Ion Channels Involved in Substance P-Mediated Nociception and Antinociception



1
Institute of Biomedical Sciences, Academia Sinica, Taipei 115, Taiwan
2
Taiwan Mouse Clinic, National Comprehensive Mouse Phenotyping and Drug Testing Center, Taipei 115, Taiwan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(7), 1596; https://doi.org/10.3390/ijms20071596
Submission received: 5 March 2019
/
Revised: 26 March 2019
/
Accepted: 27 March 2019
/
Published: 30 March 2019
(This article belongs to the Special Issue Ion Channels of Nociception)
Abstract
:Substance P (SP), an 11-amino-acid neuropeptide, has long been considered an effector of pain. However, accumulating studies have proposed a paradoxical role of SP in anti-nociception. Here, we review studies of SP-mediated nociception and anti-nociception in terms of peptide features, SP-modulated ion channels, and differential effector systems underlying neurokinin 1 receptors (NK1Rs) in differential cell types to elucidate the effect of SP and further our understanding of SP in anti-nociception. Most importantly, understanding the anti-nociceptive SP-NK1R pathway would provide new insights for analgesic drug development.
1. Background
Substance P (SP) was first described by von Euler and Gaddum, in 1931 [1]. The authors observed an unknown substance that stimulated contraction of the intestine ex vivo. This substance was identified and Euler and Gaddum named it substance P, from the bottle containing it, labeled P1, P2 etc., meaning “powder”. In the 1970s, SP was homogeneously purified by Chang and Leeman [2] and was later determined to be an 11 amino-acid peptide, H-Arg Pro Lys Pro Gln Gln Phe Phe Gly Leu Met-NH2 (RPKPQQFFGLM), with an amidation at the C-terminus [3]. SP belongs to the tachykinin family and serves as a neurotransmitter and a neuromodulator. It is encoded by preprotachykinin-1 (or tachykinin 1 (TAC1)) that produces SP and neurokinin A via alternative slicing and post-translational modifications [4].
SP is widely distributed in the human body, especially in nervous systems and inflammatory cells. A general assumption of the SP action describes SP as a neuropeptide released from pain-sensing fibers (nociceptors) to increase pain sensitivity through its actions in the dorsal horn of the spinal cord [5]. SP also triggers proinflammatory cytokine release resulting in inflammation, vasodilation and plasma extravasation [6]. Although considerable evidence indicates that SP transmits pain signaling and serves as a mediator of pain [7,8,9], accumulating studies reveal that SP also has an anti-nociceptive effect [10,11]. Interestingly, evidence showing the antinociceptive role of SP has been built over several years and can be dated back to 1976 [10]. The discrepant function of SP is believed to depend on the cell type regarding the expression of neurokinin receptors with unique underlying effector systems modulating differential ion channels.
2. SP-Mediated Signaling
There are three tachykinin receptors: Neurokinin 1 (NK1), 2 (NK2), and 3 (NK3). SP preferentially binds to NK1 receptor (NK1R). NKRs are G-protein-coupled receptors located in both the central nervous system (CNS) and peripheral nervous system (PNS). G proteins have Gα, Gβ, and Gγ subunits. The Gα subunit is classified into 5 families (Gs, Gi, Go, Gq/11, and G12/13). In most cases, NK1R is coupled to the pertussis toxin-insensitive Gq/11 cascade [12,13]; nevertheless, crosstalk with other G proteins such as G12/13 [14], Go, and Gs [15] has been reported. In Gq/11-mediated signaling, phospholipase C is activated by Gβγ binding, which in turn results in hydrolysis of membrane phospholipid PtdIns(4,5)P2 to form diacylglycerol (DAG) and inositol triphosphate (IP3). IP3 further triggers calcium release in cytoplasm from sarcoplasmic reticulum, whereas DAG activates protein kinase C (PKC) and results in calcium influx from the L-type calcium channel in the plasma membrane [16]. The increase in calcium ion in the cytoplasm further leads to an array of cell responses.
There are two natural forms of NK1R: A full-length receptor with 407 amino acids and a truncated receptor with 311 amino acids, lacking 96 amino acids in the carboxyl terminal [17,18]. The human NK1R gene contains five exons. The truncated version is generated when the intron between exons 4 and 5 is not removed that encounters a premature stop codon before the start of exon 5. These two types of NK1Rs have differential features. The binding affinity of SP is 10 times less to the truncated NK1R than the full-length form. Furthermore, the underlying signaling of the two different NK1Rs differs. The carboxyl terminus of full-length NK1R is a crucial element for G-protein coupling. The truncated form of NK1R impairs the ability of G-protein binding and results in G-protein-independent mechanisms. Indeed, the truncated form of NK1R fails to interact with β-arrestin, an important protein mediating the desensitization and internalization of activated G-protein-coupled receptors [19]. Furthermore, activation of the truncated NK1R has different effects on calcium mobilization, phosphorylation of PKCδ, extracellular signal-regulated kinase 1/2, and regulation of interleukin 8 mRNA expression as compared with the full-length NK1R [20].
The two NK1Rs are differentially distributed in peripheral tissues and in the nervous system. In the brain, full-length form is abundantly expressed in striatum, caudate nucleus, putamen, globus pallidus, nucleus accumbent, and hypothalamus; whereas the truncated NK1R expression is relatively low in the brain, and most represented in the PNS and peripheral tissues including heart, lung, prostate, and bone [21]. To sum up, the different NK1R subtypes could trigger differential effector systems and further result in distinct cellular responses in different tissues and organs.
3. SP and Pain
Pain is an unpleasant sensation and can be divided into two major categories: Acute and chronic pain. Acute pain lasts only short time and can be ameliorated over time. Acute pain helps us avoid the physical damage to our body and serves as a warning signal. In contrast, chronic pain has no biological function and is a disease itself rather than symptom of a disease. Chronic pain usually lasts for more than six months. People with chronic pain represent about 10.1% to 55.2% of the population in various countries according to epidemiological study [22]. People with chronic pain often show other symptoms, such as fatigue, sleep disorder, memory problems, anxiety, and depression, which greatly affect their quality of life.
Chronic pain can be further divided into two subtypes: Inflammatory and neuropathic pain. Inflammatory pain is related to tissue injuries, which lead to inflammatory reactions. In contrast, neuropathic pain results from nerve injury and neuronal sensitization in the CNS and PNS. However, inflammatory pain and neuropathic pain are also closely correlated. Nerve injuries result in tissue inflammatory responses, which lead to inflammatory pain. Likewise, tissue inflammation triggers an inflammatory cascade releasing a variety of inflammatory mediators such as SP, calcitonin gene-related peptide, and neurokinin A to damage nerves [23,24]. Clinical studies have demonstrated that many forms of chronic pain have mixed components of inflammation and neuropathy [25].
Substantial evidence suggests that SP is the key element in neurogenic inflammation and has an important role in eliciting pain sensation in both the CNS and PNS. In the CNS, SP results in central sensitization by activating excitatory post-synaptic potential [26]. In the PNS, releasing SP from peripheral nociceptive nerve fibers can cause neurogenic inflammation in the skin [27,28]. In addition, people with fibromyalgia, a chronic pain disorder, showed elevated SP level in cerebrospinal fluid [29,30]. Chemical ablation of neurons expressing SP receptors in lamina I [31] and genetic disruption of the encoding gene of SP [32] or its receptor [33] reduced pain responses. Furthermore, a recent study showed that selectively ablating TAC1-expressing neurons in the spinal cord abolished the sustained pain but not the reflexive defensive response [34]. Together, these studies support that SP is an important signal molecule in pain transmission.
