Novel Benzene-Based Carbamates for AChE/BChE Inhibition: Synthesis and Ligand/Structure-Oriented SAR Study

 , , , ,
, , , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
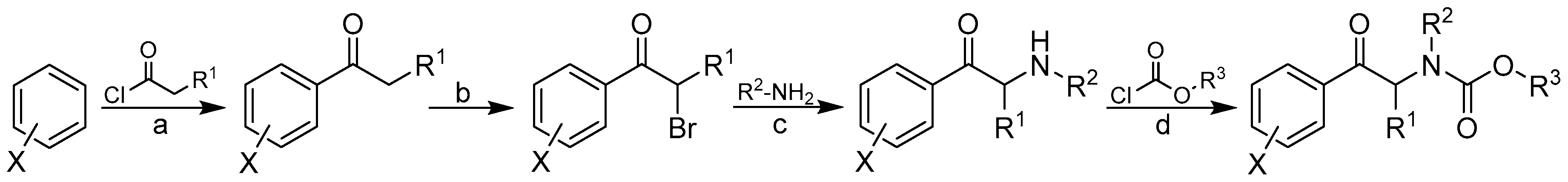
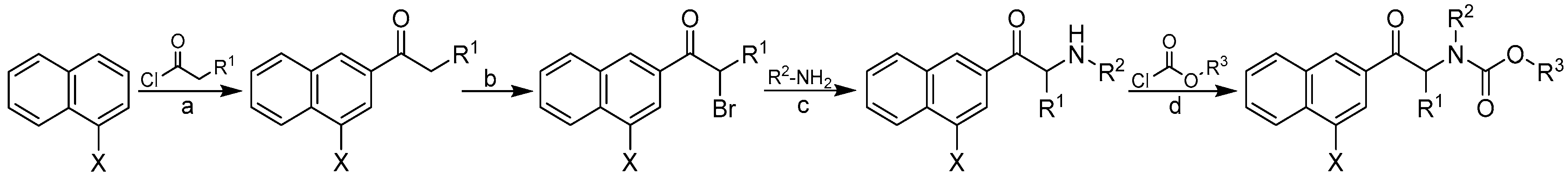
2.1. Design and Synthesis
2.2. In Vitro Assessment of the AChE- and BChE-Inhibitory Profiles
2.3. In Silico Evaluation of AChE- and BChE-Inhibitory Profile
2.4. Probability-Guided Pharmacophore Mapping
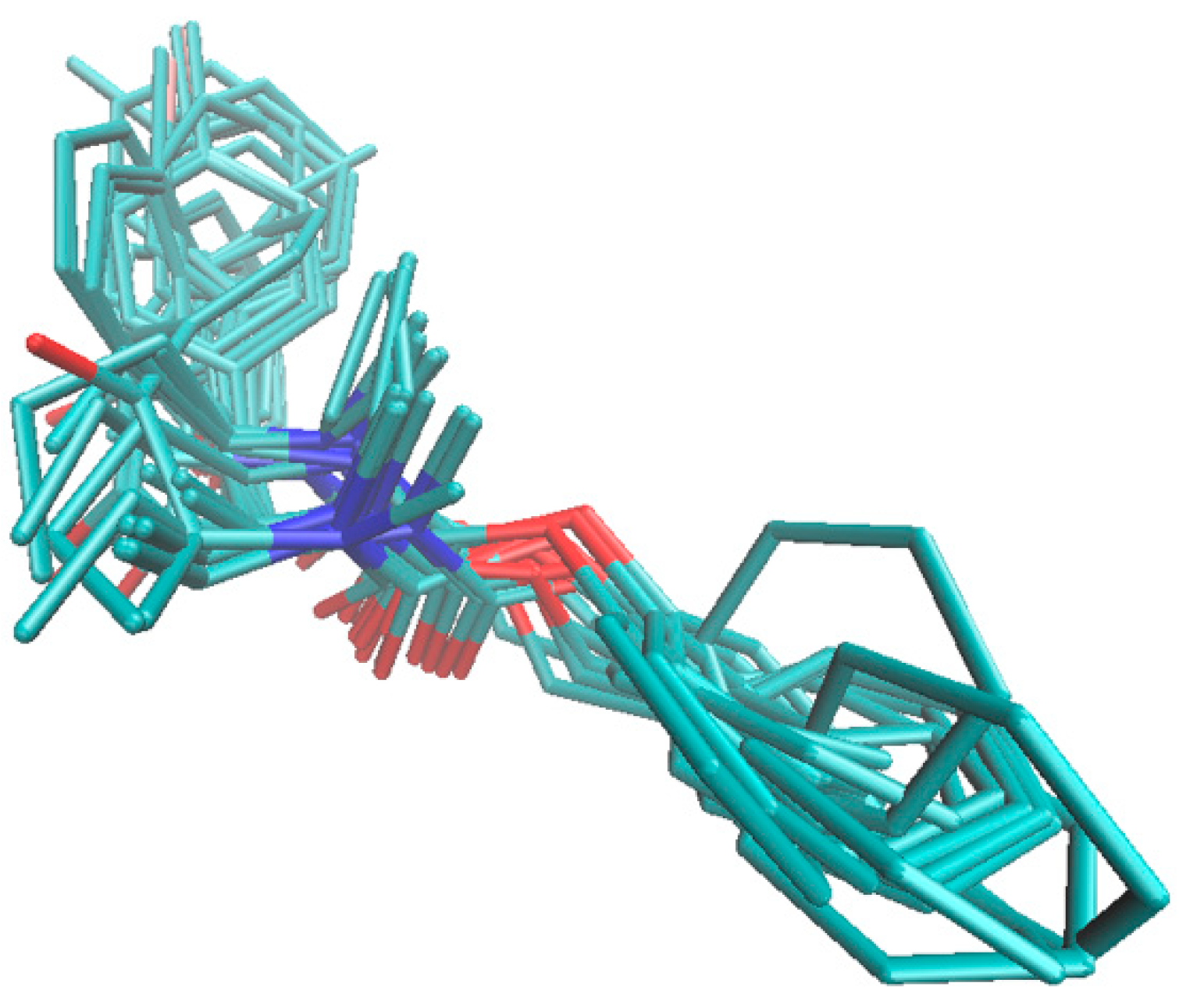
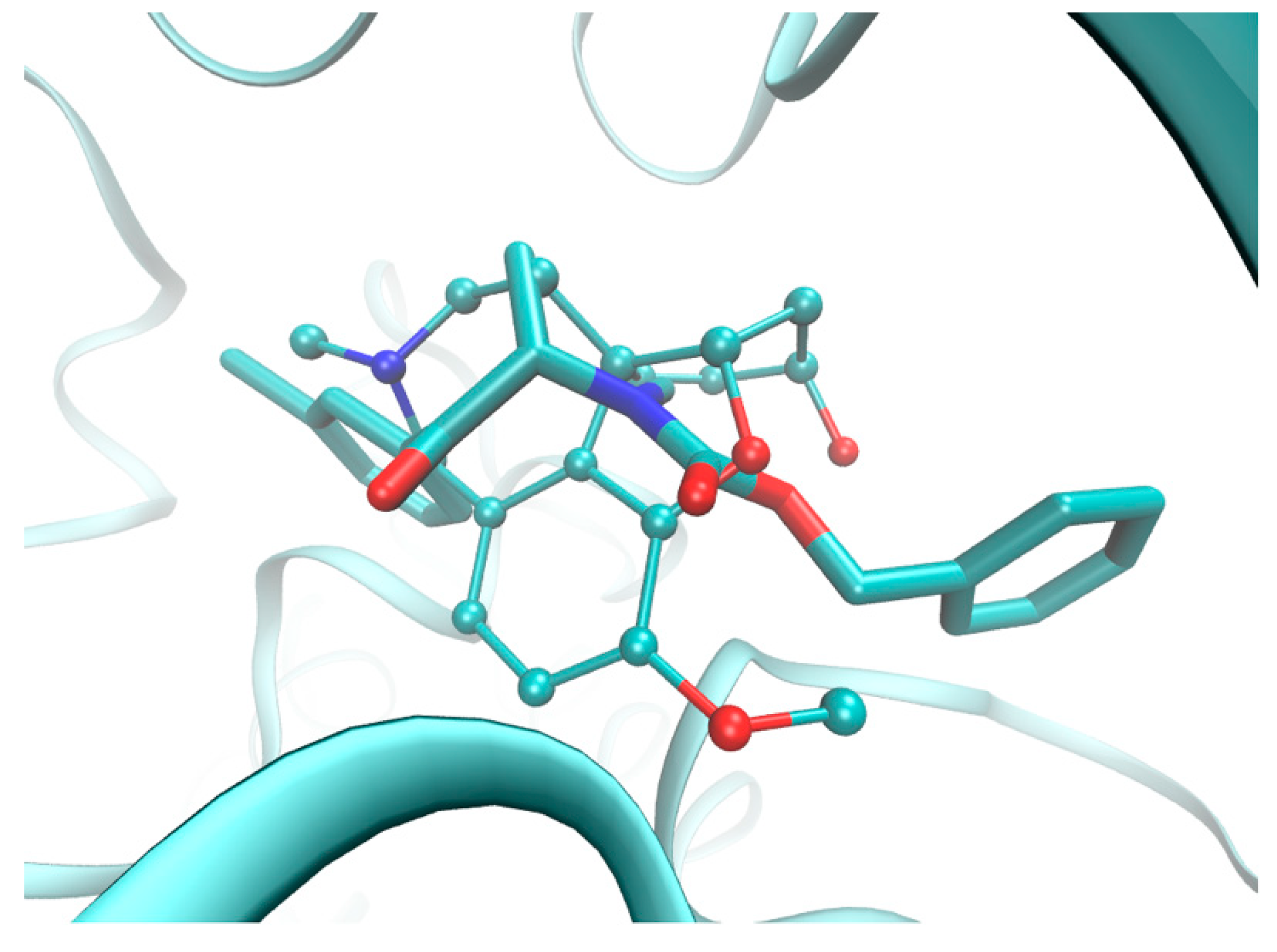
2.5. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure Used to Synthesize Carbamates 1–41
3.3. Evaluating the In Vitro AChE- and BChE-Inhibition Potency
3.4. Comparative Molecular Surface Analysis using Iterative PLS-Based Variable Elimination
3.5. Building the Model and Molecular Modeling
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| BChE | Butyrylcholinesterase |
| CAMD | Computer Asisted Molecular Design |
| ADMET | Absorption Distribution Metabolism Excretion Toxicity |
| CoMSA | Comparative Molecular Surface Analysis |
| SMV | Stochastic Model Validation |
| RIV | Rivastigmine |
| GLT | Galanthamine |
| PCA | Principal Component Analysis |
| IVE-PLS | Iterative Variable Elimination Partial Least Squares |
| PLIP | Protein Ligand Interaction Profiler |
References
- Lemke, T.L.; Williams, D.A. Foye’s Principles of Medicinal Chemistry, 7th ed.; Lippincott Williams & Wilkins and Wolters Kluwer: Baltimore, MD, USA, 2013. [Google Scholar]
- Ghosh, A.K.; Brindisi, M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J.; Brychtova, K. Azone analogues: Classification, design, and transdermal penetration principles. Med. Res. Rev. 2012, 32, 907–947. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Angelina, E.; Lima, S.; Gonec, T.; Otevrel, J.; Marvanova, P.; Padrtova, T.; Mokry, P.; Bobal, P.; Acosta, L.M.; et al. Search of new structural scaffolds for sphingosine kinase 1 inhibitors. Eur. J. Med. Chem. 2017, 139, 461–481. [Google Scholar] [CrossRef]
