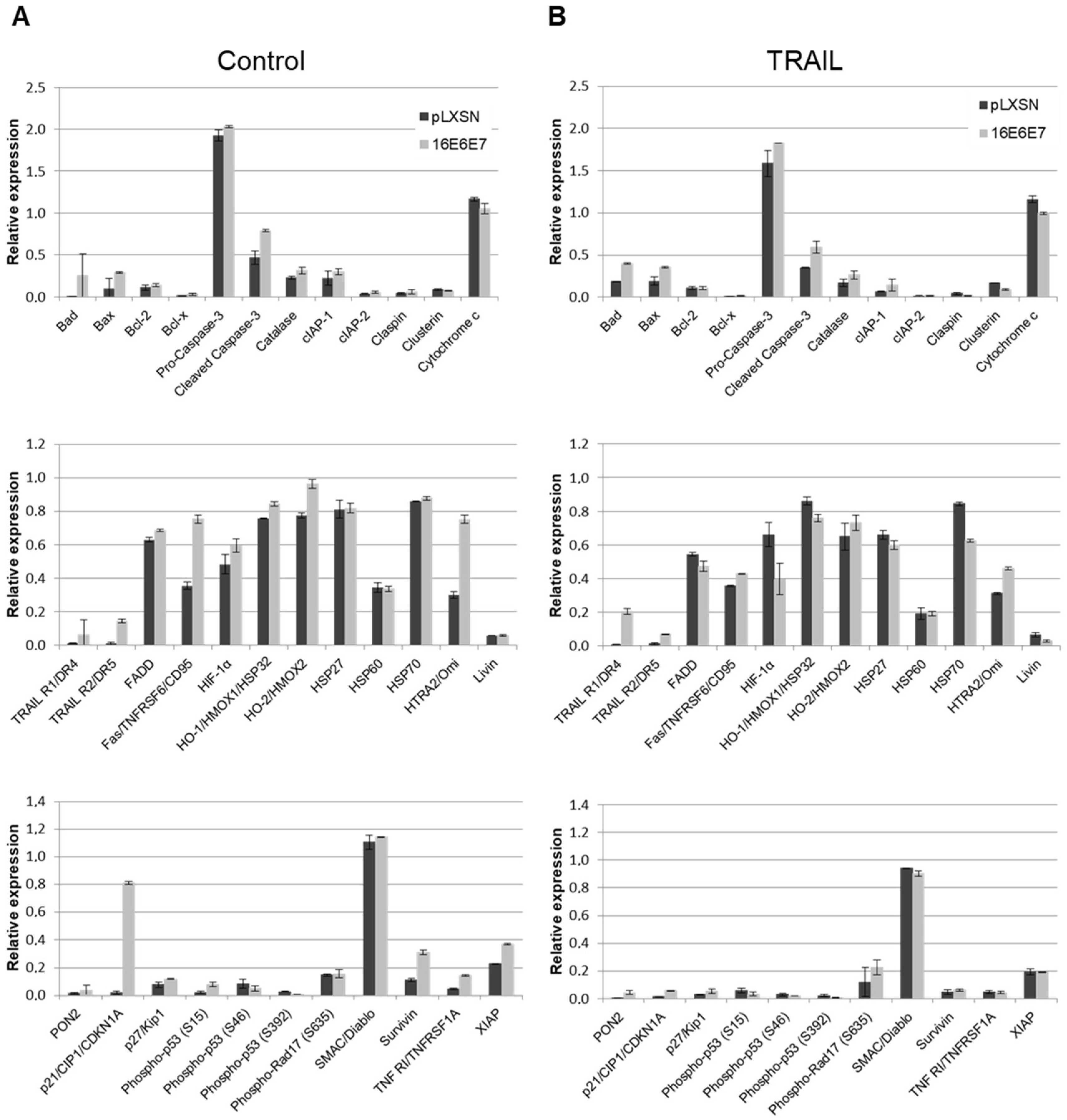
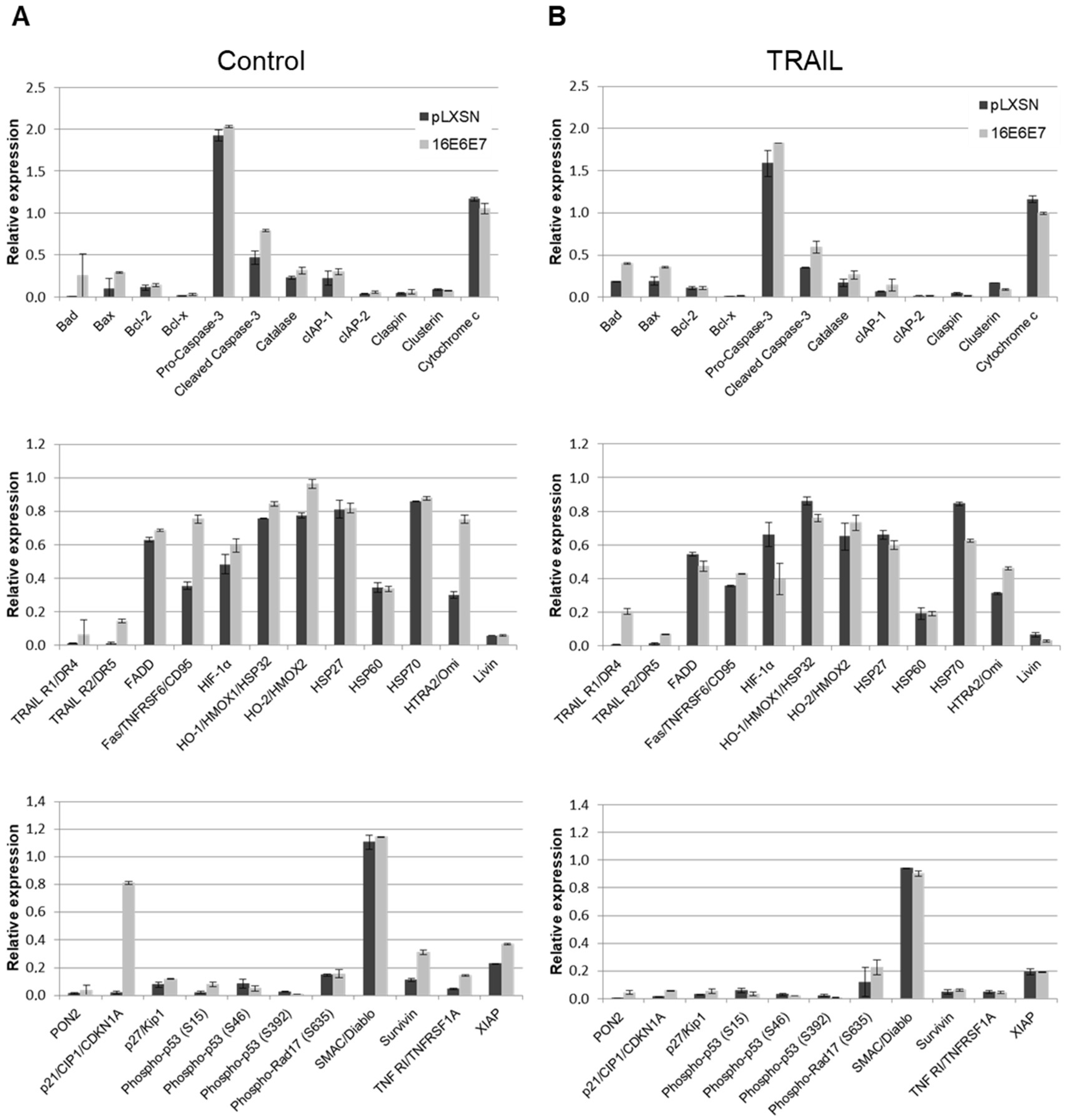
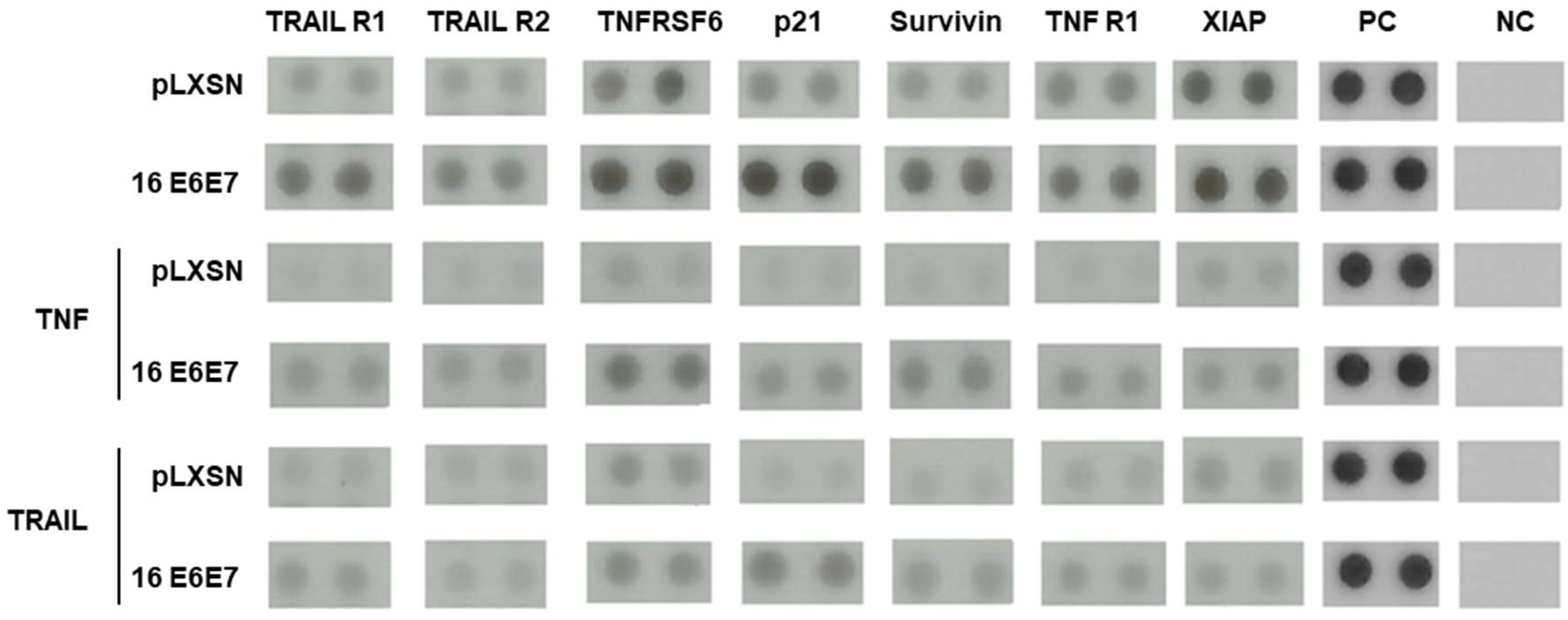
2.3. HPV11 and HPV16 E6 and E7 Differentially Regulate the Expression of Proteins Involved in Apoptosis Regulation/Execution
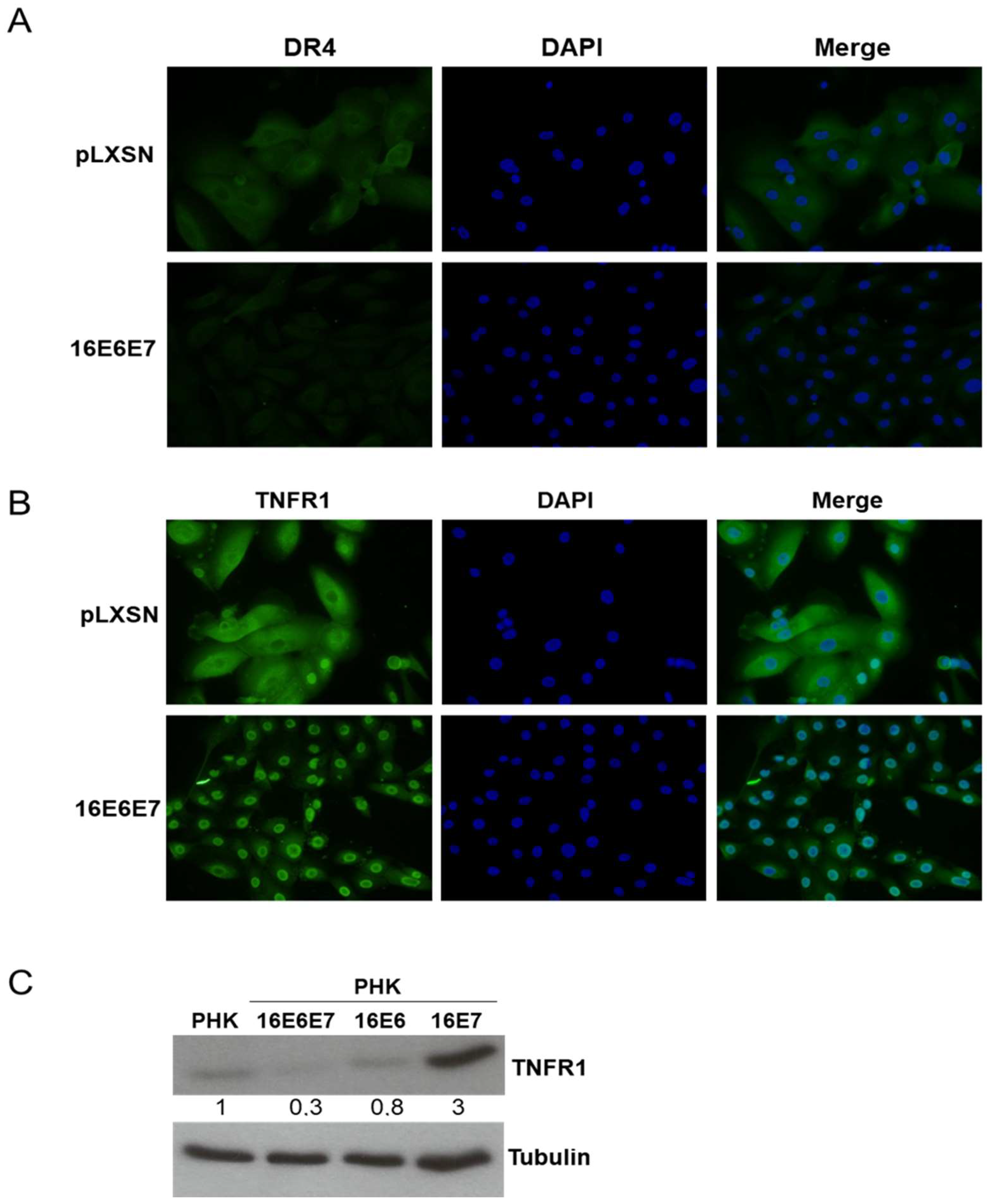
2.4. HPV16 Oncogenes Regulate TNFR1 Expression in Human Keratinocytes
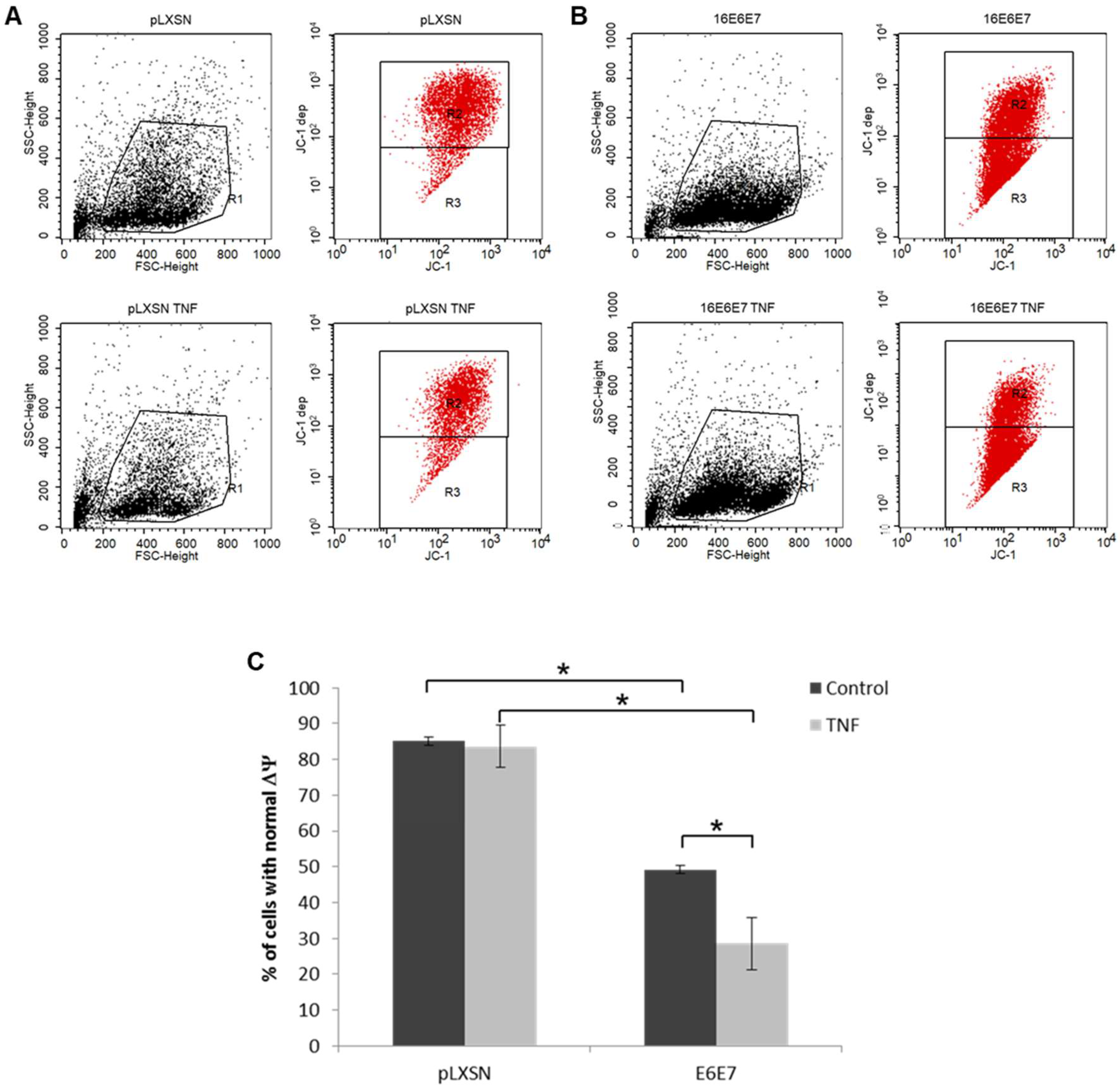
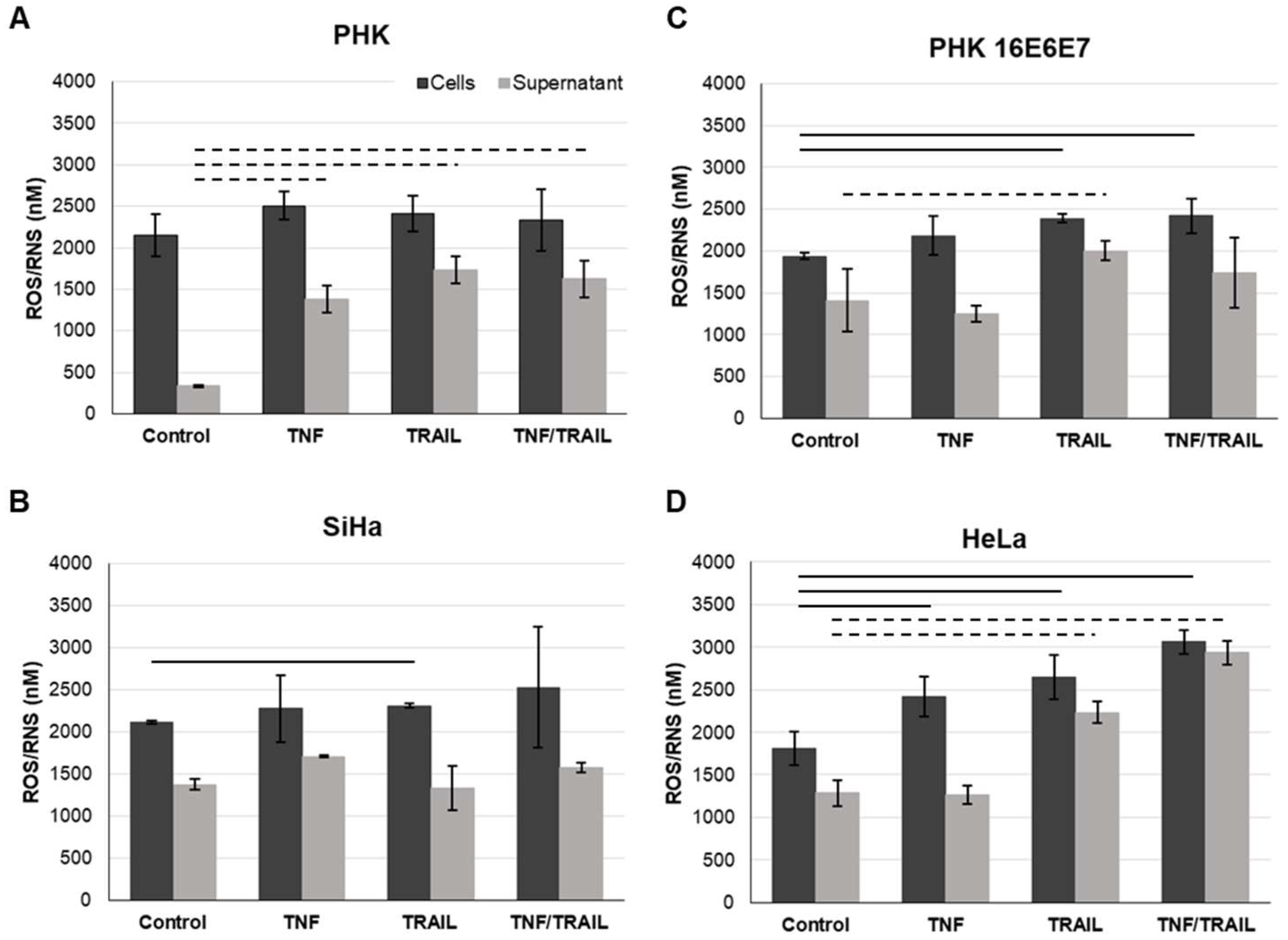
2.5. Treatment with TNF and/or TRAIL Induces ROS/RNS in Cells Expressing HPV Genes
3. Discussion
4. Materials and Methods
4.1. Cells, Retroviruses and Organotypic Raft Cultures
4.2. Treatment with Cytokines and Proliferation Analysis
4.3. Analysis of the Expression of Genes Involved in NFκB Signaling Pathway
4.4. Protein Extraction, Quantification and Expression Analysis
4.5. Immunofluorescence
4.6. Analysis of the Production of ROS and RNS
4.7. Analysis of Mitochondrial Membrane Potential (ΔΨ)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.; Mattarollo, S.R.; Gosmann, C.; Frazer, I.H.; Leggatt, G.R. Regulation of immune responses to HPV infection and during HPV-directed immunotherapy. Immunol. Rev. 2011, 239, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Sauder, D.N. Tumor necrosis factor (TNF) receptor type 1 (p55) is a main mediator for TNF-alpha-induced skin inflammation. Eur. J. Immunol. 1997, 27, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, L.L.; Vieira, K.B.; Pei, X.F.; Schlegel, R. Differential effect of tumor necrosis factor on proliferation of primary human keratinocytes and cell lines containing human papillomavirus types 16 and 18. Mol. Carcinog. 1992, 6, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Basile, J.R.; Zacny, V.; Münger, K. The cytokines tumor necrosis factor-alpha (TNF-alpha) and TNF-related apoptosis-inducing ligand differentially modulate proliferation and apoptotic pathways in human keratinocytes expressing the human papillomavirus-16 E7 oncoprotein. J. Biol. Chem. 2001, 276, 22522–22528. [Google Scholar] [CrossRef] [PubMed]
