Proteomics Analysis to Identify Proteins and Pathways Associated with the Novel Lesion Mimic Mutant E40 in Rice Using iTRAQ-Based Strategy

 , and
, and
Abstract
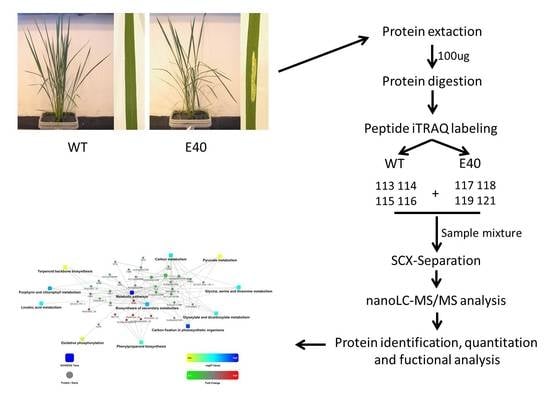
:
1. Introduction
2. Results
2.1. Phenotypic Characterization of E40
2.2. Proteomics Analysis of Differentially Abundant Proteins between E40 and WT
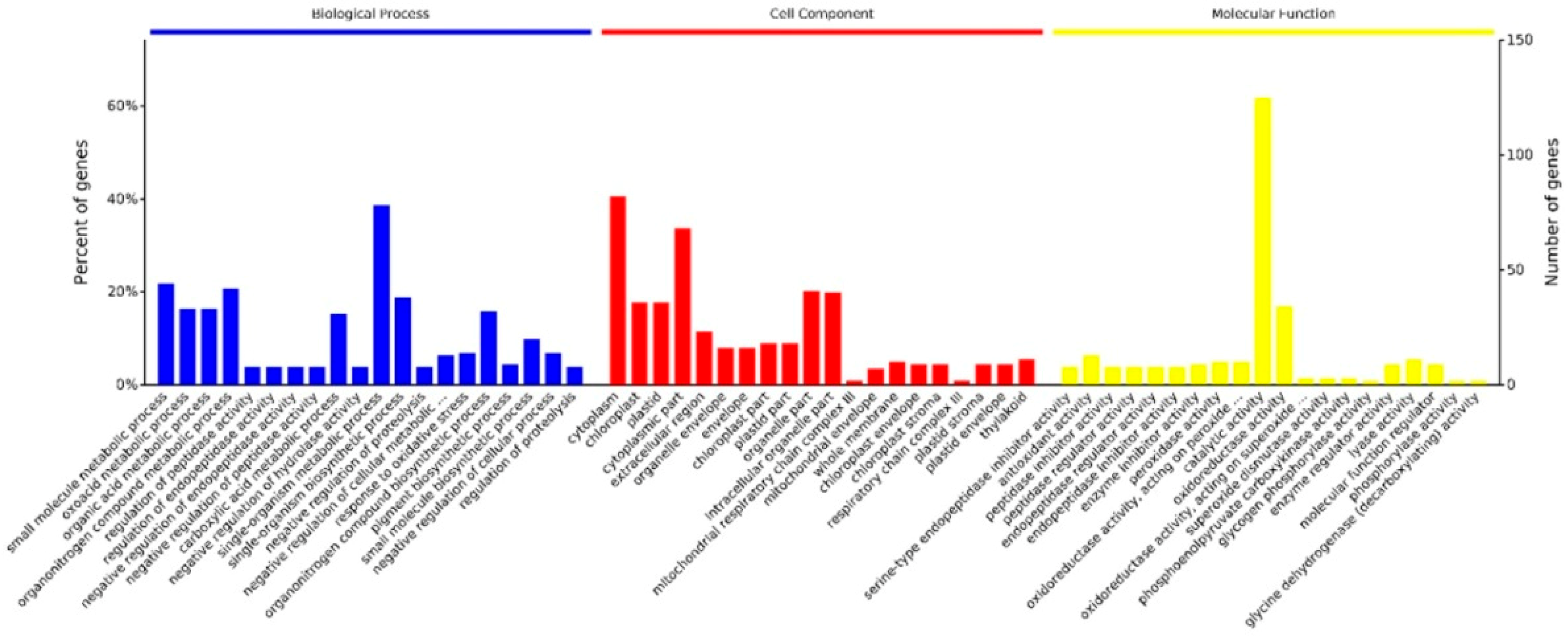
2.3. Gene Ontology (GO) Analysis of Altered Proteins
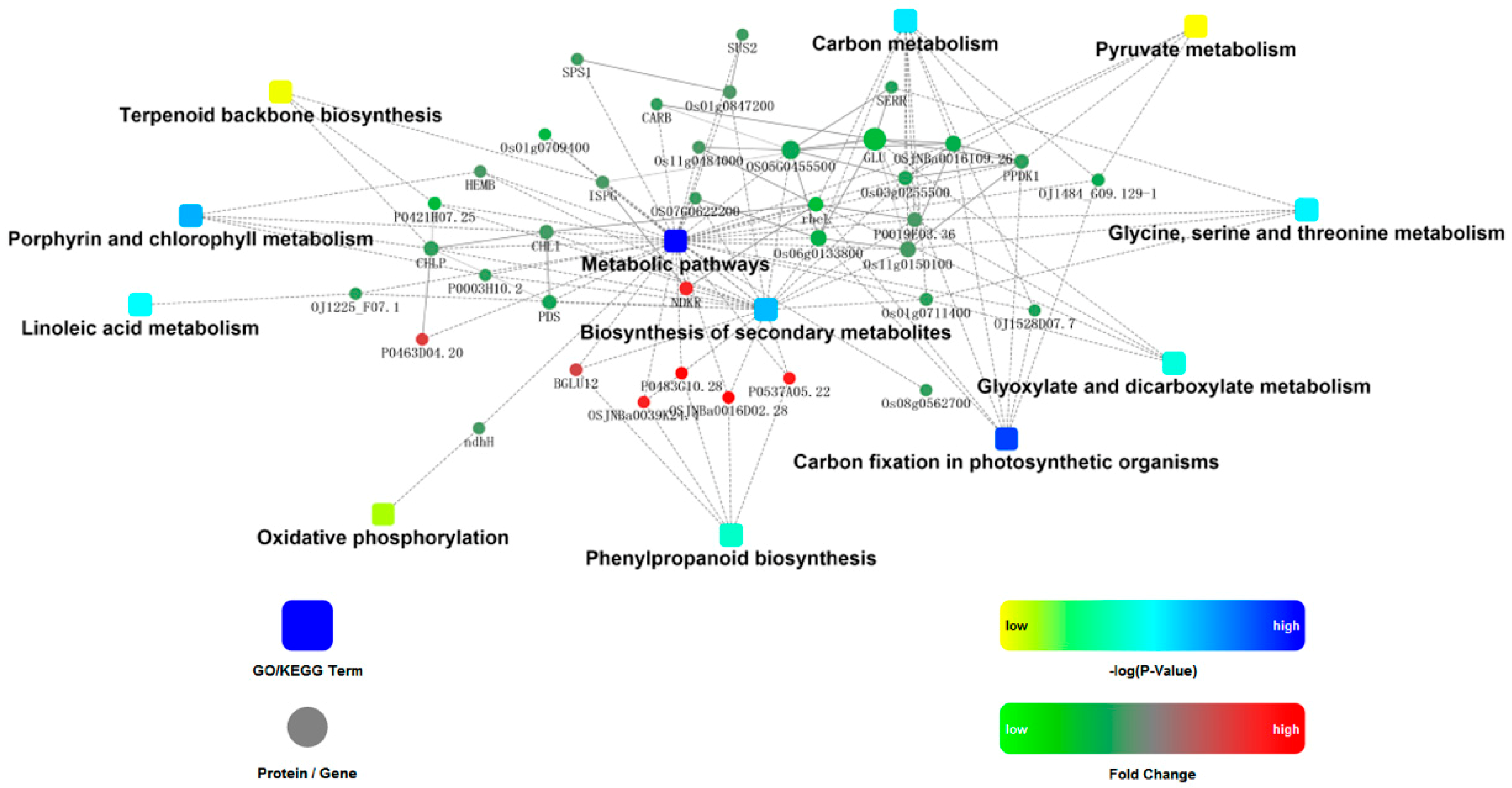
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
2.5. Protein-Protein Interaction Analysis
2.6. qRT-PCR Data are Consistent with Protein Abundance Analysis Revealed by iTRAQ
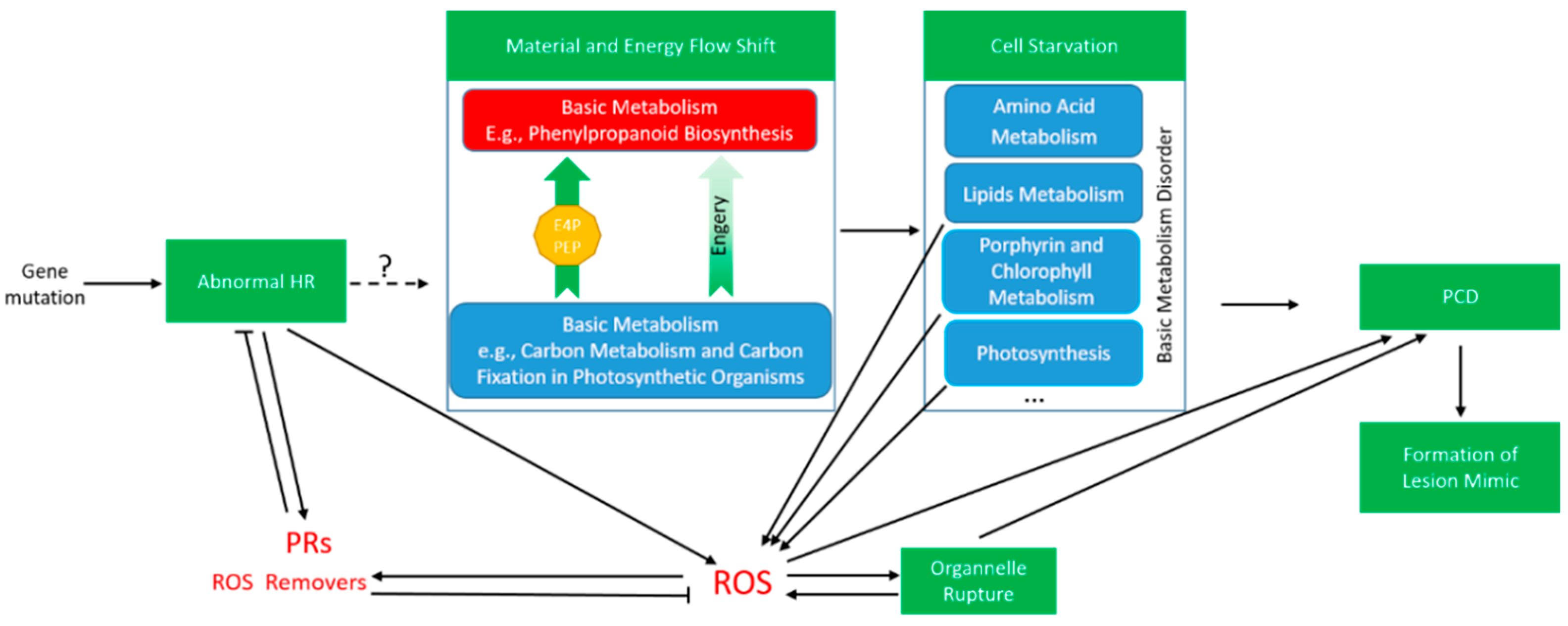
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Protein Extraction
4.3. Trypsin Digestion and iTRAQ Labeling
4.4. LC-MS/MS and Bioinformatics Analysis
4.5. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kiyosawa, S. Inheritance of a particular sensitivity of the Rice variety, Sekiguchi Asahi, to pathogens and chemicals, and linkage relationship with blast resistance genes. Tokyo Nat. Inst. Agric. Sci. Bull. Ser. D 1970, 21, 61–72. [Google Scholar]
- Badigannavar, A.; Kale, D.; Eapen, S.; Murty, G. Inheritance of disease lesion mimic leaf trait in groundnut. J. Hered. 2002, 93, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Büschges, R.; Hollricher, K.; Panstruga, R.; Simons, G.; Wolter, M.; Frijters, A.; van Daelen, R.; van der Lee, T.; Diergaarde, P.; Groenendijk, J. The barley Mlo gene: A novel control element of plant pathogen resistance. Cell 1997, 88, 695–705. [Google Scholar] [CrossRef]
- Dietrich, R.A.; Delaney, T.P.; Uknes, S.J.; Ward, E.R.; Ryals, J.A.; Dangl, J.L. Arabidopsis mutants simulating disease resistance response. Cell 1994, 77, 565–577. [Google Scholar] [CrossRef]
- Fekih, R.; Tamiru, M.; Kanzaki, H.; Abe, A.; Yoshida, K.; Kanzaki, E.; Saitoh, H.; Takagi, H.; Natsume, S.; Undan, J.R. The rice (Oryza sativa L.) LESION MIMIC RESEMBLING, which encodes an AAA-type ATPase, is implicated in defense response. Mol. Genet. Genom. 2015, 290, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.T. Programmed cell death in plant-pathogen interactions. Annu. Rev. Plant Biol. 1997, 48, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.T.; Guo, A.; Klessig, D.F.; Ausubel, F.M. Programmed cell death in plants: A pathogen-triggered response activated coordinately with multiple defense functions. Cell 1994, 77, 551–563. [Google Scholar] [CrossRef]