- Imramovsky, A.; Pesko, M.; Monreal-Ferriz, J.; Kralova, K.; Vinsova, J.; Jampilek, J. Photosynthesis-inhibiting efficiency of 4-chloro-2-(chlorophenylcarbamoyl)phenyl alkylcarbamates. Bioorg. Med. Chem. Lett. 2011, 21, 4564–4567. [Google Scholar] [CrossRef] [PubMed]
- Zadrazilova, I.; Pospisilova, S.; Masarikova, M.; Imramovsky, A.; Monreal-Ferriz, J.; Vinsova, J.; Cizek, A.; Jampilek, J. Salicylanilide Carbamates: Promising antibacterial agents with high in vitro activity against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Pharm. Sci. 2015, 77, 197–207. [Google Scholar] [CrossRef]
- Kos, J.; Nevin, E.; Soral, M.; Kushkevych, I.; Gonec, T.; Bobal, P.; Kollar, P.; Coffey, A.; O’Mahony, J.; Liptaj, T.; et al. Synthesis and antimycobacterial properties of ring-substituted 6-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2015, 23, 2035–2043. [Google Scholar] [CrossRef]
- Kauerova, T.; Kos, J.; Gonec, T.; Jampilek, J.; Kollar, P. Antiproliferative and pro-apoptotic effect of novel nitro-substituted hydroxynaphthanilides on human cancer cell lines. Int. J. Mol. Sci. 2016, 17, 1219. [Google Scholar] [CrossRef] [PubMed]
- Pospisilova, S.; Kos, J.; Michnova, H.; Kapustikova, I.; Strharsky, T.; Oravec, M.; Moricz, A.M.; Bakonyi, J.; Kauerova, T.; Kollar, P.; et al. Synthesis and spectrum of biological activities of novel N-arylcinnamamides. Int. J. Mol. Sci. 2018, 19, 2318. [Google Scholar] [CrossRef]
- Pizova, H.; Havelkova, M.; Stepankova, S.; Bak, A.; Kauerova, T.; Kozik, V.; Oravec, M.; Imramovsky, A.; Kollar, P.; Bobal, P.; et al. Proline-based carbamates as cholinesterase inhibitors. Molecules 2017, 14, 1969. [Google Scholar] [CrossRef]
- Moss, D.E.; Perez, R.G.; Kobayashi, H. Cholinesterase inhibitor therapy in Alzheimer’s disease: The limits and tolerability of irreversible CNS-selective acetylcholinesterase inhibition in primates. J. Alzheimers Dis. 2017, 55, 1285–1294. [Google Scholar] [CrossRef]
- Bajic, V.; Milovanovic, E.S.; Spremo-Potparevic, B.; Zivkovic, L.; Miliccivc, Z.; Stanimirovic, J.; Bogdanovic, N.; Isenovic, E.R. Treatment of Alzheimer’s Disease: Classical therapeutic approach. Curr. Pharm. Anal. 2016, 12, 82–90. [Google Scholar] [CrossRef]