- Boccardo, E.; Manzini Baldi, C.V.; Carvalho, A.F.; Rabachini, T.; Torres, C.; Barreta, L.A.; Brentani, H.; Villa, L.L. Expression of human papillomavirus type 16 E7 oncoprotein alters keratinocytes expression profile in response to tumor necrosis factor-alpha. Carcinogenesis 2010, 31, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Vanamee, É.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, eaao4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natoli, G.; Costanzo, A.; Guido, F.; Moretti, F.; Bernardo, A.; Burgio, V.L.; Agresti, C.; Levrero, M. Nuclear factor kB-independent cytoprotective pathways originating at tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 1998, 273, 31262–31272. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-κB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Benedict, C.A.; Norris, P.S.; Ware, C.F. To kill or be killed: Viral evasion of apoptosis. Nat. Immunol. 2002, 3, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.A. Viruses and the TNF-related cytokines, an evolving battle. Cytokine Growth Factor Rev. 2003, 14, 349–357. [Google Scholar] [CrossRef]
- Hawley-Nelson, P.; Vousden, K.H.; Hubbert, N.L.; Lowy, D.R.; Schiller, J.T. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989, 8, 3905–3910. [Google Scholar] [CrossRef]
- Münger, K.; Phelps, W.C.; Bubb, V.; Howley, P.M.; Schlegel, R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 1989, 63, 4417–4421. [Google Scholar] [PubMed]