Thus, many studies have focused on developing selective NK1R antagonists as a potential analgesic drug. Although several preclinical studies showed an anti-nociceptive effect of NK1R antagonists, most clinical trials failed to show analgesia effects. The reasons for failure of most NK1R antagonists in clinical trials are still elusive. Several reasons for discrepancy between preclinical and clinical results have been proposed such as species differences in NKRs distribution [35], species differences in affinities to antagonists for NK1R [36,37,38], and the ability of animal models in predicting clinical pain [39]. The discovery of an antinociceptive effect of SP may also explain in part the failure of those clinical trials [10]. One hypothesis is that neurons innervating distinct locations may respond differently to SP—some neurons excited by SP and some inhibited by SP. Thus, elucidation of SP-mediated responses in different tissues and organs becomes a crucial step in developing specific NK1 antagonists as analgesic drugs.
4. SP-Mediated Anti-Nociception
The antinociceptive effect of SP was first reported in 1976 [10]. Although SP had been identified for more than four decades, in the early time, the impure natural SP contaminated by bradykinin or some other kinin-like compounds impeded SP research. In the 1970s, SP was homogenously synthesized. With purely synthetic SP, the effect of SP could be clearly deciphered. Stewart et al. first reported that SP treatment by intracerebral and intraperitoneal injection could produce naloxone-reversible analgesia, and the site of action was in the CNS [9]. Subsequently, several studies confirmed the SP-mediated anti-nociception via opioid receptors in the CNS, and SP seemed to be a regulatory peptide to normalize the responses to pain stimuli. In 1978, Frederickson et al. claimed that a small amount of SP (1.25 to 5 ng per mouse) by intracerebroventricular injection produced a naloxone-reversed anti-nociception effect in mice [32]. However, higher doses (>50 ng per mouse) caused hyperalgesia. The authors also reported that although the C-terminus of SP (SP6–11) is very similar to that of endogenous opioid peptides, neither SP nor the SP6–11 acted on opioid receptors. At low doses, SP triggered the release of endorphins but at higher doses, directly excited neurons in the brain [40]. In 1980, Oehme et al. suggested that SP produced naloxone-reversed analgesia in mice with high sensitivity to thermal stimulation but induced hyperalgesia in mice with low sensitivity to thermal stimulation [41]. In addition, SP has been found to effectively reduce neuropathic pain [42] and inflammatory pain [43]. To sum up, these studies demonstrated that SP can regulates opioid-dependent analgesic effects in distinct cell types, probably via different receptors.
Because it has also been reported that the C-terminus of SP is sufficient for biological activities in nociception [44], a certain active fragment of SP may be essential for anti-nociception. Indeed, evidence has shown that the N-terminal and C-terminal domains of SP have opposite functions. Hall and Stewart demonstrated that the N-terminus of SP (SP1–7) was related to naloxone-reversible anti-nociceptive and anti-aggressive actions, whereas the C-terminus (SP7-11) was thought to mediate pain transmission [45,46]. Furthermore, Skilling et al. showed that N-terminus of SP (SP1–7) but not the C-terminus (SP5–11) inhibited the release of excitatory neurotransmitters into spinal-cord extracellular fluid, which was reversed by naloxone [47]. These studies agree with the findings of the naloxone-reversible analgesic effect by SP in the CNS and PNS. The 11 amino-acid SP may be cleaved by enzymatic degradation into differential fragments. Those fragments could interact with differential receptors to induce an anti-nociceptive effect possibly via release of met-enkephalin or other endogenous opioid peptides [48,49,50].
Other evidence has shown that SP could act on NK1R to modulate opioid receptors [51,52]. Bowman and colleagues showed that SP increased the recycling of mu-opioid receptors in sensory neurons and led to elevated sensitivity of opioids [53]. Together, these studies suggest that the anti-nociceptive role of SP could act via opioid signaling.
5. SP-Mediated Anti-Nociception in Muscle
Accumulating evidence has shown a role for SP in anti-nociception in the PNS, especially in muscle. Despite much evidence indicating that SP can cause cutaneous pain, applying SP to muscle induced neither neurogenic inflammation nor painful perception in humans and rats [54,55,56]. In contrast, Lin et al. showed that SP had an anti-nociceptive role in muscle rather than causing pain [57]. With whole-cell patch clamp recordings on dissociated muscle-afferent dorsal root ganglion (DRG) neurons, the authors revealed that SP attenuated the acid sensing ion channel 3 (ASIC3)-induced inward current by enhancing M-channel-like potassium current. ASIC3 is a voltage-independent sodium channel activated by the extracellular protons. It has been found as a molecular determinant involved in pain-associated tissue acidosis [58,59]. As well, a recent study showed that ASIC3 can detect extracellular acidification and also respond to mechanical stimuli [60,61].
The in vivo antinociceptive role of SP was demonstrated in a rodent model of chronic widespread muscle pain induced by dual intramuscular acid injections, one of the fibromyalgia pain models developed by Sluka et al., in 2001 [62]. Two injections, separated by one to five days, of pH-4 acidic saline in the unilateral gastrocnemius muscle in rodents produced chronic and bilateral mechanical hyperalgesia of hind paws and muscle that required activation of ASIC3 [63]. Blocking ASIC3 activation, at the first or the second or both acid injections, abolished the induction and development of chronic muscle hyperalgesia. Furthermore, in the dual acid-injection model, the first acid injection could depolarize ASIC3-expressing muscle nociceptors and also simultaneously trigger SP release, which further enhanced the M-channel-like potassium current to attenuate ASIC3-induced depolarization in gastrocnemius muscle-afferent DRG neurons. In mice lacking TAC1 (no SP and neurokinin A production), chronic pain could be induced by a single acid injection, which suggests that the anti-nociceptive effect was produced by the first acid injection but was diminished with the second injection [57]. The reason for ineffective SP in a second acid injection is still unclear and requires further investigation.
Regarding the acid-induced anti-nociception via SP release, the other important question is what types of acid sensors contribute to the release of SP as an anti-nociceptive acid sensor. The anti-nociceptive acid sensors are still unknown. Although previous study indicated that an acid sensor other than ASIC3 and transient receptor potential cation channel subfamily V member 1 (TRPV1) could trigger SP release [64], the possibility of the co-contribution of ASIC3, TRPV1, and other acid sensors such as other ASIC subtypes and/or proton-sensing G protein-coupled receptors is still not excluded. A recent study demonstrated that low-level laser therapy (LLLT) was effective in reducing mechanical hyperalgesia in the dual acid-injection model. The analgesic mechanism is associated with activation of TRPV1 to release SP in muscle [65]. This study provides new insights regarding the involvement of TRPV1 in acid-mediated anti-nociception. Furthermore, it reveals the involvement of SP in LLLT analgesia, which is widely used in pain control for musculoskeletal pain in the field of physical medicine and rehabilitation. In light of the antinociceptive role of SP in muscle, NK1R agonists might be promising candidates for pain relief in intractable musculoskeletal pain, such as fibromyalgia.
6. Ion Channels Involved in SP Signaling
SP can modulate a variety of ion channels (Table 1) resulting in an increase or decrease of neuronal excitability [66]. In most studies, SP excites neurons by increasing the function of excitatory ion channels and decreasing that of inhibitory ion channels. For example, SP has been shown to excite neurons by elevating the conductance of sodium channels and decreasing that of potassium channels in locus coeruleus neurons [67]. SP also inhibits inwardly rectifying K+ channels in nucleus basalis neurons via Gq/11 [68,69], and inhibits Ca2+-activated potassium channels [IK(Ca)] in stellate ganglion neurons via pertussis toxin-insensitive G proteins [70]. Other studies showed that SP can inhibit the N-type calcium channel in sympathetic neurons via pertussis toxin-insensitive G proteins [71,72]. The above studies suggest that SP mainly modulates ion channel activity via the G-protein-dependent pathway. However, non-G-protein effector systems are also reported in SP-mediated signaling. Lu and colleagues revealed that SP-induced increase of sodium conductance was mediated by activating the sodium ion-permeable cation channel complex of NALCN (sodium leak channel, non-selective) and UNC-80 in mouse hippocampal and ventral tegmental area neurons independent of G-protein but mediated by Src family tyrosine kinase [73]. Accordingly, SP can modulate diverse channels and activate the neurons by G-protein-dependent or -independent signaling.