- Skrzypek, A.; Matysiak, J.; Niewiadomy, A.; Bajda, M.; Szymański, P. Synthesis and biological evaluation of 1,3,4-thiadiazole analogues as novel AChE and BuChE inhibitors. Eur. J. Med. Chem. 2013, 62, 311–319. [Google Scholar] [CrossRef]
- Kumar, J.; Meena, P.; Singh, A.; Jameel, E.; Maqbool, M.; Mobashir, M.; Shandilya, A.; Tiwari, M.; Hoda, N.; Jayaram, B. Synthesis and screening of triazolopyrimidine scaffold as multi-functional agents for Alzheimer’s disease therapies. Eur. J. Med. Chem. 2016, 119, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Rao, P.P. 2,4-Disubstituted quinazolines as amyloid-β aggregation inhibitors with dual cholinesterase inhibition and antioxidant properties: Development and structure-activity relationship (SAR) studies. Eur. J. Med. Chem. 2017, 126, 823–843. [Google Scholar] [CrossRef] [PubMed]
- Knez, D.; Brus, B.; Coquelle, N.; Sosic, I.; Sink, R.; Brazzolotto, X.; Mravljak, J.; Colletier, J.P.; Gobec, S. Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorg. Med. Chem. 2015, 23, 4442–4452. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.A.; Gutierrez, M.; Ramirez, D.; Alzate-Morales, J.; Bernal, C.C.; Guiza, F.M.; Romero Bohorquez, A.R. Novel N-allyl/propargyl tetrahydroquinolines: Synthesis via three-component cationic imino Diels-Alder reaction, binding prediction, and evaluation as cholinesterase inhibitors. Chem. Biol. Drug Des. 2016, 88, 498–510. [Google Scholar] [CrossRef]
- Kozakiewicz, D.; Polanski, J.; Jampilek, J.; Imramovsky, A.; Stepankova, S. New Carbamate Derivatives and Their Application. U.S. Patent 420626, 23 February 2017. [Google Scholar]
- Davis, B.J.; Erlanson, D.A. Learning from our mistakes: The ‘unknowns’ in fragment screening. Bioorg. Med. Chem. Lett. 2013, 23, 2844–2852. [Google Scholar] [CrossRef]
- Kenny, P.W. Comment on the ecstasy and agony of assay interference compounds. J. Chem. Inf. Model. 2017, 57, 2640–2645. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Salvino, J.M.; Mallamo, J.P. Knowledge-based chemoinformatic approaches to drug discovery. Drug Discov. Today 2006, 11, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Stanton, D.T. QSAR and QSPR model interpretation using partial least squares (PLS) analysis. Curr. Comput. Aided Drug Des. 2012, 8, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. Multidimensional (3D/4D-QSAR) probability-guided pharmacophore mapping: Investigation of activity profile for a series of drug absorption promoters. RSC Adv. 2016, 6, 76183–76205. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Zentgraf, M.; Steuber, H.; Koch, C.; La Motta, C.; Sartini, S.; Sotriffer, C.A.; Klebe, G. How reliable are current docking approaches for structure-based drug design? Lessons from aldose reductase. Angew. Chem. Int. Ed. Engl. 2007, 46, 3575–3580. [Google Scholar] [CrossRef]
- Mazur, P.; Magdziarz, T.; Bak, A.; Chilmonczyk, Z.; Kasprzycka-Guttman, T.; Misiewicz-Krzeminska, I.; Skupinska, K.; Polanski, J. Does molecular docking reveal alternative chemopreventive mechanism of activation of oxidoreductase by sulforaphane isothiocyanates? J. Mol. Model. 2010, 16, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Irwin, J.J.; Webb, B.M.; Klebe, G.; Shoichet, B.K.; Sali, A. Molecular docking screens using comparative models of proteins. J. Chem. Inf. Model. 2009, 49, 2512–2527. [Google Scholar] [CrossRef]
- Soler, M.A.; de Marco, A.; Fortunac, S. Molecular dynamics simulations and docking enable to explore the biophysical factors controlling the yields of engineered nanobodies. Sci Rep. 2016, 6, 34869. [Google Scholar] [CrossRef] [Green Version]
- Pejchal, V.; Stepankova, S.; Padelkova, Z.; Imramovsky, A.; Jampilek, J. 1,3-Substituted Imidazolidine-2,4,5-triones: Synthesis and inhibition of cholinergic enzymes. Molecules 2011, 16, 7565–7582. [Google Scholar] [CrossRef]
- Imramovsky, A.; Stepankova, S.; Vanco, J.; Pauk, K.; Monreal-Ferriz, J.; Vinsova, J.; Jampilek, J. Acetylcholinesterase-inhibiting activity of salicylanilide N-alkylcarbamates and their molecular docking. Molecules 2012, 17, 10142–10158. [Google Scholar] [CrossRef] [PubMed]