- Goodwin, E.C.; Naeger, L.K.; Breiding, D.E.; Androphy, E.J.; DiMaio, D. Transactivation-competent bovine papillomavirus E2 protein is specifically required for efficient repression of human papillomavirus oncogene expression and for acute growth inhibition of cervical carcinoma cell lines. J. Virol. 1998, 72, 3925–3934. [Google Scholar]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef]
- Hagari, Y.; Budgeon, L.R.; Pickel, M.D.; Kreider, J.W. Association of tumor necrosis factor-alpha gene expression and apoptotic cell death with regression of Shope papillomas. J. Investig. Dermatol. 1995, 104, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, K.; Mossadegh, N.; Kohl, A.; Komposch, G.; Schenkel, J.; Alonso, A.; Tomakidi, P. The HPV-16 E5 protein inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte raft cultures. Intervirology 2004, 47, 48–56. [Google Scholar] [CrossRef]
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ. 2006, 13, 1915–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerksen-Hughes, P.J.; Yang, J.; Schwartz, S.B. HPV 16 E6 blocks TNF-mediated apoptosis in mouse fibroblast LM cells. Virology 1999, 264, 55–65. [Google Scholar] [CrossRef]
- Boccardo, E.; Noya, F.; Broker, T.R.; Chow, L.T.; Villa, L.L. HPV-18 confers resistance to TNF-alpha in organotypic cultures of human keratinocytes. Virology 2004, 328, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef] [PubMed]
- Vieira, K.B.; Goldstein, D.J.; Villa, L.L. Tumor necrosis factor alpha interferes with the cell cycle of normal and papillomavirus-immortalized human keratinocytes. Cancer Res 1996, 56, 2452–2457. [Google Scholar] [PubMed]
- Jian, Y.; Schmidt-Grimminger, D.C.; Chien, W.M.; Wu, X.; Broker, T.R.; Chow, L.T. Post-transcriptional induction of p21cip1 protein by human papillomavirus E7 inhibits unscheduled DNA synthesis reactivated in differentiated keratinocytes. Oncogene 1998, 17, 2027–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Dürr, P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 1996, 13, 2323–2330. [Google Scholar]
