Dietary Macronutrient Management to Treat Mitochondrial Dysfunction in Parkinson’s Disease


1
School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW 2052, Australia
2
School of Medical Sciences, University of New South Wales, Sydney, NSW 2052, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(8), 1850; https://doi.org/10.3390/ijms20081850
Submission received: 27 February 2019
/
Revised: 26 March 2019
/
Accepted: 7 April 2019
/
Published: 15 April 2019
(This article belongs to the Special Issue Mitochondrial Dysfunction and Oxidative Damage)
{kind=link}
{kind=link}
Abstract
:Mitochondrial dysfunction has been demonstrated to play an important role in the pathogenesis of Parkinson’s disease (PD). The products of several PD-associated genes, including alpha-synuclein, parkin, pink1, protein deglycase DJ-1, and leucine rich repeat kinase 2, have important roles in mitochondrial biology. Thus, modifying mitochondrial function could be a potential therapeutic strategy for PD. Dietary management can alter mitochondrial function as shifts in dietary macronutrients and their ratios in food can alter mitochondrial energy metabolism, morphology and dynamics. Our studies have established that a low protein to carbohydrate (P:C) ratio can increase lifespan, motor ability and mitochondrial function in a parkin mutant Drosophila model of PD. In this review, we describe mitochondrial dysfunction in PD patients and models, and dietary macronutrient management strategies to reverse it. We focus on the effects of protein, carbohydrate, fatty acids, and their dietary ratios. In addition, we propose potential mechanisms that can improve mitochondrial function and thus reverse or delay the onset of PD.
1. Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease in ageing individuals [1]. The classic motor system disturbances such as resting tremor, difficulty initiating movements, and postural instability of the disease [2] are mostly attributed to selective degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta and the development of neuronal Lewy bodies [3]. The neuronal degeneration is associated with biochemical defects, many of which can be attributed to mitochondrial dysfunction [4]. The role of mitochondria in PD itself came into focus with an observation in the 1980s suggesting that an inhibitor of complex I of the electron transport chain can induce Parkinsonism [5]. Aberrant mitochondrial forms and functions have been identified in a subset of patients with PD [6,7], which demonstrates that mitochondrial dysfunction and oxidative stress have a central role in PD pathogenesis. Mitochondrial dysfunction in the dopaminergic neurons of idiopathic and familial PD is well known although the underlying mechanisms are not clear. Mitochondria are highly dynamic organelles which facilitate many cellular functions such as energy metabolism, the stress response, and cell death. Considering their central role in cellular biochemistry it is not surprising that dysfunctional mitochondria can result in cellular damage and can be linked to a large variety of diseases (e.g., cancer, diabetes, epilepsy) and ageing. In this review, we wish to describe the role of mitochondria in PD pathology and potential treatment via dietary approaches.
Diet is a major contributor to health and disease. A healthy diet can defend the body against certain types of diseases, such as obesity, diabetes, and cardiovascular diseases. Various nutrition guides are published by medical and governmental institutions to educate the public [8,9] on what they should be eating to promote health and even prevent some diseases. However, no such official guidelines have been formed to potentially delay the onset or slow the progression of PD. Accordingly, this is an active area of current research and many studies have examined dietary components that either protect from, or increase the risk of PD. While there have been several reviews on how dietary macronutrients, micronutrients, or supplements can influence PD [10,11,12,13,14,15,16,17,18,19], few of these adequately describe the mechanisms (known or hypothetical) of mitochondrial dysfunction that underlie disease pathology or are reversed by treatment. The goal of this review is to report the current information on how dietary macronutrient management could play a role in maintaining mitochondrial health in PD. We start the discussion by considering animal models of PD because much of the information in this area is from fundamental research that was undertaken in models or in vitro systems. To understand the role of dietary management in mitochondrial functions, we then briefly discuss how dietary macronutrients can influence oxidative stress, defects in respiratory complexes, and alter mitochondrial morphology and mitophagy. In this review, we also wish to extend this research area by proposing two potential approaches through which macronutrient composition in the diet could improve PD outcomes via alteration of mitochondria. We note that the interactions between diet and gut microbiota is a developing area of interest [20,21] and these interactions likely influence development of PD [22,23,24,25,26,27]. In the next section, we discuss animal models for PD research, focusing on the Drosophila and mouse models. We note other models, including zebrafish (Danio rerio) [28,29] as well as non-human primate systems (e.g., reviewed in [30]) have been developed and have provided insight into PD.
2. Animal Models for Parkinson’s Disease Research
It is our view that there is no “best model of PD,” as none is a true pathocopy of the human condition. Each of the models is an approximation, each likely holding a certain degree of relevance in a specific circumstance. Thus, translational researchers should: (i) understand the strengths and the weaknesses of each model, (ii) select the most appropriate model for addressing the experimental question, and (iii) apply clinically-relevant designs [31].
2.1. Drosophila Models for PD
The fruit fly Drosophila melanogaster has been crucial for mitochondrial research. The ability to tightly control the reproduction, genetics, and physiology of an animal that shares 75% of its genes with humans has led to many discoveries into normal mitochondrial function as well as its role in disease [32]. Accordingly, Drosophila has been an excellent model for studying the role of mitochondria in PD pathology and treatment [33,34,35].
Mutations in Drosophila of orthologs of genes that are responsible for human familial PD have recapitulated mitochondrial dysfunction as well as higher order pathologies such as neurodegeneration, motor symptoms and shortened lifespan [36]. Examples of these, gene mutations in the PINK1/Parkin pathway, DJ-1, and LRRK2 are discussed further below. Additionally, chemically-induced Drosophila PD models have been useful, particularly for examination of environmental factors that contribute to disease risk and progression [37]. Further, a combination of these drugs, such as rotenone, MPTP and paraquat, with flies that harbour the PD-associated gene mutations has allowed powerful examination of gene-environment interactions in the disease [38,39,40,41]. Finally, Drosophila models have been used effectively in PD-therapy research in drug screens, identification of genetic modifiers that suppress PD, or testing of lifestyle modifications such as diet for management of the disease [41,42,43].
Of course, weaknesses exist in Drosophila PD models. Differences in brain anatomy, cardiovascular and respiratory systems, as well as in behaviour and complex motor functions exist. Also, drug metabolism differences restrict the potential for some areas of pharmacological research. In this review, we are focussing on diet–mitochondrial–PD mechanisms which are well conserved between Drosophila and humans, particularly in macronutrient utilisation for energy production and potential for generation of oxidative stress.
2.2. Mouse Models for PD
Mice are more closely related to humans than Drosophila, and so it may be expected that they would be a better model for human health and disease. In terms of PD, the most important signs to replicate are motor symptoms, pathologic accumulation and aggregation of α-synuclein, neuronal cell loss in the basal ganglia, age-related disease progression, and nonmotor symptoms. Unfortunately, none of the current models satisfies all of these criteria. Fortunately, many models fulfil a subset. Empirical choices of adequate models to answer specific questions in translational research need to consider, case by case, the model-specific advantages and limitations [44,45,46].
Recent advances in understanding the pathobiology of PD have been obtained in mice. Kam et al. [47] used an experimentally powerful animal model of Parkinson’s disease and related α-synucleinopathies in which α-synuclein fibrils that are formed in vitro (preformed fibrils) are injected directly into the mouse brain. The authors identified a feed-forward loop in which α-synuclein preformed fibrils increased nitric oxide-mediated DNA damage, which in turn activated PAR polymerase 1 (PARP1), generating poly(adenosine 5′-diphosphate-ribose) (PAR) causing cell death through the parthenatos cell-death pathway.
Mouse models have also been instrumental in determining the potential beneficial effects of specific dietary interventions in PD. Perez-Pardo et al. [48] employed a mouse model of PD that injected rotenone or vehicle into the striatum of mice and then started a dietary intervention when motor symptoms were observed. The authors found the rotenone-induced motor and non-motor problems were alleviated by the active diet (AD), which contained precursors and cofactors required for membrane phospholipid synthesis, as well as prebiotic fibres. Most of the nutrients in the AD are part of the nutritional combination known as Fortasyn Connect [49]. Subsequently, the same group showed the combination of the AD and Levodopa showed an additive beneficial effect on rotarod performance [50]. In the next section, we consider oxidative stress in PD. Oxidative stress is the imbalance between the levels of reactive oxygen species (ROS) produced and the ability of a biological system to detoxify these reactive intermediates. Oxidative stress is a key link between PD and diet as most ROS in the cell are generated through mitochondrial metabolism of dietary macronutrients.