A few studies showed that SP hyperpolarizes neurons in the PNS. SP hyperpolarized vagal sensory neurons of ferrets by inducing a Ca2+-dependent outward potassium current [74]. SP decreased non-selective cation channel conductance in outer hair cells of guinea pig cochlea [75]. SP enhanced the M-type potassium current independent of G-protein but dependent on tyrosine kinase in half of muscle-afferent DRG neurons [57]. Similarly, an SP-mediated Gi/o-dependent pathway could augment the M-type potassium current in DRG neurons and trigeminal ganglion (TG) neurons [76]. Finally, SP could inhibit T-type calcium channels in DRG and TG neurons [77]. Together, SP can modulate a variety of ion channels via different signaling pathways, which are cell type-specific.
7. Ion Channels Involved in SP-Mediated Nociception
SP has a well-known role in the transmission of nociceptive information in the spinal cord. In the supraspinal level, microinjection of SP in the rostral ventromedial medulla (RVM) has also been found to induce hyperalgesia via descending facilitation mechanisms in a glutamate- and GABAA-dependent manner [92,93]. Furthermore, SP release is increased in the spinal dorsal horn after peripheral nociceptive stimulation [94]. Overall, the amount of nociceptive neurotransmitter released by primary afferent nerve terminals determines the level of pain. The release of SP in the spinal dorsal horn would interact with glutamate to enhance peripheral inputs. Previously, SP and glutamate were found to co-exist in small-diameter DRG neurons and their nerve terminals in the spinal dorsal horn [95]. Glutamate acts as the molecule transmitting the fast excitatory signal, whereas SP modulates relatively slow excitatory synapse responses [96]. Besides, SP signaling can enhance the N-methyl-D-aspartate (NMDA) channel function leading to greater pain sensitivity [78,79,80,81]. Thus, the two transmitters, glutamate and SP, are considered to interact and convey the nociceptive information in the spinal cord.
In the peripheral system, SP is an important element in neurogenic inflammation causing extravasation and sensory neuron sensitization. During inflammatory processes, inflammatory cells and peripheral nerve terminals release SP, which, in turn, modulates a variety of ion channels rendering sensitization of sensory neurons in an autocrine or paracrine manner. In the PNS, SP mainly exists in the small sensory nociceptors. Release of SP can act on NK1R via differential intracellular mechanisms to potentiate the channel activities of TRPV1 [84,85,86], Nav1.8 [83], and l- and N-type calcium channels [87] in a subset of small-diameter DRG neurons, thereby resulting in hyperalgesia. SP could also decease the activity of low-threshold potassium channel (kv4) in capsaicin-sensitive DRG neurons and thus sensitize the nociceptors [88]. In the orofacial region, SP can potentiate the P2X3 receptor in TG neurons, leading to elevated pain sensitivity [89]. In summary, SP predominantly acts on peripheral sensory small neurons (presumably nociceptors) to excite the neurons, thereby increasing nociceptive responses.
8. Ion Channels Involved in SP-Mediated Anti-Nociception
The role of SP in anti-nociception has been confirmed in both the CNS and the PNS. In the CNS, the analgesic effect of SP is mainly associated with opioid-dependent pathways, although other studies also demonstrated the involvement of GABAAR and glycine receptors in the lamina V of the spinal cord [90,97].
In the PNS, SP can act on NK1R via either tyrosine kinase or Gi/o effector system to potentiate the inhibitory M-type potassium channels and inhibit excitatory T-type calcium channels in a specific subset of sensory neurons [76,77]. In muscle afferent DRG neurons, the SP-NK1R signaling is coupled with tyrosine kinase to enhance M-type potassium channel activity [57]. Enhancing the M-type potassium current and inhibiting the T-type calcium current results in dampening neuronal excitability, in turn further leading to an anti-nociceptive effect (Figure 1).
9. M-type Potassium Channels
M-type potassium channels are voltage-gated potassium channels (M for muscarine) encoded by KCNQ genes. KCNQ genes encode five Kv7 subunits (Kv7.1–7.5) [98]. Kv7.2, 7.3, 7.4, and 7.5 are expressed in the nervous system. The M-type potassium channel mainly features Kv7.2 and Kv7.3, although other subunits can also contribute to the formation of M channels in some locations.
M channels were first discovered in sympathetic neurons [99]. When muscarinic agonists activate the muscarinic acetylcholine receptors, sympathetic neurons become more responsive to the synaptic inputs and can become burst firing, rather than fire a single spike. This situation is due to the suppression of a unique channel, which led to the name “M” channel.
M-type channels are able to conduct a non-inactivating outward current with a threshold of about −60 to −80 mV and regulated by many neurotransmitters such as SP and bradykinin. The biophysical features of this current include slow activation and deactivation potassium current. Because M channels can be opened near the resting membrane potential, they also play a role in clamping the resting membrane potential, so they are ideally suited to control neuronal excitability.
M channels are functionally expressed in various central and peripheral neurons including hippocampal [100] and DRG neurons [101]. KCNQ2, 3, and 5 are variably expressed in sensory neurons, including small-diameter nociceptors and large-diameter proprioceptors. Because pathological pain such as inflammatory and neuropathic pain features for neuronal hypersensitivity responds to nociceptive inputs, M-type potassium channels are considered a potential analgesic target in controlling nociceptive excitability. Opening of M channels results in hyperpolarization of the neurons, which decreases cell membrane excitability. Accordingly, the M-channel openers retigabine and flupirtine can effectively attenuate muscle pain [102] and inflammatory pain [101].
In 2012, Lin and colleagues showed that SP-enhanced M channel activity in muscle-afferent DRG neurons via an unconventional signal pathway by activating NK1R coupled with phosphotyrosine kinase to attenuate the mechanical hyperalgesia [57]. Then, Gamper’s group demonstrated that SP could augment the M channel in a subset DRG neurons mainly by acting on NK1R via Gi/o and a redox-dependent pathway [76]. The similar result but discrepant cellular mechanisms could be due to the different subset of DRG neurons examined. The SP-enhanced M current via the phosphotyrosine kinase pathway is prominent on medium to large DRG neurons expressing ASIC3 innervating muscle, whereas the SP-enhanced M current via the Gi/o pathway acts mainly on small neurons expressing TRPV1 innervating skin.
These studies provide insight into SP-mediated anti-nociception in local tissues. Activating the SP-NK1R pathway specifically in muscle afferents becomes an attractive therapeutic target to treat chronic pain. Although the pro-nociceptive effect of SP in the spinal cord has been well documented, local peripheral application of SP can be diluted and has little effect on the spinal cord. Thus, targeting the SP-NK1R pathway in the PNS creates a strategy for pain relief without producing severe side effects in the CNS.
10. T-type Calcium Channels
T-type calcium channels (Cav3) are low-voltage activated calcium channels opened with small membrane depolarization and can generate Ca2+-dependent burst firing, pacemaker activity, and low-amplitude neuronal oscillation [103]. In contrast to high-threshold L-type calcium channels (long-lasting calcium channels), T-type calcium channels only mediate a “transient” calcium influx, with the voltage threshold about −60 with slower activation. T channels also form a window current in the resting membrane potential controlling the sub-threshold neuronal excitability.