- Imramovsky, A.; Pejchal, V.; Stepankova, S.; Vorcakova, K.; Jampilek, J.; Vanco, J.; Simunek, P.; Kralovec, K.; Bruckova, L.; Mandikova, J.; et al. Synthesis and in vitro evaluation of new derivatives of 2-substituted-6-fluorobenzo[d]thiazoles as cholinesterase inhibitors. Bioorg. Med. Chem. 2013, 21, 1735–1748. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.; Polanski, J. Modeling robust QSAR 3: SOM-4D-QSAR with iterative variable elimination IVE-PLS: Application to steroid, azo dye, and benzoic acid series. J. Chem. Inf. Model. 2007, 47, 1469–1480. [Google Scholar] [CrossRef]
- Bak, A.; Kozik, V.; Walczak, M.; Fraczyk, J.; Kaminski, Z.; Kolesinska, B.; Smolinski, A.; Jampilek, J. Towards intelligent drug design system: Application of artificial dipeptide receptor library in QSAR-oriented studies. Molecules 2018, 23, 1964. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.; Oprea, T. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef]
- Kubinyi, H. Hansch Analysis and Related Approaches; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1993. [Google Scholar]
- Bak, A.; Kozik, V.; Malik, I.; Jampilek, J.; Smolinski, A. Probability-driven 3D pharmacophore mapping of antimycobacterial potential of hybrid molecules combining phenylcarbamoyloxy and N-arylpiperazine fragments. SAR QSAR Environ. Res. 2018, 29, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2010. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2000. [Google Scholar]
- Polanski, J.; Bak, A.; Gieleciak, R.; Magdziarz, T. Modeling robust QSAR. J. Chem. Inf. Model. 2003, 46, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Irwin, J.J. Docking screens: Right for the right reasons? Curr. Top. Med. Chem. 2009, 9, 755–770. [Google Scholar] [CrossRef]
- Colquhoun, D. The quantitative analysis of drug–receptor interactions: A short history. Trends Pharmacol. Sci. 2006, 27, 149–157. [Google Scholar] [CrossRef]
- Levoin, N.; Calmels, T.; Poupardin-Olivier, O.; Labeeuw, O.; Danvy, D.; Robert, P.; Berrebi-Bertrand, I.; Ganellin, C.R.; Schunack, W.; Stark, H.; et al. Refined docking as a valuable tool for lead optimization: Application to histamine H3 receptor antagonists. Arch. Pharm. Chem. Life Sci. 2008, 341, 610–623. [Google Scholar] [CrossRef]
- Devillers, J. Methods for building QSARs. Methods Mol. Biol. 2013, 930, 3–27. [Google Scholar]