- Nakamura, M.; Bodily, J.M.; Beglin, M.; Kyo, S.; Inoue, M.; Laimins, L.A. Hypoxia-specific stabilization of HIF-1alpha by human papillomaviruses. Virology 2009, 387, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Bodily, J.M.; Mehta, K.P.; Laimins, L.A. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. 2011, 71, 1187–1195. [Google Scholar] [CrossRef]
- Rodolico, V.; Arancio, W.; Amato, M.C.; Aragona, F.; Cappello, F.; Di Fede, O.; Pannone, G.; Campisi, G. Hypoxia inducible factor-1 alpha expression is increased in infected positive HPV16 DNA oral squamous cell carcinoma and positively associated with HPV16 E7 oncoprotein. Infect. Agent Cancer 2011, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Michels, C.L.; Leung, M.K.; Arany, Z.P.; Kung, A.L.; Livingston, D.M. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999, 13, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Freedman, S.J.; Sun, Z.Y.; Kung, A.L.; France, D.S.; Wagner, G.; Eck, M.J. Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nat. Struct. Biol. 2003, 10, 504–512. [Google Scholar] [CrossRef]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- De Marco, F. Oxidative stress and HPV carcinogenesis. Viruses 2013, 5, 708–731. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; El-Deiry, W.S. Distinct signaling pathways in TRAIL—Versus tumor necrosis factor-induced apoptosis. Mol. Cell. Biol. 2006, 26, 8136–8148. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Spitkovsky, D.; Hehner, S.P.; Hofmann, T.G.; Möller, A.; Schmitz, M.L. The human papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the Ikappa B kinase complex. J. Biol. Chem. 2002, 277, 25576–25582. [Google Scholar] [CrossRef]