3. Oxidative Stress in Parkinson’s Disease
Elevated intracellular ROS levels can be toxic and can damage lipids, proteins, and DNA. But low levels of ROS can function as a signalling molecule that activates pathways necessary for normal cell homeostasis [51,52]. Numerous studies have indicated that mitochondrial damage induced by oxidative stress contributes to a series of events leading to degeneration of dopaminergic neurons in PD [53,54]. Also, in both sporadic and genetic causes of PD, oxidative stress is thought to be a common underlying mechanism.
Mitochondria are the primary site of ROS generation within a cell, responsible for more than 90% of the total [24]. Mitochondrial ROS production occurs when electrons leak out of the electron transport chain and react with dioxygen and form superoxide. In response, superoxide dismutase (SOD) in the mitochondria and cytosol converts superoxide to diatomic oxygen and hydrogen peroxide (H2O2) and is released in the cytosol. The cytosol has peroxisomes containing the enzyme catalase which converts H2O2 to water thus preventing accumulation of ROS [55]. Mitochondria also contribute to H2O2 disposal by glutathione peroxidase and glutathione reductase [56,57]. However, an increase in ROS production beyond a threshold, or a failure of the anti-ROS processes contribute to oxidative stress causing cellular and mitochondrial damage. Damage to mitochondria themselves exacerbates the process and can cause a dramatic increase in ROS which can overwhelm the cellular antioxidant mechanisms [58,59]. It has been proposed that enhancing antioxidant function may be useful in treating PD [60]. When used at the appropriate levels the mineral selenium is an important antioxidant and may be involved in combating oxidative stress [61]. Studies found reduced selenium levels in PD patients [62] and selenium reduces bradykinesia, a well-described symptom of PD, in rats [63] suggesting that higher selenium levels may be beneficial for PD patients. More recently, high-throughput allele-specific expression analyses suggested that selenium is beneficial for PD patients through its influence on allelic expression in LAMP3 [64]. In contrast the benefit of coenzyme Q10 is less clear. Two meta-analyses of randomized, placebo-controlled trials found no evidence that coenzyme Q10 improved motor-related symptoms or delayed the progression of the disease when compared to placebo [65,66].
The presence of ROS generating enzymes in dopaminergic neurons makes them particularly prone to oxidative stress which may lead to PD. Additionally, dopaminergic neurons contain iron, which can catalyse a Fenton reaction generating superoxide radicals that also can contribute to oxidative stress [67]. This intrinsic sensitivity of dopaminergic neurons to reactive species means that moderate oxidative stress can trigger a series of events that lead to cellular demise. However, importantly, different dietary macronutrients have differing potential for generating oxidative stress, therefore PD could be potentially managed through dietary strategies.
Dietary management has been shown to play a vital role in modulating oxidative stress in various organs. The relative ratio of macronutrients can alter the production of ATP and ROS in mitochondria. Intake of excess protein has been linked to oxidative damage in the pancreas of the mouse by reducing levels of antioxidants such as superoxide dismutase (SOD) [68]. A similar observation of oxidative stress has been reported in rat brains with high protein intake [69]. Conversely, protein restriction reduces mitochondrial ROS levels and DNA and lipid damage in rat livers [70]. These studies suggest the consumption of higher protein causes oxidative damage.
Carbohydrate metabolism will release less ROS than protein and fatty acids for the same amount of ATP produced [71]. However, a high glucose level is generally associated with increased TCA cycle flux and increased ROS levels [72,73], whereas inhibition of mitochondrial pyruvate uptake lowered ROS levels [74,75]. When ROS levels are too high or remain increased during a prolonged period, a vicious circle of ROS-stimulated glucose uptake and glucose-stimulated ROS production can be triggered [71].
The role of fatty acids in the generation of ROS is complicated. One study reported that fatty acid stimulates ROS production through its oxidation [76]. However, ROS generation varies with different types of fatty acids. Medium chain fatty acids were found to induce more mitochondrial ROS generation than long chain fatty acid in rats [77]. The same study also reported differences in saturated or unsaturated fatty acids with unsaturated fatty acids producing more ROS [77]. Moreover, some polyunsaturated fatty acids have anti-inflammatory and neuroprotective properties and have been shown to reduce oxidative stress in various animal models [78,79,80]. Fatty acid availability also alters the lipid packing state of membranes [81] and thus can influence membrane ion permeability. Increased mitochondrial membrane potential is an indication of a less leaky membrane and less ROS production [82,83,84]. Our studies have shown lower levels of basal ROS production in stearic acid (saturated long chain fatty acid) fed diet in Drosophila [85]. There are also reports suggesting fatty acid oxidation can drive reverse electron transport and lead to higher mitochondrial generation of ROS [86]. All these studies show different roles of various fatty acids on ROS generation. We propose that the dual function of fatty acids, in meeting energy demand by fatty acid oxidation and through contributing to the composition of cellular and mitochondrial membranes is central to their importance for dietary strategies for management of PD.
4. Mitochondrial Respiratory Complex I Deficiency and ROS in Parkinson’s Disease
Studies in the early 1980s provided evidence that substances which inhibit complex I of the mitochondrial respiratory chain (MRC) can cause nigrostriatal degeneration and parkinsonism [87,88,89]. Subsequent studies have found decreased complex I activity in frontal cortex homogenate from individuals with PD [87,90,91]. Further evidence of mitochondrial dysfunction in PD comes from the findings that mutations in genes associated with mitochondrial proteins (parkin, pink1, and leucine rich repeat kinase 2 (lrrk2)) that cause a respiratory complex deficiency, are linked to familial forms of PD [92,93]. Cells derived from patients with parkin gene mutations show decreased complex I activity [94]. Mutations in the pink1 gene also reduce complex I activity [95] and PINK1 is important in remodelling of mitochondrial cristae junctions as well as dopaminergic neuronal survival in vivo [96]. Furthermore, α-synuclein, although mostly a cytosolic protein, has been shown to interact with mitochondrial membranes and inhibit complex I in neuronal cultures from a PD patient brain [97]. Changes in subunit proteins and function of mitochondrial complexes II, III, IV, and V have also been reported in PD. However, they are not consistently observed and could be a secondary effect due to some other pathogenic mechanism [94,98,99,100,101]. Therefore, respiratory complex dysfunction has been widely found in PD. Respiratory complex I is the first enzyme of the respiratory chain and contributes to ATP synthesis and maintenance of mitochondrial membrane potential [102,103]. Defects in complex I activity can be expected to produce depressed rates of ATP and cause mitochondrial membrane depolarization leading to excessive generation of ROS [104]. Thus, it would be expected that substantial complex I defects would cause severe, early-onset, rapidly progressive neurological diseases such as PD. Because inhibition of complex I activity is the most consistent finding between human PD and altered mitochondrial respiratory function, dietary modulation of complex I may provide the highest potential for nutritional interventions in PD.
5. Relative Macronutrient Content in Diets and Metabolic Flexibility to Reduce Mitochondrial ROS From Complex I
Metabolic flexibility is the capacity of an organism to modify energy oxidation in response to nutrient availability, thus allowing it to adapt to a variety of physiological conditions [105]. Concerning dietary macronutrients, metabolic flexibility enables a cell to favour one substrate (e.g., lipid) over another (e.g., carbohydrate) for energy generation in response to substrate availability, or due to dysfunction in the biochemical pathways of one substrate’s metabolism. The concept of metabolic flexibility has been proposed in the restoration of mitochondrial function in Type 2 Diabetes [106]. A similar approach can be recommended to alleviate complex I defect in PD. Mitochondrial energy (ATP) production from lipid and protein has less reliance on complex I than carbohydrate metabolism [107]. Therefore, strategies that increase fat and protein while reducing carbohydrate metabolism could be considered to be useful for PD that is associated with complex I dysfunction. However, as we discuss later the picture is not that clear as there is evidence that conversely suggests that high carbohydrate diets could be beneficial and high protein diets detrimental for PD. One dietary intervention that involves metabolic flexibility that has been tested in disease with mitochondrial defects is the ketogenic diet i.e., high fat, moderate protein, and low carbohydrate. In balanced diets, the brain generally relies on the metabolism of glucose for energy. However, in the ketogenic diet, ketone bodies produced through fat metabolism in the liver can be used as an alternative fuel for the brain. MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) inhibits complex I activity [5], and a study shows that in MPTP mice model of PD, ketone bodies can exert neuroprotective activity within the brain by promoting antioxidant activity [108]. The ketogenic diet has been shown to increase both glucose metabolism and ATP production in postnatal day 35 male rats [109]. Largely, these improvements have been attributed to ketone bodies being able to act as an “alternative fuel”, ultimately bypassing glycolysis and providing Acetyl-CoA to enter the citric acid cycle and facilitate ATP production [110].