- Johal, G.S.; Hulbert, S.H.; Briggs, S.P. Disease lesion mimics of maize: A model for cell death in plants. Bioessays 1995, 17, 685–692. [Google Scholar] [CrossRef]
- Takahashi, A.; Kawasaki, T.; Henmi, K.; Shii, K.; Kodama, O.; Satoh, H.; Shimamoto, K. Lesion mimic mutants of rice with alterations in early signaling events of defense. Plant J. 1999, 17, 535–545. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.-H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Chen, J.; Zeng, L.; Goh, M.; Leung, H.; Khush, G.S.; Wang, G.-L. Characterizing rice lesion mimic mutants and identifying a mutant with broad-spectrum resistance to rice blast and bacterial blight. Mol. Plant-Microbe Interact. 2000, 13, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Mizobuchi, R.; Hirabayashi, H.; Kaji, R.; Nishizawa, Y.; Yoshimura, A.; Satoh, H.; Ogawa, T.; Okamoto, M. Isolation and characterization of rice lesion-mimic mutants with enhanced resistance to rice blast and bacterial blight. Plant Sci. 2002, 163, 345–353. [Google Scholar] [CrossRef]
- Jung, Y.-H.; Lee, J.-H.; Agrawal, G.K.; Rakwal, R.; Kim, J.-A.; Shim, J.-K.; Lee, S.-K.; Jeon, J.-S.; Koh, H.-J.; Lee, Y.-H. The rice (Oryza sativa) blast lesion mimic mutant, blm, may confer resistance to blast pathogens by triggering multiple defense-associated signaling pathways. Plant Physiol. Biochem. 2005, 43, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Xu, W.G.; Wang, H.W.; Hu, L.; Li, Y.; Qi, X.L.; Zhang, L.; Li, C.X.; Hua, X. Pyramiding expression of maize genes encoding phosphoenolpyruvate carboxylase (PEPC) and pyruvate orthophosphate dikinase (PPDK) synergistically improve the photosynthetic characteristics of transgenic wheat. Protoplasma 2014, 251, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, P.; Li, C.; Wang, Y.; Guo, L.; Jiang, G.; Zhai, W. LMM5. 1 and LMM5. 4, two eukaryotic translation elongation factor 1A-like gene family members, negatively affect cell death and disease resistance in rice. J. Genet. Genom. 2017, 44, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, J.; Bao, F.; Zhang, X.; Yang, S. A gain-of-function mutation in the Arabidopsis disease resistance gene RPP4 confers sensitivity to low temperature. Plant Physiol. 