The α1 subunit consists of the major channel pore of T-type calcium channels, allowing for calcium influx through it. The channel pore-forming α subunits include Cav3.1, Cav3.2, and Cav3.3 encoded by calcium voltage-gated channel subunit alpha1 G (CACNA1G), H, and I, respectively. T-type calcium channels were first found in small sensory neurons by Carbone and Lux in 1984 [104]. Subsequently, functional expression of T-type channels was confirmed in nociceptive DRG neurons, with Cav3.2 as the predominant isoform. Nelson et al. further characterized the expression profile of T channels and indicated that a subtype of DRG neurons highly expressed T channels, named T-rich cells, which were also highly sensitive to capsaicin and ATP stimuli [105]. Those T-rich cells are mainly small-diameter neurons with narrower APs and after depolarizing potentials during the action potential falling phase. Altering gating parameters or directly increasing the amplitude of the T current can result in burst firing and neuronal excitability [105]. Inhibiting the T-type channels can effectively reduce the neuropathic and inflammatory pain [106,107,108].
Recently, Huang et al. demonstrated that T channels were modulated by SP [77]. The authors found that SP release in damaging tissue would act on NK1R via Gi/o to release mitochondria reactive oxygen species, which further inhibited the T channels. This study revealed SP-mediated anti-nociception in a subtype of small-diameter sensory neurons. Further studies are required to determine whether medium- to large-diameter sensory neurons also show the same modulation of SP in T-type calcium channels.
11. Conclusions
Accumulating evidence has shown SP-mediated anti-nociception in both the CNS and PNS. However, SP has a mixed effect of pro-nociception and anti-nociception in the nervous system. Especially, blocking NK1R signaling in the CNS can result in adverse outcomes. In contrast, the anti-nociceptive effect of SP in the PNS has been well characterized, especially in muscle. Applying SP to muscle-afferent DRG neurons enhances the M-channel outward current, which further hyperpolarizes the neurons. Accordingly, locally targeting the SP-NK1R pathway on muscle afferent neurons could be a promising analgesic strategy.
Author Contributions
C.-C.C. composed the idea and wrote the manuscript. C.-T.C. drafted the manuscript and figure. B.-Y.J. prepared the information of table.
Funding
This work was supported by a grant from the Institute of Biomedical Sciences, Academia Sinica (IBMS-CRC107-P01), and grants from the Ministry of Science and Technology, Taiwan (MOST105-2320-B-001-018-MY3, MOST107-2319-B-001-002, and MOST108-2321-B-001-005).
Acknowledgments
We thank the staff of the Medical Art Room at Institute of Biomedical Sciences, Academia Sinica, for the scientific illustration in Figure 1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Euler, U.S.; Gaddum, J.H. An unidentified depressor substance in certain tissue extracts. J. Physiol. 1931, 72, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.M.; Leeman, S.E. Isolation of a sialogogic peptide from bovine hypothalamic tissue and its characterization as substance P. J. Biol. Chem. 1970, 245, 4784–4790. [Google Scholar] [PubMed]
- Chang, M.M.; Leeman, S.E.; Niall, H.D. Amino-acid sequence of substance P. Nat. New Biol. 1971, 232, 86–87. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.E.; Chirgwin, J.M.; Carter, M.S.; Xu, Z.S.; Hershey, A.D. Three rat preprotachykinin mRNAs encode the neuropeptides substance P and neurokinin A. Proc. Natl. Acad. Sci. USA 1987, 84, 881–885. [Google Scholar] [CrossRef]
- Jessell, T.M. Substance P in Nociceptive Sensory Neurons. In Substance P in the Nervous System; Porter, R., O’Connor, M., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1982. [Google Scholar]
- Rameshwar, P.; Gascon, P.; Ganea, D. Immunoregulatory effects of neuropeptides. Stimulation of interleukin-2 production by substance p. J. Neuroimmunol. 1992, 37, 65–74. [Google Scholar] [CrossRef]
- Potter, G.D.; Guzman, F.; Lim, R.K. Visceral pain evoked by intra-arterial injection of substance P. Nature 1962, 193, 983–984. [Google Scholar] [CrossRef] [PubMed]
- Jessell, T.M. Neurotransmitters and CNS disease. Pain. Lancet 1982, 2, 1084–1088. [Google Scholar] [CrossRef]
- Iversen, L. Substance P equals pain substance? Nature 1998, 392, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M.; Getto, C.J.; Neldner, K.; Reeve, E.B.; Krivoy, W.A.; Zimmermann, E. Substance P and analgesia. Nature 1976, 262, 784–785. [Google Scholar] [CrossRef]
- Hall, M.E.; Stewart, J.M. Substance P and antinociception. Peptides 1983, 4, 31–35. [Google Scholar] [CrossRef]
- Macdonald, S.G.; Dumas, J.J.; Boyd, N.D. Chemical cross-linking of the substance P (NK-1) receptor to the alpha subunits of the G proteins Gq and G11. Biochemistry 1996, 35, 2909–2916. [Google Scholar] [CrossRef]
- Meza, U.; Thapliyal, A.; Bannister, R.A.; Adams, B.A. Neurokinin 1 receptors trigger overlapping stimulation and inhibition of CaV2.3 (R-type) calcium channels. Mol. Pharmacol. 2007, 71, 284–293. [Google Scholar] [CrossRef]
- Meshki, J.; Douglas, S.D.; Lai, J.P.; Schwartz, L.; Kilpatrick, L.E.; Tuluc, F. Neurokinin 1 receptor mediates membrane blebbing in HEK293 cells through a Rho/Rho-associated coiled-coil kinase-dependent mechanism. J. Biol. Chem. 2009, 284, 9280–9289. [Google Scholar] [CrossRef]
- Roush, E.D.; Kwatra, M.M. Human substance P receptor expressed in Chinese hamster ovary cells directly activates G(alpha q/11), G(alpha s), G(alpha o). FEBS Lett. 1998, 428, 291–294. [Google Scholar] [CrossRef]
- Khawaja, A.M.; Rogers, D.F. Tachykinins: Receptor to effector. Int. J. Biochem. Cell Biol. 1996, 28, 721–738. [Google Scholar] [CrossRef]
- Fong, T.M.; Anderson, S.A.; Yu, H.; Huang, R.R.; Strader, C.D. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol. Pharmacol. 1992, 41, 24–30. [Google Scholar] [PubMed]