Another potential dietary strategy for management of respiratory complex I defect is to reduce intake of the essential amino acid methionine. Higher intake of methionine has been associated with higher ROS generation in complex I as well as damage to mitochondrial DNA [111]. Amino acids can only generate ATP in a mitochondrial-dependent manner which means that more ROS is produced when proteins are used as the primary fuel for energy. So, methionine restrictive diets or diets with low protein content could be a potential dietary management strategy to reduce complex I defects in PD.
Numerous studies have shown that dysfunction of mitochondria can cause a switch from mitochondrial respiration to aerobic glycolysis, which is also known as a Warburg-like effect [112,113,114]. With regard to PD, a study has shown that parkin deficiency activates glycolysis and reduces mitochondrial respiration [115]. Aw et al., [107] proposed that a diet high in carbohydrate could potentially provide advantages for an organism that harbors a complex I defect if the metabolic substrates of carbohydrates are converted to fatty acids before entry into the mitochondria. In this way, a high carbohydrate diet could meet ATP requirements and also bypass complex I by lipid oxidation. If this is true, it would suggest a potential route for dietary management in diseases with mitochondrial complex I defects such as PD.
6. Mitochondrial Dynamics and Morphology in Parkinson’s Disease
Another important mechanistic link between diet and PD is mitochondrial dynamics. Mitochondrial fusion and fission contribute to the maintenance of mitochondrial morphology, function, and help optimize their bioenergetic capacity. Fusion allows the spreading of metabolites, enzymes, and mitochondrial gene products throughout the entire mitochondrial compartment. Mitochondrial fission also plays a major role in the removal of damaged organelles by autophagy [116]. Achieving a balance between fusion and fission allows a cell to have the proper organization of its mitochondrial network during biogenesis and has an important role in muscle adaptation to changing physiological conditions. The ketogenic diet, as well as possessing ketones, also contains medium chain fatty acids and the medium chain fatty acid, decanoic acid, has been associated with an improvement in mitochondrial function/mitochondrial biogenesis as the result of its peroxisome proliferator-activated receptor agonist activity [117]. More recently, a ketogenic diet has also been shown to increase mitochondrial mass and functional competence apparently via peroxisome proliferator-activated receptor γ-coactivator-1α, which regulates mitochondrial biogenesis, mitochondrial sirtuins (such as SIRT3) and the uncoupling protein UCP2 [118].
The main machinery of mitochondrial dynamics consists of three large GTPases that fuse and divide the mitochondrial membranes. They are mitofusin 1 and 2 (mfn1 and mfn2) (outer membrane fusion), opa1 (inner membrane fusion) and dynamin related protein 1 (drp1) (fission) [119,120,121,122,123]. Several studies show alteration of mitochondrial morphology in cases of PD. One early study reported a reduction in mitochondrial number and disruption of mitochondrial membrane in muscle tissues of PD patients [124]. There are reports of abnormal mitochondrial distribution, variation in mitochondrial size and swelling in biopsies taken from PD patients [125,126,127,128]. In recent years, PD related genes have been shown to have a critical role in the maintenance of normal mitochondrial morphology with a balance between fusion and fission essential for healthy organelle function. Mutation in parkin causes abnormal mitochondrial morphology in fibroblast cells of PD patients [129]. Detailed studies in Drosophila have demonstrated that pink1 and parkin act to promote mitochondrial fission or inhibit fusion. And, the phenotypes of pink1 or parkin mutant flies can be dramatically improved by overexpression of drp1 or down-regulation of mfn1/mfn2 or opa1 [130,131]. Parkin and pink1 are also involved in the generation of mitochondrial-derived vesicles (MDVs) in response to mitochondrial oxidative stress. Parkin binds with MDVs in a pink1-dependent manner and targets to lysosomes for degradation in a manner independent of mitophagy [132,133,134]. These findings suggest that pink1 and parkin operate in an early stage to salvage mitochondria by selective extraction of damaged components via vesicular carriers. Similarly, DJ-1 KO mice accumulate more ROS and have fragmented mitochondria [135]. It has also been shown that overexpressing a mutant form of α-synuclein in mice causes abnormalities in mitochondrial structure and function [136].
Fusion and fission machinery plays a vital role in maintaining a healthy mitochondrial population [137]. Mitochondrial dynamics and morphology have a strict interconnection with energy balance and diet [138]. Therefore, the maintenance of mitochondrial morphology could be a key area where dietary management can play an important role. The roles of protein and carbohydrate in mitochondrial morphology have been illustrated by studying morphological changes induced by caloric restriction [139] and starvation [140]. Caloric restriction in mice (40% for 6 months) has been shown to increase mitochondrial fission with no changes in total ATP production [139]. Nutrient starvation of glucose increases fission [140]. However, a very high glucose concentration can also elevate fission through high ROS production [74].
The role of fatty acids and lipid in mitochondrial fusion/fission activity has been widely studied and has revealed differences due to the specific type of fatty acids. A high-fat diet rich in saturated fatty acid (high lard diet) has been reported to facilitate mitochondrial fission by elevating levels of drp1 and decreasing mfn2 [141]. However, stearic acid which is also a saturated fatty acid has been reported to promote mitochondrial fusion by stearoylation of transferrin receptor which inhibits JNK signalling leading to reduced ubiquitination of mitofusin [142]. In contrast to the effect of saturated fatty acids (except stearic acid), omega 3 polyunsaturated fatty acids have shown to improve mitochondrial function by reducing ROS production and promoting mitochondrial fusion both with in vitro and in vivo experiments by increased levels of mfn2 and ATP levels [143].
7. Potential Roles of Macronutrients on the Progression of Parkinson’s Disease
In the above section, we described evidence for how different macronutrients can affect PD, mainly through mitochondrial dysfunction. In this section, we propose that a low protein to carbohydrate (P:C) ratio, and diets rich in certain fatty acids may have high potential for management of mitochondrial dysfunction in PD. We have focussed on these new proposals, and not on previously described diets for management of PD such as the ketogenic diet and caloric restriction as these has been well covered elsewhere [144,145]. Our hypotheses stem from our recent work in Drosophila parkin mutants which were initially constructed by Greene et al. [36]. Flies bearing null alleles of parkin are viable but exhibit reduced longevity, flight and climbing defects, and male sterility [36]. The locomotor phenotypes result from apoptotic muscle degeneration, while the male sterile phenotype results from a defect in spermatogenesis [36]. One limitation of this model is that it is based on an early onset form of PD known as autosomal recessive juvenile parkinsonism (AR-JP) [146]. AR-JP patients display many of the clinical features of idiopathic PD; however, most cases identified lack Lewy body pathology. This observation has led to the suggestion that Parkin may be required for Lewy body formation, or that dopaminergic neuron loss in idiopathic PD and AR-JP individuals proceed through distinct mechanisms [36].