- Seitz, C.S.; Lin, Q.; Deng, H.; Khavari, P.A. Alterations in NF-kappaB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-kappaB. Proc. Natl. Acad. Sci. USA 1998, 95, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Seitz, C.S.; Deng, H.; Hinata, K.; Lin, Q.; Khavari, P.A. Nuclear factor kappaB subunits induce epithelial cell growth arrest. Cancer Res. 2000, 60, 4085–4092. [Google Scholar]
- Chaturvedi, V.; Qin, J.Z.; Denning, M.F.; Choubey, D.; Diaz, M.O.; Nickoloff, B.J. Apoptosis in proliferating, senescent, and immortalized keratinocytes. J. Biol. Chem. 1999, 274, 23358–23367. [Google Scholar] [CrossRef]
- Komine, M.; Rao, L.S.; Kaneko, T.; Tomic-Canic, M.; Tamaki, K.; Freedberg, I.M.; Blumenberg, M. Inflammatory versus proliferative processes in epidermis. Tumor necrosis factor alpha induces K6b keratin synthesis through a transcriptional complex containing NFkappa B and C/EBPbeta. J. Biol. Chem. 2000, 275, 32077–32088. [Google Scholar] [CrossRef]
- Hu, Y.; Baud, V.; Oga, T.; Kim, K.I.; Yoshida, K.; Karin, M. IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature 2001, 410, 710–714. [Google Scholar] [CrossRef]
- Van Hogerlinden, M.; Rozell, B.L.; Ahrlund-Richter, L.; Toftgård, R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999, 59, 3299–3303. [Google Scholar] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C.; Hehner, S.P.; Dröge, W.; Schmitz, M.L. Repression of NF-kappaB impairs HeLa cell proliferation by functional interference with cell cycle checkpoint regulators. Oncogene 1999, 18, 3213–3225. [Google Scholar] [CrossRef] [PubMed]
- Nees, M.; Geoghegan, J.M.; Hyman, T.; Frank, S.; Miller, L.; Woodworth, C.D. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J. Virol. 2001, 75, 4283–4296. [Google Scholar] [CrossRef] [PubMed]