- Baker, S.J.; Morris, J.L.; Gibbins, I.L. Cloning of a C-terminally truncated NK-1 receptor from guinea-pig nervous system. Brain Res. Mol. Brain Res. 2003, 111, 136–147. [Google Scholar] [CrossRef]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Dery, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef]
- Lai, J.P.; Lai, S.; Tuluc, F.; Tansky, M.F.; Kilpatrick, L.E.; Leeman, S.E.; Douglas, S.D. Differences in the length of the carboxyl terminus mediate functional properties of neurokinin-1 receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 12605–12610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberlotto, L.; Hurd, Y.L.; Murdock, P.; Wahlin, J.P.; Melotto, S.; Corsi, M.; Carletti, R. Neurokinin 1 receptor and relative abundance of the short and long isoforms in the human brain. Eur. J. Neurosci. 2003, 17, 1736–1746. [Google Scholar] [CrossRef] [PubMed]
- Harstall, C.; Ospina, M. How prevalent is chronic pain. Pain Clin. Updates 2003, 11, 1–4. [Google Scholar]
- Tracey, D.J.; Walker, J.S. Pain Due to Nerve Damage—Are Inflammatory Mediators Involved. Inflamm. Res. 1995, 44, 407–411. [Google Scholar] [CrossRef]
- Moalem, G.; Tracey, D.J. Immune and inflammatory mechanisms in neuropathic pain. Brain Res. Rev. 2006, 51, 240–264. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J. Can we distinguish between inflammatory and neuropathic pain? Pain Res. Manag. 2006, 11, 11A–15A. [Google Scholar] [CrossRef]
- De Koninck, Y.; Henry, J.L. Substance P-mediated slow excitatory postsynaptic potential elicited in dorsal horn neurons in vivo by noxious stimulation. Proc. Natl. Acad. Sci. USA 1991, 88, 11344–11348. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P. Neurogenic vasodilatation and plasma leakage in the skin. Gen. Pharmacol. 1998, 30, 5–11. [Google Scholar] [CrossRef]
- Otsuka, M.; Yoshioka, K. Neurotransmitter functions of mammalian tachykinins. Physiol. Rev. 1993, 73, 229–308. [Google Scholar] [CrossRef] [PubMed]
- Russell, I.J.; Orr, M.D.; Littman, B.; Vipraio, G.A.; Alboukrek, D.; Michalek, J.E.; Lopez, Y.; MacKillip, F. Elevated cerebrospinal fluid levels of substance P in patients with the fibromyalgia syndrome. Arthritis Rheumatol. 1994, 37, 1593–1601. [Google Scholar] [CrossRef]
- Vaeroy, H.; Helle, R.; Forre, O.; Kass, E.; Terenius, L. Elevated CSF levels of substance P and high incidence of Raynaud phenomenon in patients with fibromyalgia: New features for diagnosis. Pain 1988, 32, 21–26. [Google Scholar] [CrossRef]
- Mantyh, P.W.; Rogers, S.D.; Honore, P.; Allen, B.J.; Ghilardi, J.R.; Li, J.; Daughters, R.S.; Lappi, D.A.; Wiley, R.G.; Simone, D.A. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science 1997, 278, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.Q.; Mantyh, P.W.; Carlson, E.J.; Gillespie, A.M.; Epstein, C.J.; Basbaum, A.I. Primary afferent tachykinins are required to experience moderate to intense pain. Nature 1998, 392, 390–394. [Google Scholar] [CrossRef] [PubMed]
- De Felipe, C.; Herrero, J.F.; O’Brien, J.A.; Palmer, J.A.; Doyle, C.A.; Smith, A.J.; Laird, J.M.; Belmonte, C.; Cervero, F.; Hunt, S.P. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature 1998, 392, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Lin, S.H.; Malewicz, N.M.; Zhang, Y.; Zhang, Y.; Goulding, M.; LaMotte, R.H.; Ma, Q. Identifying the pathways required for coping behaviours associated with sustained pain. Nature 2019, 565, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Rigby, M.; O’Donnell, R.; Rupniak, N.M. Species differences in tachykinin receptor distribution: Further evidence that the substance P (NK1) receptor predominates in human brain. J. Comp. Neurol. 2005, 490, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Gitter, B.D.; Waters, D.C.; Bruns, R.F.; Mason, N.R.; Nixon, J.A.; Howbert, J.J. Species differences in affinities of non-peptide antagonists for substance P receptors. Eur. J. Pharmacol. 1991, 197, 237–238. [Google Scholar] [CrossRef]
- Beresford, I.; Birch, P.; Hagan, R.; Ireland, S. Investigation into species variants in tachykinin NK1 receptors by use of the non-peptide antagonist, CP-96,345. Br. J. Pharmacol. 1991, 104, 292–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appell, K.C.; Fragale, B.J.; Loscig, J.; Singh, S.; Tomczuk, B.E. Antagonists That Demonstrate Species-Differences in Neurokinin-1 Receptors. Mol. Pharmacol. 1992, 41, 772–778. [Google Scholar] [PubMed]
- Mogil, J.S. Animal models of pain: Progress and challenges. Nat. Rev. Neurosci. 2009, 10, 283–294. [Google Scholar] [CrossRef]
- Frederickson, R.C.; Burgis, V.; Harrell, C.E.; Edwards, J.D. Dual actions of substance P on nociception: Possible role of endogenous opioids. Science 1978, 199, 1359–1362. [Google Scholar] [CrossRef] [PubMed]
- Oehme, P.; Hilse, H.; Morgenstern, E.; Gores, E. Substance-P—Does It Produce Analgesia or Hyperalgesia. Science 1980, 208, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Yoon, T.G.; Kim, S.; Kang, M.; Kim, H.J.; Son, Y. Intravenous Administration of Substance P Attenuates Mechanical Allodynia Following Nerve Injury by Regulating Neuropathic Pain-Related Factors. Biomol. Ther. (Seoul) 2017, 25, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Parenti, C.; Arico, G.; Ronsisvalle, G.; Scoto, G.M. Supraspinal injection of Substance P attenuates allodynia and hyperalgesia in a rat model of inflammatory pain. Peptides 2012, 34, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Bury, R.W.; Mashford, M.L. Biological activity of C-terminal partial sequences of substance P. J. Med. Chem. 1976, 19, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.E.; Stewart, J.M. Substance P and behavior: Opposite effects of N-terminal and C-terminal fragments. Peptides 1983, 4, 763–768. [Google Scholar] [CrossRef]
- Stewart, J.M.; Hall, M.E.; Harkins, J.; Frederickson, R.C.; Terenius, L.; Hokfelt, T.; Krivoy, W.A. A fragment of substance P with specific central activity: SP(1-7). Peptides 1982, 3, 851–857. [Google Scholar] [CrossRef]