7.1. Protein to Carbohydrate Ratio
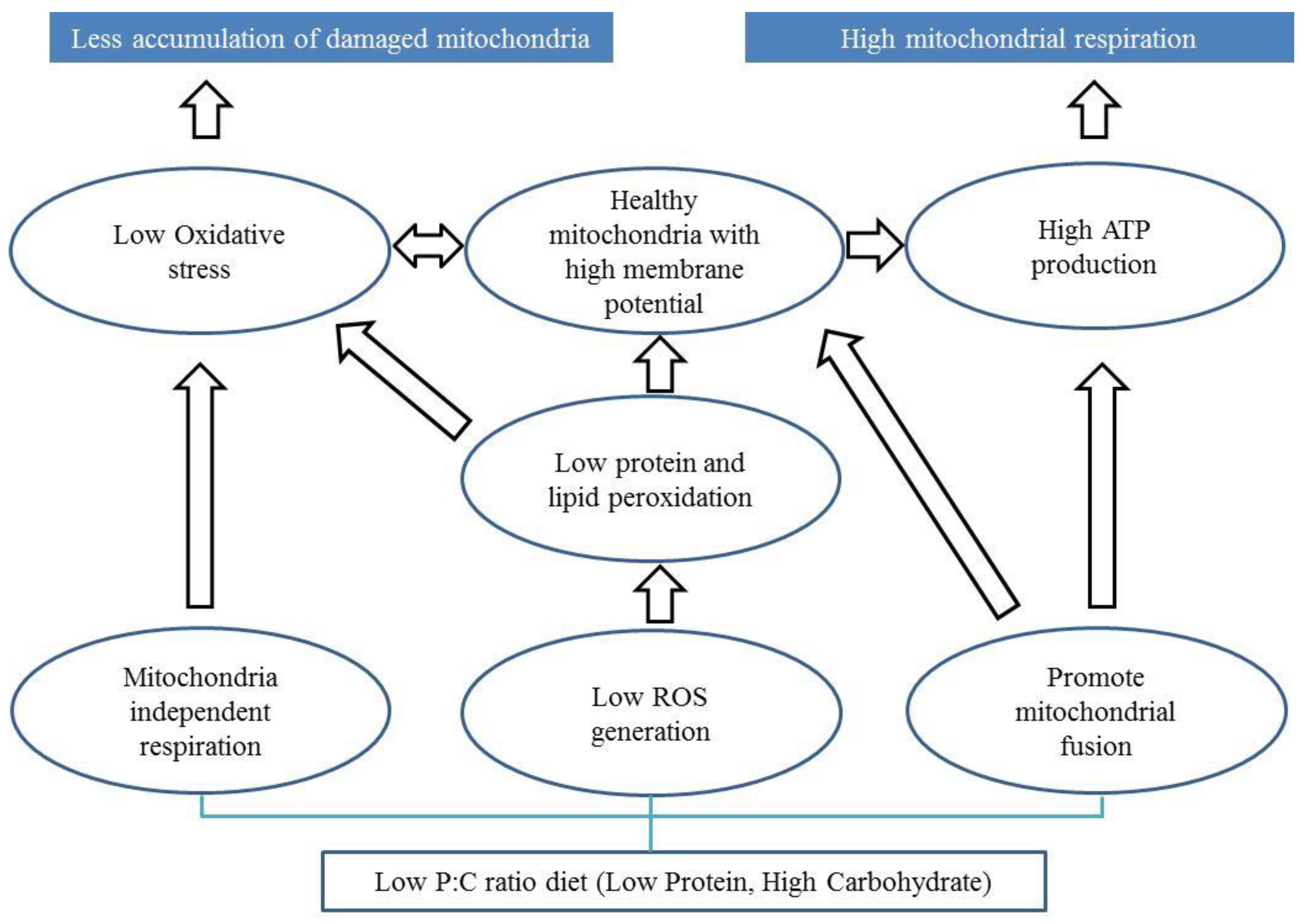
It has been argued that the ratio or balance of protein to carbohydrate in the food we consume influences ageing [147,148,149,150]. Here we discuss potential mechanisms by which the protein to carbohydrate ratio in the diet may influence the progression of PD. Restricting dietary protein or a balanced diet of carbohydrate and protein has been proposed to improve motor performance and levodopa treatment in PD patients [151,152,153]. However, the role of carbohydrate consumption and PD is still debated. Several lines of evidence suggest the potential for low protein and high carbohydrate diet in improving mitochondrial functions [70,149,154]. However, it is not fully understood if this can improve mitochondrial function in PD patients. We propose three mitochondrial-associated mechanisms that plausibly underlie the benefits of a low P:C ratio diet for PD (Figure 1).
First, a low P:C ratio diet can facilitate lower ROS levels through a switch in energy production from the mitochondrial-dependent (mitochondrial OXPHOS) to the mitochondrial-independent (glycolysis) pathway. In this way metabolic flexibility therefore avoids the greater potential for carbohydrate metabolism to produce mitochondrial ROS than fat or protein metabolism, and could result in an advantage to patients with a mitochondrial complex I defect [107]. In our recent publication, we showed that a low P:C ratio diet results in low ROS levels and improved mitochondrial functions in a Drosophila PD model [42]. However, low ATP generation is a potential limitation of mitochondrial independent respiration.
Second, low ROS generation can lead to low oxidative stress, which can prevent mitochondrial membranes from peroxidation [155,156]. Low lipid peroxidation may prevent the formation of a leaky mitochondrial membrane, which in turn maintains ATP generation [83,157,158].
Third, a low P:C diet may facilitate mitochondrial fusion allowing a greater ATP generation and respiration. Fusion of mitochondria enables the spreading of metabolites, enzymes, and mitochondrial gene products throughout the entire mitochondrial compartment. It can facilitate the extension of mitochondrial health by improving function and ATP generation [116,142,159]. Mitochondrial fusion requires and maintains potential across the inner membrane of mitochondria [160], which can also preserve ATP generation.
In addition to the potential mechanisms mentioned above, there are studies that show the importance of P:C ratio in determining the composition of gut microbes in dogs and mice [161,162]. Reports have now shown that gut microbes are linked with α-synuclein mediated motor deficits and brain pathology in Parkinson’s disease [163]. Scientists found that the human gut microbes from PD patients can induce motor dysfunction in murid models of PD and concluded that alteration of microbiome can represent a risk factor for PD [162]. The low protein high carbohydrate diet appears to support the growth of Bacteroides uniformis and Clostridium butyricum, while the high protein low carbohydrate diet increased the abundance of Clostridium hiranonis, Clostridium perfringens, and Ruminococcus gnavus in dogs [161]. But, how these microbes relate to the risk of PD is still unknown, which makes further research into understanding the microbial community a compelling area of future study.
7.2. Diets Rich in Saturated Fatty Acids
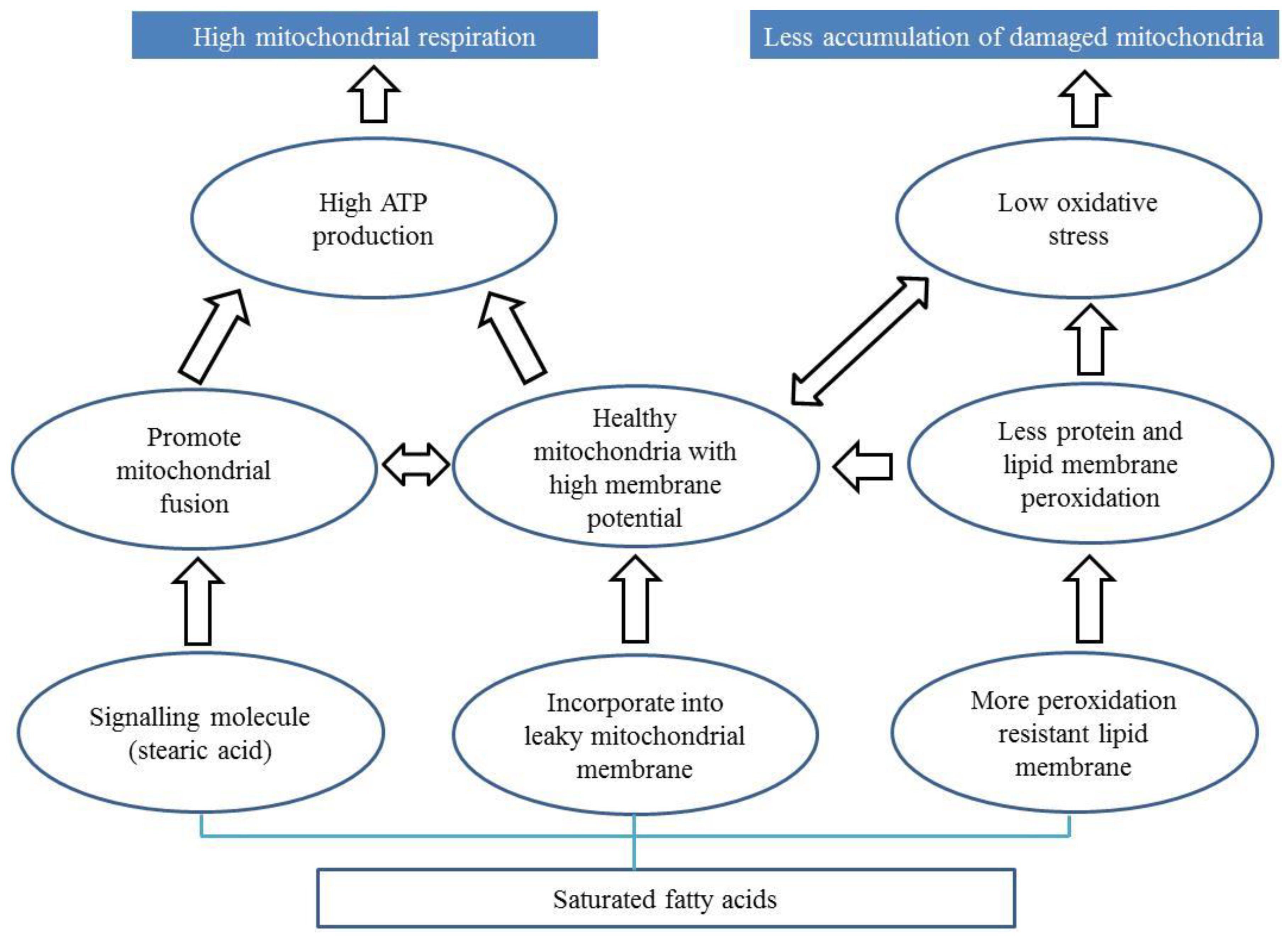
The role of fatty acids in the progression of PD is debated in the literature. Here we propose three potential mechanisms by which fatty acids can affect the progression of PD (Figure 2) via alteration of mitochondria.