2010, 154, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, U.; Yano, M.; Lin, H.; Ashikari, M.; Yamada, K. A Rice Spotted Leaf Gene, Spl7, Encodes a Heat Stress Transcription Factor Protein. Proc. Natl. Acad. Sci. USA. 2002, 99, 7530–7535. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.-R.; Qu, S.; Bordeos, A.; Yang, C.; Baraoidan, M.; Yan, H.; Xie, Q.; Nahm, B.H.; Leung, H.; Wang, G.-L. Spotted leaf11, a negative regulator of plant cell death and defense, encodes a U-box/armadillo repeat protein endowed with E3 ubiquitin ligase activity. Plant Cell 2004, 16, 2795–2808. [Google Scholar] [CrossRef]
- Lorrain, S.; Lin, B.; Auriac, M.C.; Kroj, T.; Saindrenan, P.; Nicole, M.; Balague, C.; Roby, D. vascular associated death1, a novel GRAM domain–containing protein, is a regulator of cell death and defense responses in vascular tissues. Plant Cell 2004, 16, 2217–2232. [Google Scholar] [CrossRef] [PubMed]
- Mosher, S.; Moeder, W.; Nishimura, N.; Jikumaru, Y.; Joo, S.H.; Urquhart, W.; Klessig, D.F.; Kim, S.K.; Nambara, E.; Yoshioka, K. The lesion-mimic mutant cpr22 shows alterations in abscisic acid signaling and abscisic acid insensitivity in a salicylic acid-dependent manner. Plant Physiol. 2010, 152, 1901–1913. [Google Scholar] [CrossRef]
- Kachroo, A.; Lapchyk, L.; Fukushige, H.; Hildebrand, D.; Klessig, D.; Kachroo, P. Plastidial fatty acid signaling modulates salicylic acid–and jasmonic acid–mediated defense pathways in the Arabidopsis ssi2 mutant. Plant Cell 2003, 15, 2952–2965. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Agrawal, G.K.; Yamazaki, M.; Onosato, K.; Miyao, A.; Kawasaki, T.; Shimamoto, K.; Hirochika, H. Rice Pti1a negatively regulates RAR1-dependent defense responses. Plant Cell 2007, 19, 2940–2951. [Google Scholar] [CrossRef]
- Qiao, Y.; Jiang, W.; Lee, J.; Park, B.; Choi, M.S.; Piao, R.; Woo, M.O.; Roh, J.H.; Han, L.; Paek, N.C. SPL28 encodes a clathrin-associated adaptor protein complex 1, medium subunit μ1 (AP1M1) and is responsible for spotted leaf and early senescence in rice (Oryza sativa). New Phytol. 2010, 185, 258–274. [Google Scholar] [CrossRef]
- Chen, X.; Hao, L.; Pan, J.; Zheng, X.; Jiang, G.; Jin, Y.; Gu, Z.; Qian, Q.; Zhai, W.; Ma, B. SPL5, a cell death and defense-related gene, encodes a putative splicing factor 3b subunit 3 (SF3b3) in rice. Mol. Breed. 2012, 30, 939–949. [Google Scholar] [CrossRef]