- Skilling, S.R.; Smullin, D.H.; Larson, A.A. Differential effects of C- and N-terminal substance P metabolites on the release of amino acid neurotransmitters from the spinal cord: Potential role in nociception. J. Neurosci. 1990, 10, 1309–1318. [Google Scholar] [CrossRef]
- Tang, J.; Chou, J.; Yang, H.Y.; Costa, E. Substance P stimulates the release of Met5-enkephalin-Arg6-Phe7 and Met5-enkephalin from rat spinal cord. Neuropharmacology 1983, 22, 1147–1150. [Google Scholar] [CrossRef]
- Naranjo, J.R.; Arnedo, A.; De Felipe, M.C.; Del Rio, J. Antinociceptive and Met-enkephalin releasing effects of tachykinins and substance P fragments. Peptides 1986, 7, 419–423. [Google Scholar] [CrossRef]
- Naranjo, J.R.; Sanchez-Franco, F.; Garzon, J.; del Rio, J. Analgesic activity of substance P in rats: Apparent mediation by met-enkephalin release. Life Sci. 1982, 30, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, T.; Sasaki, M.; Sanai, K.; Kuwahata, H.; Sakurada, C.; Tsuzuki, M.; Iwata, Y.; Sakurada, S.; Sakurada, T. Intrathecal substance P augments morphine-induced antinociception: Possible relevance in the production of substance P N-terminal fragments. Peptides 2009, 30, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.G.; Yu, L.C. Alterations in the substance P-induced anti-nociception in the central nervous system of rats after morphine tolerance. Neurosci. Lett. 2005, 381, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Bowman, S.L.; Soohoo, A.L.; Shiwarski, D.J.; Schulz, S.; Pradhan, A.A.; Puthenveedu, M.A. Cell-autonomous regulation of Mu-opioid receptor recycling by substance P. Cell Rep. 2015, 10, 1925–1936. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, U.; Nielsen, L.B.; Jensen, K.; Edvinsson, L.; Jansen, I.; Olesen, J. Algesia and local responses induced by neurokinin A and substance P in human skin and temporal muscle. Peptides 1989, 10, 1147–1152. [Google Scholar] [CrossRef]
- Babenko, V.V.; Graven-Nielsen, T.; Svensson, P.; Drewes, A.M.; Jensen, T.S.; Arendt-Nielsen, L. Experimental human muscle pain induced by intramuscular injections of bradykinin, serotonin, and substance P. Eur. J. Pain 1999, 3, 93–102. [Google Scholar] [CrossRef]
- Jensen, K.; Tuxen, C.; Pedersen-Bjergaard, U.; Jansen, I. Pain, tenderness, wheal and flare induced by substance-P, bradykinin and 5-hydroxytryptamine in humans. Cephalalgia 1991, 11, 175–182. [Google Scholar] [CrossRef]
- Lin, C.C.; Chen, W.N.; Chen, C.J.; Lin, Y.W.; Zimmer, A.; Chen, C.C. An antinociceptive role for substance P in acid-induced chronic muscle pain. Proc. Natl. Acad. Sci. USA 2012, 109, E76–E83. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.L.; Cheng, C.F.; Sun, W.H.; Wong, C.W.; Chen, C.C. Targeting ASIC3 for pain, anxiety, and insulin resistance. Pharmacol. Ther. 2012, 134, 127–138. [Google Scholar] [CrossRef]
- Lin, J.H.; Hung, C.H.; Han, D.S.; Chen, S.T.; Lee, C.H.; Sun, W.Z.; Chen, C.C. Sensing acidosis: Nociception or sngception? J. Biomed. Sci. 2018, 25, 85. [Google Scholar] [CrossRef]
- Lin, S.H.; Cheng, Y.R.; Banks, R.W.; Min, M.Y.; Bewick, G.S.; Chen, C.C. Evidence for the involvement of ASIC3 in sensory mechanotransduction in proprioceptors. Nat. Commun. 2016, 7, 11460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.R.; Jiang, B.Y.; Chen, C.C. Acid-sensing ion channels: Dual function proteins for chemo-sensing and mechano-sensing. J. Biomed. Sci. 2018, 25, 46. [Google Scholar] [CrossRef] [PubMed]
- Sluka, K.A.; Kalra, A.; Moore, S.A. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle Nerve 2001, 24, 37–46. [Google Scholar] [CrossRef]
- Sluka, K.A.; Price, M.P.; Breese, N.M.; Stucky, C.L.; Wemmie, J.A.; Welsh, M.J. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain 2003, 106, 229–239. [Google Scholar] [CrossRef]
- Chen, W.N.; Chen, C.C. Acid mediates a prolonged antinociception via substance P signaling in acid-induced chronic widespread pain. Mol. Pain 2014, 10, 30. [Google Scholar] [CrossRef]
- Han, D.S.; Lee, C.H.; Shieh, Y.D.; Chen, C.C. Involvement of Substance P in the Analgesic Effect of Low-levle Laser Therapy in a Mouse Model of Chronic Widespread Muscle Pain. Pain Med. 2019. [Google Scholar] [CrossRef]
- Moraes, E.R.; Kushmerick, C.; Naves, L.A. Characteristics of dorsal root ganglia neurons sensitive to Substance P. Mol. Pain 2014, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.Z.; North, R.A. Substance P opens cation channels and closes potassium channels in rat locus coeruleus neurons. Neuroscience 1992, 50, 345–353. [Google Scholar] [CrossRef]
- Nakajima, Y.; Nakajima, S.; Inoue, M. Pertussis toxin-insensitive G protein mediates substance P-induced inhibition of potassium channels in brain neurons. Proc. Natl. Acad. Sci. USA 1988, 85, 3643–3647. [Google Scholar] [CrossRef]
- Takano, K.; Yasufuku-Takano, J.; Kozasa, T.; Singer, W.D.; Nakajima, S.; Nakajima, Y. Gq/11 and PLC-beta 1 mediate the substance P-induced inhibition of an inward rectifier K+ channel in brain neurons. J. Neurophysiol. 1996, 76, 2131–2136. [Google Scholar] [CrossRef]
- Gilbert, R.; Ryan, J.S.; Horackova, M.; Smith, F.M.; Kelly, M.E. Actions of substance P on membrane potential and ionic currents in guinea pig stellate ganglion neurons. Am. J. Physiol. 1998, 274, C892–C903. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.S.; Hille, B. Substance P and somatostatin inhibit calcium channels in rat sympathetic neurons via different G protein pathways. Neuron 1993, 10, 11–20. [Google Scholar] [CrossRef]
- Bley, K.R.; Tsien, R.W. Inhibition of Ca2+ and K+ channels in sympathetic neurons by neuropeptides and other ganglionic transmitters. Neuron 1990, 4, 379–391. [Google Scholar] [CrossRef]
- Lu, B.; Su, Y.; Das, S.; Wang, H.; Wang, Y.; Liu, J.; Ren, D. Peptide neurotransmitters activate a cation channel complex of NALCN and UNC-80. Nature 2009, 457, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.S.; Weinreich, D. Substance P hyperpolarizes vagal sensory neurones of the ferret. J. Physiol. 1996, 493 Pt 1, 157–166. [Google Scholar] [CrossRef]