First, some fatty acids (e.g., stearic acid) may prevent mitochondrial dysfunction in PD by promoting mitochondrial fusion [142] Stearic acid acts a signalling molecule which stearoylates transferrin receptor thereby inhibiting the activation of JNK signalling pathway [142]. This leads to ubiquitination of mitofusin promoting mitochondrial fusion and function. Interestingly, stearoylation of the transferrin receptor has been shown to be specific to stearic acid.
Second, saturated fatty acids may bind to leaky mitochondrial membranes promoting mitochondrial function. Stearic acid is known to be converted to oleic acid [164], which is incorporated into the mitochondrial phospholipid membrane in rats [165]. Plausibly, oleic acid derived from stearic acid may be integrated into these leaky membranes promoting mitochondrial respiration and function. Phospholipids make up the characteristic outer and inner membranes that give mitochondria their shape [166]. Leaky membranes are indicative of lower membrane potential, which in turn can reduce ATP generation [82,83]. Dysfunctional and leaky mitochondria also produce more ROS [84], which can cause an imbalance that can compromise the ability of an organism to detoxify these reactive intermediates causing oxidative stress. So, incorporation of saturated fatty acids likely results in increased membrane potential, improved ATP generation, and reduced oxidative stress.
Third, saturated fatty acid can bind to lipid membranes making them more resistant to peroxidation [81,167]. Membrane peroxidation is observed in parkin-deficient mice [168]. Peroxidation resistant membranes are found to be correlated with extended lifespan and mitochondrial function in many organisms such as birds and rats [51,169,170,171,172,173,174]. Supplementation of saturated fatty acid to reduce membrane peroxidation can be a promising approach to mitigate mitochondrial defects in PD patients.
8. Conclusions
Improvement of mitochondrial function through dietary approaches is promoted for the management of diseases such as epilepsy, cancer, and diabetes. Considering the growing evidence that mitochondrial dysfunction plays a crucial role in the pathophysiology of PD, it is time to ponder how diets could aid in its management. We suggest that low protein/high carbohydrate diets, or diets enriched with certain fat species could reverse many of the mitochondrial-associated cellular changes in PD. However, as many of the observations that support these dietary approaches stem from animal models or fundamental biochemical research, a cautious approach to human testing is essential. Further, it is not known whether partial nutritional supplementation with specific foods will have therapeutic value. For example, is it possible that regular consumption of a banana or a mango (which have a P:C value of ~1:16) has the potential to delay the progression of PD?
Funding
Funding was received from a University of New South Wales Goldstar award.
Acknowledgments
We would like to acknowledge David Le Couteur for suggestions to improve the manuscript.
Conflicts of Interest
The authors have no competing interests to declare.
References
- Mata, I.F.; Lockhart, P.J.; Farrer, M.J. Parkin genetics: One model for Parkinson’s disease. Hum. Mol. Genet. 2004, 13, R127–R133. [Google Scholar] [CrossRef] [PubMed]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Muqit, M.M.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef]
- Jankovic, N.; Geelen, A.; Streppel, M.T.; de Groot, L.C.; Kiefte-de Jong, J.C.; Orfanos, P.; Bamia, C.; Trichopoulou, A.; Boffetta, P.; Bobak, M.; et al. WHO guidelines for a healthy diet and mortality from cardiovascular disease in European and American elderly: The CHANCES project. Am. J. Clin. Nutr. 2015, 102, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Breda, J.; Jewell, J.; Keller, A. The importance of the World Health Organization sugar guidelines for dental health and obesity prevention. Caries Res. 2018, 53, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.; Checkoway, H.; Franklin, G.M.; Beresford, S.; Smith-Weller, T.; Swanson, P.D. Dietary factors in Parkinson’s disease: The role of food groups and specific foods. Mov. Disord. 1999, 14, 21–27. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhang, S.M.; Hernán, M.A.; Willett, W.C.; Ascherio, A. Dietary intakes of fat and risk of Parkinson’s disease. Am. J. Epidemiol. 2003, 157, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Willett, W.C.; Ascherio, A. Diet and Parkinson’s disease: A potential role of dairy products in men. Ann. Neurol. 2002, 52, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Hellenbrand, W.; Boeing, H.; Robra, B.P.; Seidler, A.; Vieregge, P.; Nischan, P.; Joerg, J.; Oertel, W.H.; Schneider, E.; Ulm, G. Diet and Parkinson’s disease. II: A possible role for the past intake of specific nutrients. Results from a self-administered food-frequency questionnaire in a case-control study. Neurology 1996, 47, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Lau, R.C.; Bennett, R.D. Role of diet and nutritional supplements in Parkinson’s disease progression. Oxid. Med. Cell. Longev. 2017, 2017, 6405278. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Tanaka, K.; Fukushima, W.; Sasaki, S.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Lack of association of dairy food, calcium, and vitamin D intake with the risk of Parkinson’s disease: A case-control study in Japan. Parkinsonism Relat. Disord. 2011, 17, 112–116. [Google Scholar] [CrossRef]
- Saaksjarvi, K.; Knekt, P.; Lundqvist, A.; Mannisto, S.; Heliovaara, M.; Rissanen, H.; Jarvinen, R. A cohort study on diet and the risk of Parkinson’s disease: The role of food groups and diet quality. Br. J. Nutr. 2013, 109, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Seidl, S.E.; Santiago, J.A.; Bilyk, H.; Potashkin, J.A. The emerging role of nutrition in Parkinson’s disease. Front. Aging Neurosci. 2014, 6, 36. [Google Scholar] [CrossRef]
- Zhang, S.M.; Hernan, M.A.; Chen, H.; Spiegelman, D.; Willett, W.C.; Ascherio, A. Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology 2002, 59, 1161–1169. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.; Seo, M.; Berk, M.; Carvalho, A.F.; Maes, M. Gut permeability and microbiota in Parkinson’s disease: Role of depression, tryptophan catabolites, oxidative and nitrosative stress and melatonergic pathways. Curr. Pharm. Des. 2016, 22, 6142–6151. [Google Scholar] [CrossRef]
- Aydin, M.A.; Agirbas, E.P.; Aydin, S. Can disruption of microbiota composition be the chemical basis of Parkinson’s disease and schizophrenia? Biosci. Microbiota Food Health 2019, 38, 1–2. [Google Scholar] [CrossRef]
- Mulak, A.; Bonaz, B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015, 21, 10609–10620. [Google Scholar] [CrossRef]
- Parashar, A.; Udayabanu, M. Gut microbiota: Implications in Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 38, 1–7. [Google Scholar] [CrossRef]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of gut microbiota in patients with Parkinson’s disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef]
- Ünal, I.; Emekli-Alturfan, E. Fishing for Parkinson’s Disease: A review of the literature. J. Clin. Neurosci. 2019, 62, 1–6. [Google Scholar] [CrossRef]
- Vaz, R.L.; Outeiro, T.F.; Ferreira, J.J. Zebrafish as an animal model for drug discovery in Parkinson’s disease and other movement disorders: A systematic review. Front. Neurol. 2018, 9, 347. [Google Scholar] [CrossRef]
- Metzger, J.M.; Emborg, M.E. Autonomic dysfunction in Parkinson disease and animal models. Clin. Auton. Res. 2019, 29, 1–18. [Google Scholar] [CrossRef]
- Bezard, E.; Yue, Z.; Kirik, D.; Spillantini, M.G. Animal models of Parkinson’s disease: Limits and relevance to neuroprotection studies. Mov. Disord. 2013, 28, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martinez, A.; Luo, N.; Clemente, P.; Adan, C.; Hernandez-Sierra, R.; Ochoa, P.; Fernandez-Moreno, M.A.; Kaguni, L.S.; Garesse, R. Modeling human mitochondrial diseases in flies. Biochim. Biophys. Acta 2006, 1757, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayajuddin, M.; Abhik, D.; Prom, L.; Modi, P.; Chaurasia, R.; Koza, Z.; Thepa, A.; Jamir, N.; Singh, P.R.; Longkumer, S.; et al. Parkinson’s Disease: Insights from Drosophila Model; IntechOpen: London, UK, 2018. [Google Scholar]