- Brodersen, P.; Petersen, M.; Pike, H.M.; Olszak, B.; Skov, S.; Ødum, N.; Jørgensen, L.B.; Brown, R.E.; Mundy, J. Knockout of Arabidopsis accelerated-cell-death11 encoding a sphingosine transfer protein causes activation of programmed cell death and defense. Genes Dev. 2002, 16, 490–502. [Google Scholar] [CrossRef]
- Zeng, Y.; Ma, L.; Ji, Z.; Wen, Z.; Li, X.; Shi, C.; Yang, C. Fine mapping and candidate gene analysis of LM3, a novel lesion mimic gene in rice. Biologia 2013, 68, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Liu, L.; Tang, J.; Lin, A.; Zhang, F.; Fang, J.; Zhang, G.; Chu, C. RLIN1, encoding a putative coproporphyrinogen III oxidase, is involved in lesion initiation in rice. J. Genet. Genom./Yi Chuan Xue Bao 2011, 38, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Araji, S.; Grammer, T.A.; Gertzen, R.; Anderson, S.D.; Mikulic-Petkovsek, M.; Veberic, R.; Phu, M.L.; Solar, A.; Leslie, C.A.; Dandekar, A.M.; et al. Novel roles for the polyphenol oxidase enzyme in secondary metabolism and the regulation of cell death in walnut. Plant Physiol 2014, 164, 1191–1203. [Google Scholar] [CrossRef]
- Jung, Y.; Rakwal, R.; Agrawal, G.; Shibato, J.; Kim, J.; Lee, M.; Choi, P.; Jung, S.; Kim, S.; Koh, H. Differential expression of defense/stress-related marker proteins in leaves of a unique rice blast lesion mimic mutant (blm). J. Proteome Res. 2006, 5, 2586–2598. [Google Scholar] [CrossRef]
- Kim, S.; Kim, S.; Kang, Y.; Wang, Y.; Kim, J.; Yi, N.; Kim, J.; Rakwal, R.; Hj Kang, K. Proteomics analysis of rice lesion mimic mutant (spill) reveals tightly localized Probenazole-Induced protein (PBZ1) in cells undergoing programmed cell death. J. Proteome Res. 2008, 7, 1750–1760. [Google Scholar] [CrossRef]
- Tsunezuka, H.; Fujiwara, M.; Kawasaki, T.; Shimamoto, K. Proteome analysis of programmed cell death and defense signaling using the rice lesion mimic mutant cdr2. Mol. Plant-Microbe Interact. 