- Kakehata, S.; Akaike, N.; Takasaka, T. Substance P decreases the non-selective cation channel conductance in dissociated outer hair cells of guinea pig cochlea. Ann. N. Y. Acad. Sci. 1993, 707, 476–479. [Google Scholar] [CrossRef]
- Linley, J.E.; Ooi, L.; Pettinger, L.; Kirton, H.; Boyle, J.P.; Peers, C.; Gamper, N. Reactive oxygen species are second messengers of neurokinin signaling in peripheral sensory neurons. Proc. Natl. Acad. Sci. USA 2012, 109, E1578–E1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Huang, S.; Gao, H.; Liu, Y.; Qi, J.; Chen, P.; Wang, C.; Scragg, J.L.; Vakurov, A.; Peers, C.; et al. Redox-Dependent Modulation of T-Type Ca(2+) Channels in Sensory Neurons Contributes to Acute Anti-Nociceptive Effect of Substance P. Antioxid. Redox Signal. 2016, 25, 233–251. [Google Scholar] [CrossRef]
- Rusin, K.I.; Bleakman, D.; Chard, P.S.; Randic, M.; Miller, R.J. Tachykinins Potentiate N-Methyl-D-Aspartate Responses in Acutely Isolated Neurons from the Dorsal Horn. J. Neurochem. 1993, 60, 952–960. [Google Scholar] [CrossRef]
- Dougherty, P.M.; Willis, W.D. Enhancement of Spinothalamic Neuron Responses to Chemical and Mechanical Stimuli Following Combined Micro-Iontophoretic Application of N-Methyl-d-Aspartic Acid and Substance-P. Pain 1991, 47, 85–93. [Google Scholar] [CrossRef]
- Randic, M.; Hecimovic, H.; Ryu, P.D. Substance P modulates glutamate-induced currents in acutely isolated rat spinal dorsal horn neurones. Neurosci. Lett. 1990, 117, 74–80. [Google Scholar] [CrossRef]
- Rusin, K.I.; Ryu, P.D.; Randic, M. Modulation of Excitatory Amino-Acid Responses in Rat Dorsal Horn Neurons by Tachykinins. J. Neurophysiol. 1992, 68, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.J.; Gover, T.D.; Cordoba-Rodriguez, R.; Weinreich, D. Substance P evokes cation currents through TRP channels in HEK293 cells. J. Neurophysiol. 2003, 90, 2069–2073. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.L.; Zhang, H.; Zhang, Y.Q.; Zhao, Z.Q. PKCepsilon-dependent potentiation of TTX-resistant Nav1.8 current by neurokinin-1 receptor activation in rat dorsal root ganglion neurons. Mol. Pain 2009, 5, 33. [Google Scholar] [CrossRef]
- Zhang, H.; Cang, C.L.; Kawasaki, Y.; Liang, L.L.; Zhang, Y.Q.; Ji, R.R.; Zhao, Z.Q. Neurokinin-1 receptor enhances TRPV1 activity in primary sensory neurons via PKCepsilon: A novel pathway for heat hyperalgesia. J. Neurosci. 2007, 27, 12067–12077. [Google Scholar] [CrossRef] [PubMed]
- Sculptoreanu, A.; Aura Kullmann, F.; de Groat, W.C. Neurokinin 2 receptor-mediated activation of protein kinase C modulates capsaicin responses in DRG neurons from adult rats. Eur. J. Neurosci. 2008, 27, 3171–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, T.K.; Basso, L.; Iftinca, M.C.; Flynn, R.; Chapman, K.; Dietrich, G.; Vergnolle, N.; Altier, C. TRPV1 sensitization mediates postinflammatory visceral pain following acute colitis. Am. J. Physiol.-Gastr. Liver Physiol. 2015, 309, G87–G99. [Google Scholar] [CrossRef] [PubMed]
- Sculptoreanu, A.; de Groat, W.C. Protein kinase C is involved in neurokinin receptor modulation of N- and L-type Ca2+ channels in DRG neurons of the adult rat. J. Neurophysiol. 2003, 90, 21–31. [Google Scholar] [CrossRef]
- Sculptoreanu, A.; Artim, D.E.; de Groat, W.C. Neurokinins inhibit low threshold inactivating K+ currents in capsaicin responsive DRG neurons. Exp. Neurol. 2009, 219, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.K.; Bae, J.H.; Kim, H.Y.; Jo, H.J.; Kim, Y.H.; Jung, S.J.; Kim, J.S.; Oh, S.B. Substance P sensitizes P2X3 in nociceptive trigeminal neurons. J. Dent. Res. 2010, 89, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Chen, M.; Takeda, D.; King, C.; Ling, J.; Xing, H.; Ataka, T.; Vierck, C.; Yezierski, R.; Gu, J.G. Substance P-driven feed-forward inhibitory activity in the mammalian spinal cord. Mol. Pain 2005, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.S.; Weinreich, D. Substance P regulates Ih via a NK-1 receptor in vagal sensory neurons of the ferret. J. Neurophysiol. 1998, 79, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Lagraize, S.C.; Guo, W.; Yang, K.; Wei, F.; Ren, K.; Dubner, R. Spinal cord mechanisms mediating behavioral hyperalgesia induced by neurokinin-1 tachykinin receptor activation in the rostral ventromedial medulla. Neuroscience 2010, 171, 1341–1356. [Google Scholar] [CrossRef] [PubMed]
- Brink, T.S.; Pacharinsak, C.; Khasabov, S.G.; Beitz, A.J.; Simone, D.A. Differential modulation of neurons in the rostral ventromedial medulla by neurokinin-1 receptors. J. Neurophysiol. 2012, 107, 1210–1221. [Google Scholar] [CrossRef] [PubMed]
- Duggan, A.W.; Hendry, I.A.; Morton, C.R.; Hutchison, W.D.; Zhao, Z.Q. Cutaneous stimuli releasing immunoreactive substance P in the dorsal horn of the cat. Brain Res. 1988, 451, 261–273. [Google Scholar] [CrossRef]
- Debiasi, S.; Rustioni, A. Glutamate and Substance-P Coexist in Primary Afferent Terminals in the Superficial Laminae of Spinal-Cord. Proc. Natl. Acad. Sci. USA 1988, 85, 7820–7824. [Google Scholar] [CrossRef]
- Urban, L.; Randic, M. Slow excitatory transmission in rat dorsal horn: Possible mediation by peptides. Brain Res. 1984, 290, 336–341. [Google Scholar] [CrossRef]
- Wu, L.J.; Xu, H.; Ko, S.W.; Yoshimura, M.; Zhuo, M. Feed-forward inhibition: A novel cellular mechanism for the analgesic effect of substance P. Mol. Pain 2005, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J. Neuronal KCNQ potassium channels: Physiology and role in disease. Nat. Rev. Neurosci. 2000, 1, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Adams, P.R. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 1980, 283, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Mistry, M.; Marsh, S.J.; Brown, D.A.; Delmas, P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J. Physiol. 2002, 544, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passmore, G.M.; Selyanko, A.A.; Mistry, M.; Al-Qatari, M.; Marsh, S.J.; Matthews, E.A.; Dickenson, A.H.; Brown, T.A.; Burbidge, S.A.; Main, M.; et al. KCNQ/M currents in sensory neurons: Significance for pain therapy. J. Neurosci. 2003, 23, 7227–7236. [Google Scholar] [CrossRef]