- Bak, A.; Wyszomirski, M.; Magdziarz, T.; Smolinski, A.; Polanski, J. Structure-based modeling of dye-fiber affinity with SOM-4D-QSAR paradigm: Application to set of anthraquinone derivatives. Comb. Chem. High Throughput Screen. 2014, 17, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, 443–447. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Mod. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Chen, Y.C. Beware of docking. Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.; Kwok, K.C.; Wang, Y.; Bao, H. An improved method to determine SH and –S–S– group content in soymilk protein. Food Chem. 2004, 88, 317–320. [Google Scholar] [CrossRef]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar] [CrossRef]
- Zdrazilova, P.; Stepankova, S.; Komers, K.; Ventura, K.; Cegan, A. Half-inhibition concentrations of new cholinesterase inhibitors. Zeitschrift für Naturforschung C 2004, 59, 293–296. [Google Scholar] [CrossRef]
- Zupan, J.; Gasteiger, J. Neural Networks and Drug Design for Chemists, 2nd ed.; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Bak, A.; Kozik, V.; Smolinski, A.; Jampilek, J. In silico estimation of basic activity-relevant parameters for a set of drug absorption promoters. SAR QSAR Environ. Res. 2017, 28, 427–449. [Google Scholar] [CrossRef]
- Centner, V.; Massart, D.L.; de Noord, O.E.; de Jong, S.; Vandeginste, B.M.V.; Sterna, C. Elimination of uninformative variables for multivariate calibration. Anal. Chem. 1996, 68, 3851–3858. [Google Scholar] [CrossRef]
- Smolinski, A.; Drobek, L.; Dombek, V.; Bak, A. Modeling of experimental data on trace elements and organic compounds content in industrial waste dumps. Chemosphere 2016, 162, 189–198. [Google Scholar] [CrossRef]
- Likus-Cieslik, J.; Smolinski, A.; Pietrzykowski, M.; Bak, A. Sulphur contamination impact on seasonal and surface water chemistry on a reforested area of a former sulphur mine. Land Degrad. Dev. 2019, 30, 212–225. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Benzene derivatives | |||||||
| No. | X | R1 | R2 | R3 | AChE IC50 [µM] | BChE IC50 [µM] | SI * |
| methyl carbamates | |||||||