2005, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Matin, M.N.; Bae, H.; Natarajan, S. Proteome analysis and characterization of phenotypes of lesion mimic mutant spotted leaf 6 in rice. Proteomics 2007, 7, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Zhao, H. Protein quantitation using iTRAQ: Review on the sources of variations and analysis of nonrandom missingness. Stat. Interface 2012, 5, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhang, L.; Shang, H.; Liu, S.; Peng, J.; Gong, W.; Shi, Y.; Zhang, S.; Li, J.; Gong, J. iTRAQ-Based Quantitative Proteomic Analysis of Cotton Roots and Leaves Reveals Pathways Associated with Salt Stress. PLoS ONE 2016, 11, e0148487. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hajano, J.U.D.; Ren, Y.; Lu, C.; Wang, X. iTRAQ-based quantitative proteomics analysis of rice leaves infected by Rice stripe virus reveals several proteins involved in symptom formation. Virol. J. 2015, 12, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.J.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.; Chiapello, M.; Montero, H.; Gehrig, P.; Grossmann, J.; O’Holleran, K.; Hartken, D.; Walters, F.; Yang, S.Y.; Hillmer, S.; et al. A rice Serine/Threonine receptor-like kinase regulates arbuscular mycorrhizal symbiosis at the peri-arbuscular membrane. Nat. Commun. 2018, 9, 4677. [Google Scholar] [CrossRef]
- Wagstaff, C.; Leverentz, M.K.; Griffiths, G.; Thomas, B.; Chanasut, U.; Stead, A.D.; Rogers, H.J. Cysteine protease gene expression and proteolytic activity during senescence of Alstroemeria petals. J. Exp. Bot. 2002, 53, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.Y.; Gao, Y.; Guo, J.; Yu, T.F.; Zheng, W.J.; Liu, Y.W.; Chen, J.; Xu, Z.S.; Ma, Y.Z. BES/BZR Transcription Factor TaBZR2 Positively Regulates Drought Responses by Activation of TaGST1. Plant Physiol. 2019. [Google Scholar] [CrossRef]
- Mauch, F.; Mauch-Mani, B.; Boller, T. Antifungal Hydrolases in Pea Tissue: II. Inhibition of Fungal Growth by Combinations of Chitinase and beta-1,3-Glucanase. Plant Physiol. 1988, 88, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q. Lignification: Flexibility, Biosynthesis and Regulation. Trends Plant Sci. 2016, 21, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.F.; Balint-Kurti, P. Maize Homologs of CCoAOMT and HCT, Two Key Enzymes in Lignin Biosynthesis, Form Complexes with the NLR Rp1 Protein to Modulate the Defense Response. Plant Physiol. 2016, 171, 2166–2177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, C.J. Multifaceted regulations of gateway enzyme phenylalanine ammonia-lyase in the biosynthesis of phenylpropanoids. Mol. Plant 2015, 8, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Le, R.J.; Brigitte, H.; Anne, C.; Simon, H.; Godfrey, N. Glycosylation Is a Major Regulator of Phenylpropanoid Availability and Biological Activity in Plants. Front. Plant Sci. 2016, 7, 735. [Google Scholar]