- Prabhavathy, D.; Subramanian, C.K.; Karunagaran, D. Re-expression of HPV16 E2 in SiHa (human cervical cancer) cells potentiates NF-κB activation induced by TNF-α concurrently increasing senescence and survival. Biosci. Rep. 2015, 35, e00175. [Google Scholar] [CrossRef] [PubMed]
- Borbély, A.A.; Murvai, M.; Kónya, J.; Beck, Z.; Gergely, L.; Li, F.; Veress, G. Effects of human papillomavirus type 16 oncoproteins on survivin gene expression. J. Gen. Virol. 2006, 87, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, M.A.; Lee, J.H.; Klingelhutz, A.J. Human papillomavirus type 16 E6 activates NF-kappaB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J. Virol. 2006, 80, 5301–5307. [Google Scholar] [CrossRef] [PubMed]
- Chamulitrat, W.; Sattayakhom, A.; Herold-Mende, C.; Herold-Mended, C.; Stremmel, W. Human papillomavirus 16 E6/E7-immortalized human gingival keratinocytes with epithelial mesenchymal transition acquire increased expression of cIAP-1, Bclx and p27(Kip1). Exp. Dermatol. 2009, 18, 1067–1069. [Google Scholar] [CrossRef]
- Termini, L.; Boccardo, E. Epithelial Organotypic Cultures: A Viable Model to Address Mechanisms of Carcinogenesis by Epitheliotropic Viruses. Curr. Top. Med. Chem. 2018, 18, 246–255. [Google Scholar] [CrossRef]
- Banerjee, N.S.; Genovese, N.J.; Noya, F.; Chien, W.M.; Broker, T.R.; Chow, L.T. Conditionally activated E7 proteins of high-risk and low-risk human papillomaviruses induce S phase in postmitotic, differentiated human keratinocytes. J. Virol. 2006, 80, 6517–6524. [Google Scholar] [CrossRef]
- Moody, C.A.; Fradet-Turcotte, A.; Archambault, J.; Laimins, L.A. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc. Natl. Acad. Sci. USA 2007, 104, 19541–19546. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J. Host control of human papillomavirus infection and disease. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, N.F.; Kulaga, S.; Robitaille, J.; Ferreira, S.; Santos, M.; Miyamura, R.A.; Duarte-Franco, E.; Rohan, T.E.; Ferenczy, A.; Villa, L.L.; et al. Persistent human papillomavirus infection as a predictor of cervical intraepithelial neoplasia. JAMA 2001, 286, 3106–3114. [Google Scholar] [CrossRef] [PubMed]
- Guess, J.C.; McCance, D.J. Decreased migration of Langerhans precursor-like cells in response to human keratinocytes expressing human papillomavirus type 16 E6/E7 is related to reduced macrophage inflammatory protein-3alpha production. J. Virol. 2005, 79, 14852–14862. [Google Scholar] [CrossRef] [PubMed]
- Helt, A.M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef]
- Ettehadi, P.; Greaves, M.W.; Wallach, D.; Aderka, D.; Camp, R.D. Elevated tumour necrosis factor-alpha (TNF-alpha) biological activity in psoriatic skin lesions. Clin. Exp. Immunol. 1994, 96, 146–151. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Description | p-Value | Fold |
|---|---|---|---|
| BCL10 | B-cell CLL/lymphoma 10 | 0.0047 | 1.53 |
| BCL3 | B-cell CLL/lymphoma 3 | 0.0248 | 1.70 |
| NOD1 | Nucleotide-binding oligomerization domain containing 1 | 0.0079 | 2.23 |
| CHUK | Conserved helix-loop-helix ubiquitous kinase | 0.0024 | 2.86 |
| CSF3 | Colony stimulating factor 3 (granulocyte) | 0.0002 | 5.81 |
| ELK1 | ELK1, member of ETS oncogene family | 0.0276 | 1.64 |
| FOS | v-fos FBJ murine osteosarcoma viral oncogene homolog | 0.0007 | −3.18 |
| GJA1 | Gap junction protein, alpha 1, 43 kDa | 0.0018 | −3.16 |
| HTR2B | 5-Hydroxytryptamine (serotonin) receptor 2B | 0.0028 | 2.34 |
| IKBKB | Inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase beta | 0.0149 | 2.23 |
| IL8 | Interleukin 8 | 0.0002 | −1.76 |
| MYD88 | Myeloid differentiation primary response gene (88) | 0.0118 | −1.89 |