| 1. | H | CH3 | C2H5 | CH3 | 245.33 ± 0.75 | 227.91 ± 0.90 | 1.08 |
| 2. | H | C2H5 | CH3 | CH3 | 328.86 ± 9.72 | 440.56 ± 7.54 | 0.75 |
| 3. | H | C2H5 | C2H5 | CH3 | 273.74 ± 2.16 | 362.87 ± 7.41 | 0.75 |
| 4. | 3-CH3 | CH3 | CH3 | CH3 | 133.98 ± 2.30 | 115.89 ± 2.16 | 1.16 |
| 5. | 3-Cl | CH3 | CH3 | CH3 | 329.68 ± 42.06 | 113.61 ± 0.29 | 2.90 |
| 6. | 4-OCH3 | CH3 | CH3 | CH3 | 146.91 ± 1.51 | 186.60 ± 1.55 | 0.79 |
| 7. | 4-CH3 | CH3 | CH3 | CH3 | 50.63 ± 0.63 | 180.77 ± 11.99 | 0.28 |
| 8. | 4-Cl | CH3 | CH3 | CH3 | 308.80 ± 6.16 | 131.57 ± 4.55 | 2.35 |
| 9. | 4-Br | CH3 | CH3 | CH3 | 282.74 ± 0.71 | 224.08 ± 2.19 | 1.26 |
| 10. | 3,4-OCH2O | CH3 | CH3 | CH3 | 225.46 ± 2.72 | 194.93 ± 1.93 | 1.16 |
| phenyl carbamates | |||||||
| 11. | H | CH3 | C2H5 | Ph | 166.72 ± 1.75 | 27.64 ± 0.55 | 6.03 |
| 12. | H | C2H5 | CH3 | Ph | 179.02 ± 6.21 | 52.96 ± 1.06 | 3.38 |
| 13. | H | C2H5 | C2H5 | Ph | 95.69 ± 5.37 | 15.62 ± 0.06 | 6.13 |
| 14. | H | C3H7 | CH3 | Ph | 97.97 ± 0.36 | 16.55 ± 0.13 | 5.92 |
| 15. | 3-CH3 | CH3 | CH3 | Ph | 165.62 ± 2.75 | 48.29 ± 0.31 | 3.43 |
| 16. | 3-Cl | CH3 | CH3 | Ph | 108.81 ± 1.10 | 14.81 ± 0.01 | 7.35 |
| 17. | 4-OCH3 | CH3 | CH3 | Ph | 90.21 ± 0.73 | 105.73 ± 1.94 | 0.85 |
| 18. | 4-CH3 | CH3 | CH3 | Ph | 138.64 ± 1.45 | 87.63 ± 2.37 | 1.58 |
| 19. | 4-Cl | CH3 | CH3 | Ph | 44.24 ± 2.35 | 38.96 ± 0.28 | 1.14 |
| 20. | 4-Br | CH3 | CH3 | Ph | 51.46 ± 1.38 | 44.05 ± 1.71 | 1.17 |
| 21. | 3,4-CH3 | CH3 | CH3 | Ph | 47.95 ± 2.09 | 26.41 ± 0.05 | 1.82 |
| 22. | 3,4-OCH2O | CH3 | CH3 | Ph | 84.00 ± 0.43 | 53.56 ± 0.72 | 1.57 |
| benzyl carbamates | |||||||
| 23. | H | CH3 | C2H5 | Bn | 158.41 ± 0.87 | 11.37 ± 0.05 | 13.93 |
| 24. | H | C2H5 | CH3 | Bn | 104.15 ± 2.91 | 21.38 ± 0.26 | 4.87 |
| 25. | H | C2H5 | C2H5 | Bn | 119.47 ± 1.23 | 21.29 ± 0.48 | 5.61 |
| 26. | H | C3H7 | CH3 | Bn | 134.65 ± 0.70 | 16.30 ± 0.17 | 8.26 |
| 27. | 3-CH3 | CH3 | CH3 | Bn | 83.32 ± 2.49 | 12.50 ± 0.24 | 6.67 |
| 28. | 3-Cl | CH3 | CH3 | Bn | 84.37 ± 2.27 | 5.51 ± 0.20 | 15.31 |
| 29. | 4-OCH3 | CH3 | CH3 | Bn | 62.57 ± 0.65 | 57.39 ± 0.02 | 1.09 |