- Ueno, M.; Kihara, J.; Arase, S. Tryptamine and sakuranetin accumulation in Sekiguchi lesions associated with the light-enhanced resistance of the lesion mimic mutant of rice to Magnaporthe oryzae. J. Gen. Plant Pathol. 2015, 81, 1–4. [Google Scholar] [CrossRef]
- Mochizuki, N.; Tanaka, R.; Grimm, B.; Masuda, T.; Moulin, M.; Smith, A.G.; Tanaka, A.; Terry, M.J. The cell biology of tetrapyrroles: A life and death struggle. Trends Plant Sci. 2010, 15, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Sarmientomañús, R.; Gonzálezbayón, R.; Hricová, A.; Ponce, M.R.; Micol, J.L. PORPHOBILINOGEN DEAMINASE Deficiency Alters Vegetative and Reproductive Development and Causes Lesions in Arabidopsis. PLoS ONE 2013, 8, e53378. [Google Scholar] [CrossRef]
- Hu, G.; Yalpani, N.; Briggs, S.P.; Johal, G.S. A porphyrin pathway impairment is responsible for the phenotype of a dominant disease lesion mimic mutant of maize. Plant Cell 1998, 10, 1095–1104. [Google Scholar] [CrossRef]
- Samol, I.; Rossig, C.; Buhr, F.; Springer, A.; Pollmann, S.; Lahroussi, A.; Wettstein, D.V.; Reinbothe, C.; Reinbothe, S. The Outer Chloroplast Envelope Protein OEP16-1 for Plastid Import of NADPH:Protochlorophyllide Oxidoreductase A in Arabidopsis thaliana. Plant Cell Physiol. 2011, 52, 96–111. [Google Scholar] [CrossRef]
- Rg, O.D.C.; Przybyla, D.; Ochsenbein, C.; Laloi, C.; Kim, C.; Danon, A.; Wagner, D.; Hideg, E.; Göbel, C.; Feussner, I. Rapid induction of distinct stress responses after the release of singlet oxygen in Arabidopsis. Plant Cell 2003, 15, 2320–2332. [Google Scholar]
- Mikkelsen, M.; Jorgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Von Wettstein, D.; Gough, S.; Kannangara, C.G. Chlorophyll Biosynthesis. Plant Cell 1995, 7, 1039–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Functional Proteins | References |
|---|---|---|
| 1 | Heat stress transcription factor | [18] |
| 2 | U-Box/Armadillo repeat protein | [19] |
| 3 | Membrane-associated protein | [3,20] |
| 4 | Ion channel | [21] |
| 5 | Zinc finger protein | [21] |
| 6 | Acyltransferase | [22] |
| 7 | Ser/Thr protein kinase | [23] |