- Dabool, L.; Juravlev, L.; Hakim-Mishnaevski, K.; Kurant, E. Modeling Parkinson’s disease in adult Drosophila. J. Neurosci. Methods 2019, 311, 89–94. [Google Scholar] [CrossRef]
- Xiong, Y.; Yu, J. Modeling Parkinson’s Disease in Drosophila: What Have We Learned for Dominant Traits? Front. Neurol. 2018, 9, 228. [Google Scholar] [CrossRef]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Soriano, V.; Paricio, N. Drosophila models of Parkinson’s disease: Discovering relevant pathways and novel therapeutic strategies. Parkinsons Dis. 2011, 2011, 520640. [Google Scholar] [CrossRef] [PubMed]
- Varga, S.J.; Qi, C.; Podolsky, E.; Lee, D. A new Drosophila model to study the interaction between genetic and environmental factors in Parkinson’s disease. Brain Res. 2014, 1583, 277–286. [Google Scholar] [CrossRef]
- Coulom, H.; Birman, S. Chronic exposure to rotenone models sporadic Parkinson’s disease in Drosophila melanogaster. J. Neurosci. 2004, 24, 10993–10998. [Google Scholar] [CrossRef]
- Soares, J.J.; Rodrigues, D.T.; Goncalves, M.B.; Lemos, M.C.; Gallarreta, M.S.; Bianchini, M.C.; Gayer, M.C.; Puntel, R.L.; Roehrs, R.; Denardin, E.L.G. Paraquat exposure-induced Parkinson’s disease-like symptoms and oxidative stress in Drosophila melanogaster: Neuroprotective effect of Bougainvillea glabra Choisy. Biomed. Pharmacother. 2017, 95, 245–251. [Google Scholar] [CrossRef]
- Whitworth, A.J. Drosophila models of Parkinson’s disease. Adv. Genet. 2011, 73, 1–50. [Google Scholar]
- Bajracharya, R.; Ballard, J.W.O. Low protein to carbohydrate ratio diet delays onset of Parkinsonism like phenotype in Drosophila melanogaster parkin null mutants. Mech. Ageing Dev. 2016, 160, 19–27. [Google Scholar] [CrossRef] [PubMed]
- West, R.J.; Furmston, R.; Williams, C.A.; Elliott, C.J. Neurophysiology of Drosophila models of Parkinson’s disease. Parkinsons Dis. 2015, 2015, 381281. [Google Scholar] [PubMed]
- Antony, P.M.; Diederich, N.J.; Balling, R. Parkinson’s disease mouse models in translational research. Mamm. Genome 2011, 22, 401–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Dawson, T.M.; Dawson, V.L. Models of LRRK2-associated Parkinson’s disease. Adv. Neurobiol. 2017, 14, 163–191. [Google Scholar] [PubMed]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of Parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef]
- Kam, T.I.; Mao, X.B.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP–ribose) drives pathologic alpha–synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; de Jong, E.M.; Broersen, L.M.; van Wijk, N.; Attali, A.; Garssen, J.; Kraneveld, A.D. Promising effects of neurorestorative diets on motor, cognitive, and gastrointestinal dysfunction after symptom development in a mouse model of Parkinson’s disease. Front. Aging Neurosci. 2017, 9, 57. [Google Scholar] [CrossRef]
- van Wijk, N.; Broersen, L.M.; de Wilde, M.C.; Hageman, R.J.; Groenendijk, M.; Sijben, J.W.; Kamphuis, P.J. Targeting synaptic dysfunction in Alzheimer’s disease by administering a specific nutrient combination. J. Alzheimers Dis. 2014, 38, 459–479. [Google Scholar] [CrossRef]
- Perez-Pardo, P.; Broersen, L.M.; Kliest, T.; van Wijk, N.; Attali, A.; Garssen, J.; Kraneveld, A.D. Additive effects of Levodopa and a neurorestorative diet in a mouse model of Parkinson’s disease. Front. Aging Neurosci. 2018, 10, 237. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. The role of O2–in the production of HO: In vitro and in vivo. Free Radic. Biol. Med. 1994, 16, 29–33. [Google Scholar] [CrossRef]
- Antunes, F.; Han, D.; Cadenas, E. Relative contributions of heart mitochondria glutathione peroxidase and catalase to H(2)O(2) detoxification in in vivo conditions. Free Radic. Biol. Med. 2002, 33, 1260–1267. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione–dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. 2013, 18, 145–146. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010, 51, 440–450. [Google Scholar] [CrossRef]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef]
- Ferguson, L.R.; Karunasinghe, N.; Zhu, S.; Wang, A.H. Selenium and its’ role in the maintenance of genomic stability. Mutat. Res. 2012, 733, 100–110. [Google Scholar] [CrossRef]
- Shahar, A.; Patel, K.V.; Semba, R.D.; Bandinelli, S.; Shahar, D.R.; Ferrucci, L.; Guralnik, J.M. Plasma selenium is positively related to performance in neurological tasks assessing coordination and motor speed. Mov. Disord. 2010, 25, 1909–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellwanger, J.H.; Molz, P.; Dallemole, D.R.; Pereira dos Santos, A.; Muller, T.E.; Cappelletti, L.; Goncalves da Silva, M.; Franke, S.I.; Pra, D.; Pegas Henriques, J.A. Selenium reduces bradykinesia and DNA damage in a rat model of Parkinson’s disease. Nutrition 2015, 31, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Moyerbrailean, G.A.; Richards, A.L.; Kurtz, D.; Kalita, C.A.; Davis, G.O.; Harvey, C.T.; Alazizi, A.; Watza, D.; Sorokin, Y.; Hauff, N.; et al. High–throughput allele–specific expression across 250 environmental conditions. Genome Res. 2016, 26, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Negida, A.; Menshawy, A.; El Ashal, G.; Elfouly, Y.; Hani, Y.; Hegazy, Y.; El Ghonimy, S.; Fouda, S.; Rashad, Y. Coenzyme Q10 for Patients with Parkinson’s Disease: A Systematic Review and Meta–Analysis. CNS Neurol. Disord Drug Targets 2016, 15, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.G.; Sun, M.X.; Zhang, W.L.; Wang, W.W.; Jin, Y.M.; Xie, C.L. The efficacy and safety of coenzyme Q10 in Parkinson’s disease: A meta–analysis of randomized controlled trials. Neurol. Sci. 2017, 38, 215–224. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Gu, C.; Xu, H. Effect of oxidative damage due to excessive protein ingestion on pancreas function in mice. Int. J. Mol. Sci. 2010, 11, 4591–4600. [Google Scholar] [CrossRef]
- Camiletti-Moiron, D.; Aparicio, V.A.; Nebot, E.; Medina, G.; Martinez, R.; Kapravelou, G.; Andrade, A.; Porres, J.M.; Lopez-Jurado, M.; Aranda, P. High–protein diet induces oxidative stress in rat brain: Protective action of high–intensity exercise against lipid peroxidation. Nutr. Hosp. 2014, 31, 866–874. [Google Scholar]
- Ayala, V.; Naudi, A.; Sanz, A.; Caro, P.; Portero-Otin, M.; Barja, G.; Pamplona, R. Dietary protein restriction decreases oxidative protein damage, peroxidizability index, and mitochondrial complex I content in rat liver. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 352–360. [Google Scholar] [CrossRef]
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, H.; Nakazaki, M.; Kanegae, Y.; Inukai, K.; Asano, T.; Katagiri, H.; Yazaki, Y.; Kikuchi, M.; Miyazaki, J.; Saito, I.; et al. Effect of mitochondrial and/or cytosolic glycerol 3–phosphate dehydrogenase overexpression on glucose–stimulated insulin secretion from MIN6 and HIT cells. Diabetes 1996, 45, 1238–1244. [Google Scholar] [CrossRef]
- Talior, I.; Yarkoni, M.; Bashan, N.; Eldar-Finkelman, H. Increased glucose uptake promotes oxidative stress and PKC–delta activation in adipocytes of obese, insulin–resistant mice. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E295–E302. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Rosca, M.G.; Vazquez, E.J.; Chen, Q.; Kerner, J.; Kern, T.S.; Hoppel, C.L. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 2012, 61, 2074–2083. [Google Scholar] [CrossRef]
- Schonfeld, P.; Wojtczak, L. Short– and medium–chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef]
- Calon, F.; Cicchetti, F. Can we prevent Parkinson’s disease with n–3 polyunsaturated fatty acids. Futur. Lipidol. 2008, 3, 133–137. [Google Scholar] [CrossRef]
- Da Boit, M.; Hunter, A.M.; Gray, S.R. Fit with good fat? The role of n–3 polyunsaturated fatty acids on exercise performance. Metabolism 2017, 66, 45–54. [Google Scholar] [CrossRef]
- Mori, M.A.; Delattre, A.M.; Carabelli, B.; Pudell, C.; Bortolanza, M.; Staziaki, P.V.; Visentainer, J.V.; Montanher, P.F.; Del Bel, E.A.; Ferraz, A.C. Neuroprotective effect of omega–3 polyunsaturated fatty acids in the 6–OHDA model of Parkinson’s disease is mediated by a reduction of inducible nitric oxide synthase. Nutr. Neurosci. 2018, 21, 341–351. [Google Scholar] [CrossRef]
- Hazel, J.R. Thermal adaptation in biological membranes: Is homeoviscous adaptation the explanation? Annu. Rev. Physiol. 1995, 57, 19–42. [Google Scholar] [CrossRef]