- Nielsen, A.N.; Mathiesen, C.; Blackburn-Munro, G. Pharmacological characterisation of acid-induced muscle allodynia in rats. Eur. J. Pharmacol. 2004, 487, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Huguenard, J.R. Low-threshold calcium currents in central nervous system neurons. Annu. Rev. Physiol. 1996, 58, 329–348. [Google Scholar] [CrossRef]
- Carbone, E.; Lux, H.D. A Low Voltage-Activated, Fully Inactivating Ca-Channel in Vertebrate Sensory Neurons. Nature 1984, 310, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Joksovic, P.M.; Perez-Reyes, E.; Todorovic, S.M. The endogenous redox agent L-cysteine induces T-type Ca2+ channel-dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J. Neurosci. 2005, 25, 8766–8775. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, S.M.; Meyenburg, A.; Jevtovic-Todorovic, V. Mechanical and thermal antinociception in rats following systemic administration of mibefradil, a T-type calcium channel blocker. Brain Res 2002, 951, 336–340. [Google Scholar] [CrossRef]
- Berger, N.D.; Gadotti, V.M.; Petrov, R.R.; Chapman, K.; Diaz, P.; Zamponi, G.W. NMP-7 inhibits chronic inflammatory and neuropathic pain via block of Cav3.2 T-type calcium channels and activation of CB2 receptors. Mol. Pain 2014, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.F.; Scott, V.E.; McGaraughty, S.; Chu, K.L.; Xu, J.; Niforatos, W.; Milicic, I.; Joshi, S.; Zhang, Q.W.; Xia, Z.R. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem. Pharmacol. 2014, 89, 536–544. [Google Scholar] [CrossRef] [PubMed]
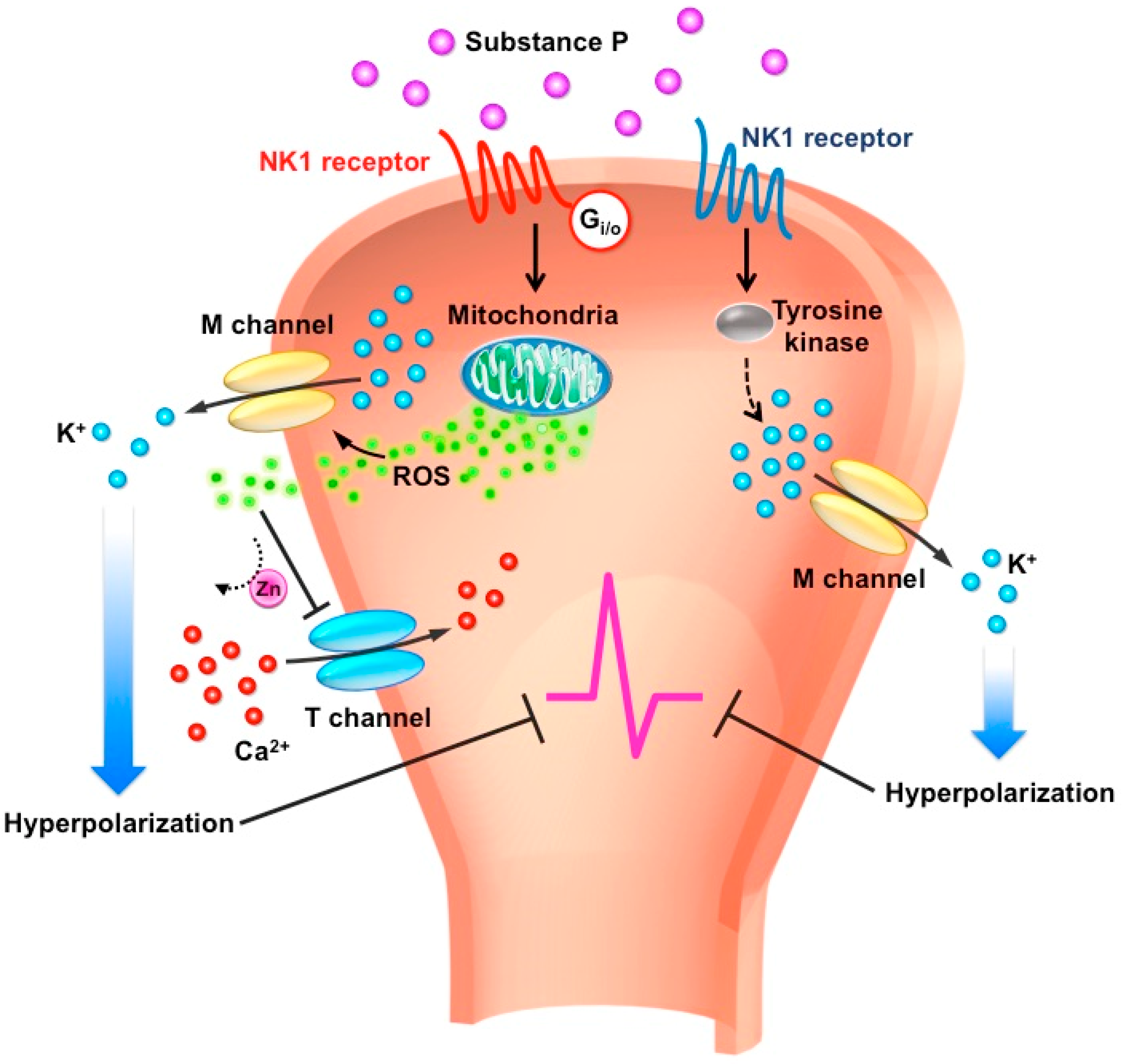
Figure 1.
Schematic diagram of substance P-mediated signaling and ion channels in the peripheral sensory neurons. Release of substance P (SP) in the nerve terminal acts on neurokinin 1 receptor (NK1R) via two different effector systems modulating M-type K+ and T-type Ca2+ channels. First, activated NK1R coupled to tyrosine kinase augments the M-type potassium channels, resulting in neuronal hyperpolarization. Second, activated NK1R coupled to Gi/o triggers reactive oxygen species (ROS) release from mitochondria simultaneously to augment M-type potassium channels and inhibit T-type calcium channels, which inhibits neural firing in peripheral sensory neurons.
Figure 1.
Schematic diagram of substance P-mediated signaling and ion channels in the peripheral sensory neurons. Release of substance P (SP) in the nerve terminal acts on neurokinin 1 receptor (NK1R) via two different effector systems modulating M-type K+ and T-type Ca2+ channels. First, activated NK1R coupled to tyrosine kinase augments the M-type potassium channels, resulting in neuronal hyperpolarization. Second, activated NK1R coupled to Gi/o triggers reactive oxygen species (ROS) release from mitochondria simultaneously to augment M-type potassium channels and inhibit T-type calcium channels, which inhibits neural firing in peripheral sensory neurons.


{kind=link}
Table 1.
Ion channels modulated by substance P.
| Channel Types | Effector System | Cell Types | Species | Effects on Current | Outcomes | References |
|---|---|---|---|---|---|---|
| NMDAR | PKC | Spinal dorsal horn neurons, spinal thalamic neurons | Rat, monkey | ↑ | Pro-nociception | [78,79,80,81] |
| Kir | Gq/11 and PLC-β1 | Locus coeruleus neurons, nucleus basalis neurons | Rat | ↓ | [67,68,69] | |
| KCa | PTX-insensitive G protein | Stellate ganglion neurons | Guinea pig | ↓ | [70] | |
| N-type Ca2+ channel | PTX-insensitive G protein | Stellate ganglion neurons, superior cervical ganglion neurons, sympathetic neurons | Guinea pig, rat, frog | ↓ | [70,71,72] | |
| NALCN | Src family kinases | Hippocampal and ventral tegmental area neurons | Mice | ↑ | [73] | |
| TRP3C | Via NK2R | HEK293 | ↑ | [82] | ||
| Nav1.8 | PKCε | DRG | Rat | ↑ | Pro-nociception | [83] |
| TRPV1 | PKCε | DRG | Rat | ↑ | Pro-nociception | [84,85,86] |
| L, N type calcium channel | PKC | DRG | Rat | ↑ | [87] | |
| Low threshold potassium channel (Kv4) | DRG | Rat | ↓ | Pro-nociception | [88] | |
| P2X3 | TG | Rat | ↑ | Pro-nociception | [89] | |
| GABAAR | Gi/o | Spinal dorsal horn neurons | Rat | ↑ | Anti-nociception | [90] |
| Glycine receptor | Gi/o | Spinal dorsal horn neurons | Rat | ↑ | Anti-nociception | [90] |
| M-type potassium channel | Tyrosine kinase | DRG | Mice | ↑ | Anti-nociception | [57] |
| M-type potassium channel | Gi/o | DRG, TG | Rat | ↑ | Anti-nociception | [76] |
| T-type calcium channel | Gi/o | DRG | Rat | ↓ | Anti-nociception | [77] |
| KCa | Vagal sensory neurons | Ferret | ↑ | [74] | ||
| Ih | Vagal sensory neurons | Ferret | ↓ | [91] | ||
| Non-selective cation channel | PTX-insensitive G protein | Outer hair cells of cochlea | Guinea pig | ↓ | [75] |
Abbreviations: NMDAR, N-methyl-D-aspartate receptor; Kir, inward rectifier potassium channel; KCa, calcium-activated potassium channel; Ih, hyperpolarization-activated channel; PLC, phospholipase C; PTX, pertussis toxin; PKC, Ca2+/phospholipid-dependent protein kinase; DRG, dorsal root ganglion; TG, trigeminal ganglion; Symbols: ↑, increase; ↓, decrease.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chang, C.-T.; Jiang, B.-Y.; Chen, C.-C. Ion Channels Involved in Substance P-Mediated Nociception and Antinociception. Int. J. Mol. Sci. 2019, 20, 1596. https://doi.org/10.3390/ijms20071596
AMA Style
Chang C-T, Jiang B-Y, Chen C-C. Ion Channels Involved in Substance P-Mediated Nociception and Antinociception. International Journal of Molecular Sciences. 2019; 20(7):1596. https://doi.org/10.3390/ijms20071596
Chicago/Turabian StyleChang, Chu-Ting, Bo-Yang Jiang, and Chih-Cheng Chen. 2019. "Ion Channels Involved in Substance P-Mediated Nociception and Antinociception" International Journal of Molecular Sciences 20, no. 7: 1596. https://doi.org/10.3390/ijms20071596
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.