| 8 | Clathrin associated adaptor protein complex 1 medium subunit 1 (AP1M1) | [24] |
| 9 | Putative splicing factor 3b subunit 3 (SF3b3) | [25] |
| 10 | Proteins involved in biosynthesis pathways of fatty acids lipids | [26] |
| 11 | Aromatic amino acid | [27] |
| 12 | Porphyrin | [28] |
| 13 | Polyphenol oxidase enzyme in secondary metabolism | [29] |
| Material | Dry Weight (g) | Plant Height (cm) | No. of Panicle | Grain Number | 1000-Grain Weight (g) | |||
|---|---|---|---|---|---|---|---|---|
| Seedling Stage | Tillering Stage | Maturity Stage | ||||||
| Field | E40 | - | - | 83.17 ± 7.71 ** | 98.7 ± 2.42 ** | 19.4 ± 4.62 * | 32.25 ± 1.71 ** | 17.68 ± 0.45 ** |
| WT | - | - | 247.62 ± 5.42 | 102.58 ± 1.05 | 23.2 ± 2.77 | 134.62 ± 2.84 | 23.6 ± 0.16 | |
| Greenhouse | E40 | 0.0224 ± 0.0017 | 4.15 ± 0.13 * | 119.36 ± 4.76 ** | 115.23 ± 1.79 ** | 21.3 ± 1.72 * | 47.05 ± 0.97 ** | 18.76 ± 0.42 ** |
| WT | 0.0235 ± 0.0021 | 5.00 ± 0.06 | 218.67 ± 5.49 | 117.58 ± 2.16 | 23.9 ± 1.05 | 118.13 ± 2.47 | 22.96 ± 0.24 | |
| Protein ID | Gene Name | Annotation | Log2 Fold Change |
|---|---|---|---|
| Q8S059 | SSI2 | Stearoyl-[acyl-carrier-protein] 9-desaturase 2 | −3.259320177 |
| Q84ZD2 | P0534A03.109 | Pentatricopeptide repeat-containing protein CRP1 homolog | −2.235369295 |
| Q2QVA7 | LOC_Os12g13460 | protein-lysine N-methyltransferase activity | −1.957389474 |
| B9F2U5 | Os02g0157700 | Promotes chloroplast protein synthesis | −1.670452917 |
| P0C512 | rbcL | Ribulose bisphosphate carboxylase large chain | −1.525042871 |
| Q69RJ0 | GLU | Ferredoxin-dependent glutamate synthase | −1.459112364 |
| O04882 | P0421H07.25 | Farnesyl diphosphate synthase | −1.418836635 |
| Q5NAI9 | P0456F08.15 | Putative OsFVE | −1.411837321 |
| Q69X42 | P0429G06.10 | glycine dehydrogenase (decarboxylating) activity | −1.372611128 |
| Q0JJY1 | Os01g0709400 | hydrolase activity | −1.329396063 |
| Q9LGB2 | P0504H10.32 | Putative wound-induced protease inhibitor | 1.251971273 |
| Q2QLS7 | LOC_Os12g43450 | P21 protein, putative | 1.325084219 |
| Q7XSU8 | OSJNBa0039K24.8 | Belongs to the peroxidase family | 1.339045312 |
| Q8W084 | OSJNBa0091E23.10 | Putative pathogenesis-related protein | 1.397933437 |
| Q0JR25 | RBBI3.3 | Bowman-Birk type bran trypsin inhibitor | 1.417877593 |
| Q9AWV5 | P0044F08.5 | serine-type endopeptidase inhibitor activity | 1.447168008 |