- Dimroth, P.; Kaim, G.; Matthey, U. Crucial role of the membrane potential for ATP synthesis by F(1)F(o) ATP synthases. J. Exp. Biol. 2000, 203, 51–59. [Google Scholar]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. JC–1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Bajracharya, R.; Bustamante, S.; Ballard, J.W.O. Stearic acid supplementation in high protein to carbohydrate (P:C) ratio diet improves physiological and mitochondrial functions of Drosophila melanogaster parkin null mutants. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 1–9. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Gatt, A.P.; Duncan, O.F.; Attems, J.; Francis, P.T.; Ballard, C.G.; Bateman, J.M. Dementia in Parkinson’s disease is associated with enhanced mitochondrial complex I deficiency. Mov. Disord. 2016, 31, 352–359. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine–analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef]
- Muftuoglu, M.; Elibol, B.; Dalmizrak, O.; Ercan, A.; Kulaksiz, G.; Ogus, H.; Dalkara, T.; Ozer, N. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov. Disord. 2004, 19, 544–548. [Google Scholar] [CrossRef]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Tsai, P.I.; Lin, C.H.; Hsieh, C.H.; Papakyrikos, A.M.; Kim, M.J.; Napolioni, V.; Schoor, C.; Couthouis, J.; Wu, R.M.; Wszolek, Z.K.; et al. PINK1 phosphorylates MIC60/Mitofilin to control structural plasticity of mitochondrial crista junctions. Mol. Cell 2018, 69, 744–756. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha–synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Bindoff, L.A.; Birch-Machin, M.A.; Cartlidge, N.E.; Parker, W.D., Jr.; Turnbull, D.M. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J. Neurol. Sci. 1991, 104, 203–208. [Google Scholar] [CrossRef]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Bioch. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef]
- Hattori, N.; Tanaka, M.; Ozawa, T.; Mizuno, Y. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann. Neurol. 1991, 30, 563–571. [Google Scholar] [CrossRef]
- Pravdic, D.; Hirata, N.; Barber, L.; Sedlic, F.; Bosnjak, Z.J.; Bienengraeber, M. Complex I and ATP synthase mediate membrane depolarization and matrix acidification by isoflurane in mitochondria. Eur. J. Pharmacol. 2012, 690, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef]
- Robinson, B.H. Human complex I deficiency: Clinical spectrum and involvement of oxygen free radicals in the pathogenicity of the defect. Biochim. Biophys. Acta 1998, 1364, 271–286. [Google Scholar] [CrossRef]
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef] [Green Version]
- Meex, R.C.R.; Schrauwen-Hinderling, V.B.; Moonen-Kornips, E.; Schaart, G.; Mensink, M.; Phielix, E.; van de Weijer, T.; Sels, J.-P.; Schrauwen, P.; Hesselink, M.K.C. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes 2010, 59, 572–579. [Google Scholar] [CrossRef]
- Aw, W.C.; Youngson, N.A.; Ballard, J.W.O. Can we alter dietary macronutrient compositions and alleviate mitochondrial disease? J. Rare Dis. Res. Treat. 2016, 1, 31–37. [Google Scholar] [Green Version]
- Yang, X.; Cheng, B. Neuroprotective and anti–inflammatory activities of ketogenic diet on MPTP–induced neurotoxicity. J. Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef]
- Deng-Bryant, Y.; Prins, M.L.; Hovda, D.A.; Harris, N.G. Ketogenic diet prevents alterations in brain metabolism in young but not adult rats after traumatic brain injury. J. Neurotrauma 2011, 28, 1813–1825. [Google Scholar] [CrossRef]
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Sanchez-Roman, I.; Gomez, A.; Gomez, J.; Suarez, H.; Sanchez, C.; Naudi, A.; Ayala, V.; Portero-Otin, M.; Lopez-Torres, M.; Pamplona, R.; et al. Forty percent methionine restriction lowers DNA methylation, complex I ROS generation, and oxidative damage to mtDNA and mitochondrial proteins in rat heart. J. Bioenerg. Biomembr. 2011, 43, 699–708. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Iommarini, L.; Kurelac, I.; Calvaruso, M.A.; Capristo, M.; Lollini, P.L.; Nanni, P.; Bergamini, C.; Nicoletti, G.; Giovanni, C.D.; et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria–defective tumor cells. Cancer Metab. 2013, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Senyilmaz, D.; Teleman, A.A. Chicken or the egg: Warburg effect and mitochondrial dysfunction. F1000Prime Rep. 2015, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lin, M.; Wu, R.; Wang, X.; Yang, B.; Levine, A.J.; Hu, W.; Feng, Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 2011, 108, 16259–16264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; O’Donnell, M.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A ketogenic diet improves mitochondrial biogenesis and bioenergetics via the PGC1alpha–SIRT3–UCP2 axis. Neurochem. Res. 2019, 44, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial inner–membrane fusion and crista maintenance requires the dynamin–related GTPase Mgm1. Cell 2006, 127, 383–395. [Google Scholar] [CrossRef]
- Rapaport, D.; Brunner, M.; Neupert, W.; Westermann, B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 20150–20155. [Google Scholar] [CrossRef]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar]
- Hermann, G.J.; Thatcher, J.W.; Mills, J.P.; Hales, K.G.; Fuller, M.T.; Nunnari, J.; Shaw, J.M. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 1998, 143, 359–373. [Google Scholar] [CrossRef]
- Ahlqvist, G.; Landin, S.; Wroblewski, R. Ultrastructure of skeletal muscle in patients with Parkinson’s disease and upper motor lesions. Lab. Investig. 1975, 32, 673–679. [Google Scholar]
- Hayashida, K.; Oyanagi, S.; Mizutani, Y.; Yokochi, M. An early cytoplasmic change before Lewy body maturation: An ultrastructural study of the substantia nigra from an autopsy case of juvenile parkinsonism. Acta Neuropathol. 1993, 85, 445–448. [Google Scholar] [CrossRef]
- Lach, B.; Grimes, D.; Benoit, B.; Minkiewicz-Janda, A. Caudate nucleus pathology in Parkinson’s disease: Ultrastructural and biochemical findings in biopsy material. Acta Neuropathol. 1992, 83, 352–360. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Trimmer, P.A.; Swerdlow, R.H.; Parks, J.K.; Keeney, P.; Bennett, J.P., Jr.; Miller, S.W.; Davis, R.E.; Parker, W.D., Jr. Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp. Neurol. 2000, 162, 37–50. [Google Scholar] [CrossRef]
- Grunewald, A.; Voges, L.; Rakovic, A.; Kasten, M.; Vandebona, H.; Hemmelmann, C.; Lohmann, K.; Orolicki, S.; Ramirez, A.; Schapira, A.H.; et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS ONE 2010, 5, e12962. [Google Scholar] [CrossRef]
- Yang, Y.; Ouyang, Y.; Yang, L.; Beal, M.F.; McQuibban, A.; Vogel, H.; Lu, B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. USA 2008, 105, 7070–7075. [Google Scholar] [CrossRef] [Green Version]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial–derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease–linked gene DJ–1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha–synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Putti, R.; Sica, R.; Migliaccio, V.; Lionetti, L. Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol. 2015, 6, 109. [Google Scholar] [CrossRef]
- Khraiwesh, H.; Lopez-Dominguez, J.A.; Lopez-Lluch, G.; Navas, P.; de Cabo, R.; Ramsey, J.J.; Villalba, J.M.; Gonzalez-Reyes, J.A. Alterations of ultrastructural and fission/fusion markers in hepatocyte mitochondria from mice following calorie restriction with different dietary fats. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 1023–1034. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, L.; Mollica, M.P.; Donizzetti, I.; Gifuni, G.; Sica, R.; Pignalosa, A.; Cavaliere, G.; Gaita, M.; De Filippo, C.; Zorzano, A.; et al. High–lard and high–fish–oil diets differ in their effects on function and dynamic behaviour of rat hepatic mitochondria. PLoS ONE 2014, 9, e92753. [Google Scholar] [CrossRef]
- Senyilmaz, D.; Virtue, S.; Xu, X.; Tan, C.Y.; Griffin, J.L.; Miller, A.K.; Vidal-Puig, A.; Teleman, A.A. Regulation of mitochondrial morphology and function by stearoylation of TFR1. Nature 2015, 525, 124–128. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, L.; Hu, W.; Zheng, Q.; Xiang, W. Mitochondrial dysfunction during in vitro hepatocyte steatosis is reversed by omega–3 fatty acid–induced up–regulation of mitofusin 2. Metabolism 2011, 60, 767–775. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [Green Version]