| NFKB1 | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 | 0.0060 | 2.00 |
| STAT1 | Signal transducer and activator of transcription 1, 91 kDa | 0.0361 | −2.00 |
| TBK1 | TANK-binding kinase 1 | 0.0301 | 1.77 |
| TLR3 | Toll-like receptor 3 | 0.0034 | −3.97 |
| TNFRSF10B | Tumor necrosis factor receptor superfamily, member 10b | 0.0138 | −3.20 |
| TNFSF14 | Tumor necrosis factor (ligand) superfamily, member 14 | 0.0294 | 2.13 |
| RPL13A | Ribosomal protein L13a | 0.0213 | 1.66 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabeça, T.K.; De Mello Abreu, A.; Andrette, R.; De Souza Lino, V.; Morale, M.G.; Aguayo, F.; Termini, L.; Villa, L.L.; Lepique, A.P.; Boccardo, E. HPV-Mediated Resistance to TNF and TRAIL Is Characterized by Global Alterations in Apoptosis Regulatory Factors, Dysregulation of Death Receptors, and Induction of ROS/RNS. Int. J. Mol. Sci. 2019, 20, 198. https://doi.org/10.3390/ijms20010198
Cabeça TK, De Mello Abreu A, Andrette R, De Souza Lino V, Morale MG, Aguayo F, Termini L, Villa LL, Lepique AP, Boccardo E. HPV-Mediated Resistance to TNF and TRAIL Is Characterized by Global Alterations in Apoptosis Regulatory Factors, Dysregulation of Death Receptors, and Induction of ROS/RNS. International Journal of Molecular Sciences. 2019; 20(1):198. https://doi.org/10.3390/ijms20010198
Chicago/Turabian StyleCabeça, Tatiane Karen, Alice De Mello Abreu, Rafael Andrette, Vanesca De Souza Lino, Mirian Galliote Morale, Francisco Aguayo, Lara Termini, Luisa Lina Villa, Ana Paula Lepique, and Enrique Boccardo. 2019. "HPV-Mediated Resistance to TNF and TRAIL Is Characterized by Global Alterations in Apoptosis Regulatory Factors, Dysregulation of Death Receptors, and Induction of ROS/RNS" International Journal of Molecular Sciences 20, no. 1: 198. https://doi.org/10.3390/ijms20010198