| Q5WMX0 | dip3 | Putative chitinase | 1.502285455 |
| Q0JMY8 | SALT | Salt stress-induced protein | 1.596525805 |
| Q75GR1 | OSJNBb0065L20.2 | * | 1.728124034 |
| Q306J3 | JAC1 | Dirigent protein | 1.770280249 |
| Pathway | Protein ID | Annotation | Sense Primer | Anti-sense Primer | Log2 Fold Change (E40/WT) | |
|---|---|---|---|---|---|---|
| iTRAQ | qRT-PCR | |||||
| Phenylpropanoid biosynthesis | Q5JMS4 | Peroxidase | GCCAACACCACCGTCAAC | TGGAAGAACGCCGACTGG | 1.16 | 0.89 |
| Q7XSV2 | Peroxidase | CTCATCCAGGCGTTCAAG | CTTCTTCACCAGCACAGG | 0.91 | 0.74 | |
| Q94DM2 | Class III peroxidase 22 | TTGTCGTTGGGCTACTAC | AACTTCTCGCTCTTCTCG | 0.71 | 0.46 | |
| Q9AS12 | Class III peroxidase 16 | TCTTCCTCTTCTTCGCCTTC | ACGCCGCTGTTGTTCTTG | 0.97 | 0.88 | |
| Porphyrin and chlorophyll metabolism | Q5Z8V9 | Delta-aminolevulinic acid dehydratase | ATTCCAGGAGACCACCATC | CATCACGAGACTTGTAGACC | −0.62 | −0.33 |
| Q6Z2T6 | Geranylgeranyl diphosphate reductase | AGGAAGGTGAGGAAGATG | CAGGAAGAGACCATTGAC | −0.75 | −0.42 | |
| Carbon fixation in photosynthetic organisms | P0C512 | Ribulose bisphosphate carboxylase large chain | GGCAGCATTCCGAGTAAC | AAGTCCATCAGTCCAAACAG | −1.53 | −1.57 |
| Q9SNK3 | Glyceraldehyde-3-phosphate dehydrogenase | GCGAAGAAGGTCATCATCAC | GAGCGAGGCAGTTGGTTG | −0.84 | −0.75 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.-B.; Meng, W.-L.; Zhao, M.-J.; Zhang, A.-X.; Liu, W.; Xu, Z.-S.; Wang, Y.-P.; Ma, J. Proteomics Analysis to Identify Proteins and Pathways Associated with the Novel Lesion Mimic Mutant E40 in Rice Using iTRAQ-Based Strategy. Int. J. Mol. Sci. 2019, 20, 1294. https://doi.org/10.3390/ijms20061294
Yang X-B, Meng W-L, Zhao M-J, Zhang A-X, Liu W, Xu Z-S, Wang Y-P, Ma J. Proteomics Analysis to Identify Proteins and Pathways Associated with the Novel Lesion Mimic Mutant E40 in Rice Using iTRAQ-Based Strategy. International Journal of Molecular Sciences. 2019; 20(6):1294. https://doi.org/10.3390/ijms20061294
Chicago/Turabian StyleYang, Xiang-Bo, Wei-Long Meng, Meng-Jie Zhao, An-Xing Zhang, Wei Liu, Zhao-Shi Xu, Yun-Peng Wang, and Jian Ma. 2019. "Proteomics Analysis to Identify Proteins and Pathways Associated with the Novel Lesion Mimic Mutant E40 in Rice Using iTRAQ-Based Strategy" International Journal of Molecular Sciences 20, no. 6: 1294. https://doi.org/10.3390/ijms20061294