| 30. | 4-CH3 | CH3 | CH3 | Bn | 40.06 ± 0.01 | 26.43 ± 0.40 | 1.52 |
| 31. | 4-Cl | CH3 | CH3 | Bn | 52.83 ± 0.34 | 7.02 ± 0.15 | 7.53 |
| 32. | 4-Br | CH3 | CH3 | Bn | 32.01 ± 0.54 | 11.28 ± 0.15 | 2.84 |
| 33. | 3,4-CH3 | CH3 | CH3 | Bn | 76.78 ± 2.76 | 16.87 ± 0.22 | 4.55 |
| 34. | 3,4-OCH2O | CH3 | CH3 | Bn | 75.57 ± 1.25 | 51.89 ± 1.18 | 1.46 |
Naphthalene derivatives | |||||||
| methyl carbamates | |||||||
| 35. | H | CH3 | CH3 | CH3 | 126.82 ± 1.74 | 150.36 ± 7.13 | 0.84 |
| phenyl carbamates | |||||||
| 36. | H | CH3 | CH3 | Ph | 99.58 ± 5.88 | 12.72 ± 0.19 | 7.83 |
| 37. | Cl | CH3 | CH3 | Ph | 30.64 ± 1.57 | 13.54 ± 0.47 | 2.26 |
| 38. | Br | CH3 | CH3 | Ph | 61.68 ± 1.42 | 12.97 ± 0.28 | 4.76 |
| benzyl carbamates | |||||||
| 39. | H | CH3 | CH3 | Bn | 105.20 ± 0.99 | 18.94 ± 0.18 | 5.55 |
| 40. | Cl | CH3 | CH3 | Bn | 103.28 ± 0.41 | 42.75 ± 0.31 | 2.42 |
| 41. | Br | CH3 | CH3 | Bn | 128.27 ± 0.88 | 235.91 ± 16.3 | 0.54 |
| RIV | 56.1 ± 1.41 | 38.4 ± 1.97 | 1.46 | ||||
| GLT | 1.54 ± 0.02 | 2.77 ± 0.15 | 0.56 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bak, A.; Kozik, V.; Kozakiewicz, D.; Gajcy, K.; Strub, D.J.; Swietlicka, A.; Stepankova, S.; Imramovsky, A.; Polanski, J.; Smolinski, A.; et al. Novel Benzene-Based Carbamates for AChE/BChE Inhibition: Synthesis and Ligand/Structure-Oriented SAR Study. Int. J. Mol. Sci. 2019, 20, 1524. https://doi.org/10.3390/ijms20071524
Bak A, Kozik V, Kozakiewicz D, Gajcy K, Strub DJ, Swietlicka A, Stepankova S, Imramovsky A, Polanski J, Smolinski A, et al. Novel Benzene-Based Carbamates for AChE/BChE Inhibition: Synthesis and Ligand/Structure-Oriented SAR Study. International Journal of Molecular Sciences. 2019; 20(7):1524. https://doi.org/10.3390/ijms20071524
Chicago/Turabian StyleBak, Andrzej, Violetta Kozik, Dariusz Kozakiewicz, Kamila Gajcy, Daniel Jan Strub, Aleksandra Swietlicka, Sarka Stepankova, Ales Imramovsky, Jaroslaw Polanski, Adam Smolinski, and et al. 2019. "Novel Benzene-Based Carbamates for AChE/BChE Inhibition: Synthesis and Ligand/Structure-Oriented SAR Study" International Journal of Molecular Sciences 20, no. 7: 1524. https://doi.org/10.3390/ijms20071524