- Wlodarek, D. Role of ketogenic diets in neurodegenerative diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; Mitchell, S.J.; Coogan, S.C.; Cogger, V.C.; Gokarn, R.; McMahon, A.C.; Raubenheimer, D.; de Cabo, R.; Simpson, S.J.; Le Couteur, D.G. Dietary protein to carbohydrate ratio and caloric restriction: Comparing metabolic outcomes in mice. Cell Rep. 2015, 11, 1529–1534. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.O.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum–fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef]
- Lee, K.P.; Simpson, S.J.; Clissold, F.J.; Brooks, R.; Ballard, J.W.O.; Taylor, P.W.; Soran, N.; Raubenheimer, D. Lifespan and reproduction in Drosophila: New insights from nutritional geometry. Proc. Natl. Acad. Sci. USA 2008, 105, 2498–2503. [Google Scholar] [CrossRef]
- Le Couteur, D.G.; Solon-Biet, S.; Cogger, V.C.; Mitchell, S.J.; Senior, A.; de Cabo, R.; Raubenheimer, D.; Simpson, S.J. The impact of low–protein high–carbohydrate diets on aging and lifespan. Cell Mol. Life Sci. 2016, 73, 1237–1252. [Google Scholar] [CrossRef]
- Berry, E.M.; Growdon, J.H.; Wurtman, J.J.; Caballero, B.; Wurtman, R.J. A balanced carbohydrate: Protein diet in the management of Parkinson’s disease. Neurology 1991, 41, 1295–1297. [Google Scholar] [CrossRef]
- Mena, I.; Cotzias, G.C. Protein intake and treatment of Parkinson’s disease with levodopa. N. Engl. J. Med. 1975, 292, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Cereda, E.; Barichella, M.; Pedrolli, C.; Pezzoli, G. Low–protein and protein–redistribution diets for Parkinson’s disease patients with motor fluctuations: A systematic review. Mov. Disord. 2010, 25, 2021–2034. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; Mitchell, S.J.; de Cabo, R.; Raubenheimer, D.; Le Couteur, D.G.; Simpson, S.J. Macronutrients and caloric intake in health and longevity. J. Endocrinol. 2015, 226, R17–R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4–hydroxy–2–nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Farmer, E.E.; Mueller, M.J. ROS–mediated lipid peroxidation and RES–activated signaling. Annu. Rev. Plant Biol. 2013, 64, 429–450. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.J.; Katunga, L.A.; Willis, M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012, 39, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Mitochondrial filaments and clusters as intracellular power–transmitting cables. Trends Biochem. Sci. 2001, 26, 23–29. [Google Scholar] [CrossRef]
- Legros, F.; Lombes, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell. 2002, 13, 4343–4354. [Google Scholar] [CrossRef]
- Li, Q.; Lauber, C.L.; Czarnecki-Maulden, G.; Pan, Y.; Hannah, S.S. Effects of the dietary protein and carbohydrate ratio on gut microbiomes in dogs of different body conditions. mBio 2017, 8, e01703. [Google Scholar] [CrossRef]
- McAllan, L.; Skuse, P.; Cotter, P.D.; O’Connor, P.; Cryan, J.F.; Ross, R.P.; Fitzgerald, G.; Roche, H.M.; Nilaweera, K.N. Protein quality and the protein to carbohydrate ratio within a high fat diet influences energy balance and the gut microbiota in C57BL/6J mice. PLoS ONE 2014, 9, e88904. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Prinz, M. Microbiology: Gut microbes augment neurodegeneration. Nature 2017, 544, 304–305. [Google Scholar] [CrossRef] [PubMed]
- Bonanome, A.; Bennett, M.; Grundy, S.M. Metabolic effects of dietary stearic acid in mice: Changes in the fatty acid composition of triglycerides and phospholipids in various tissues. Atherosclerosis 1992, 94, 119–127. [Google Scholar] [CrossRef]
- Nachbaur, J.; Colbeau, A.; Vignais, P.M. Incorporation of fatty acids into the outer and inner membranes of isolated rat liver mitochondria. FEBS Lett. 1969, 3, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Mejia, E.M.; Hatch, G.M. Mitochondrial phospholipids: Role in mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef]
- Kang, M.J.; Shin, M.S.; Park, J.N.; Lee, S.S. The effects of polyunsaturated:saturated fatty acids ratios and peroxidisability index values of dietary fats on serum lipid profiles and hepatic enzyme activities in rats. Br. J. Nutr. 2005, 94, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin–deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [PubMed]
- Ceconi, C.; Curello, S.; Albertini, A.; Ferrari, R. Effect of lipid peroxidation on heart mitochondria oxygen consuming and calcium transporting capacities. Mol. Cell Biochem. 1988, 81, 131–135. [Google Scholar] [CrossRef]
- Holmes, D.J.; Austad, S.N. Birds as animal models for the comparative biology of aging: A prospectus. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, B59–B66. [Google Scholar] [CrossRef]
- Hulbert, A.J. Explaining longevity of different animals: Is membrane fatty acid composition the missing link? Age 2008, 30, 89–97. [Google Scholar] [CrossRef]
- Madrigal, J.L.; Olivenza, R.; Moro, M.A.; Lizasoain, I.; Lorenzo, P.; Rodrigo, J.; Leza, J.C. Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 2001, 24, 420–429. [Google Scholar] [CrossRef]
- Rafique, R.; Schapira, A.H.; Cooper, J.M. Sensitivity of respiratory chain activities to lipid peroxidation: Effect of vitamin E deficiency. Biochem. J. 2001, 357, 887–892. [Google Scholar] [CrossRef]
- Pamplona, R.; Portero-Otin, M.; Riba, D.; Ruiz, C.; Prat, J.; Bellmunt, M.J.; Barja, G. Mitochondrial membrane peroxidizability index is inversely related to maximum life span in mammals. J. Lipid Res. 1998, 39, 1989–1994. [Google Scholar]
Figure 1.
Potential mechanisms by which low protein and high carbohydrate diet can prevent mitochondrial dysfunction in Parkinson’s disease. Low P:C ratio diet can enable mitochondrial independent respiration causing low reactive oxygen species (ROS) generation which may lead to low oxidative stress. Low P:C ratio diet may also facilitate lower ROS generation preventing lipid peroxidation. Low P:C ratio diet can promote mitochondrial fusion and function. The bottom rectangle is the macronutrient (low P:C ratio diet). The ovals represent the potential mechanisms and processes of how macronutrients can affect mitochondrial functions. The top blue filled rectangles are the ways that can lead to healthy and efficient mitochondria in PD.
Figure 1.
Potential mechanisms by which low protein and high carbohydrate diet can prevent mitochondrial dysfunction in Parkinson’s disease. Low P:C ratio diet can enable mitochondrial independent respiration causing low reactive oxygen species (ROS) generation which may lead to low oxidative stress. Low P:C ratio diet may also facilitate lower ROS generation preventing lipid peroxidation. Low P:C ratio diet can promote mitochondrial fusion and function. The bottom rectangle is the macronutrient (low P:C ratio diet). The ovals represent the potential mechanisms and processes of how macronutrients can affect mitochondrial functions. The top blue filled rectangles are the ways that can lead to healthy and efficient mitochondria in PD.

Figure 2.
Potential mechanisms by which fatty acid can prevent mitochondrial dysfunction in PD. Fatty acid can be involved in mitochondrial function in three possible ways. First, fatty acid (stearic acid) can act as a signalling molecule to promote mitochondrial fusion. Second, saturated fatty acids can be directly incorporated into leaky mitochondrial membranes. Third, saturated fatty acids can be incorporated into lipid membranes making them resistant to peroxidation. The bottom rectangle is the macronutrient (saturated fatty acid). The ovals represent the potential mechanisms and processes of how the macronutrient can affect mitochondrial functions. The top blue filled rectangles are the ways that can lead to healthy and efficient mitochondria in PD.
Figure 2.
Potential mechanisms by which fatty acid can prevent mitochondrial dysfunction in PD. Fatty acid can be involved in mitochondrial function in three possible ways. First, fatty acid (stearic acid) can act as a signalling molecule to promote mitochondrial fusion. Second, saturated fatty acids can be directly incorporated into leaky mitochondrial membranes. Third, saturated fatty acids can be incorporated into lipid membranes making them resistant to peroxidation. The bottom rectangle is the macronutrient (saturated fatty acid). The ovals represent the potential mechanisms and processes of how the macronutrient can affect mitochondrial functions. The top blue filled rectangles are the ways that can lead to healthy and efficient mitochondria in PD.
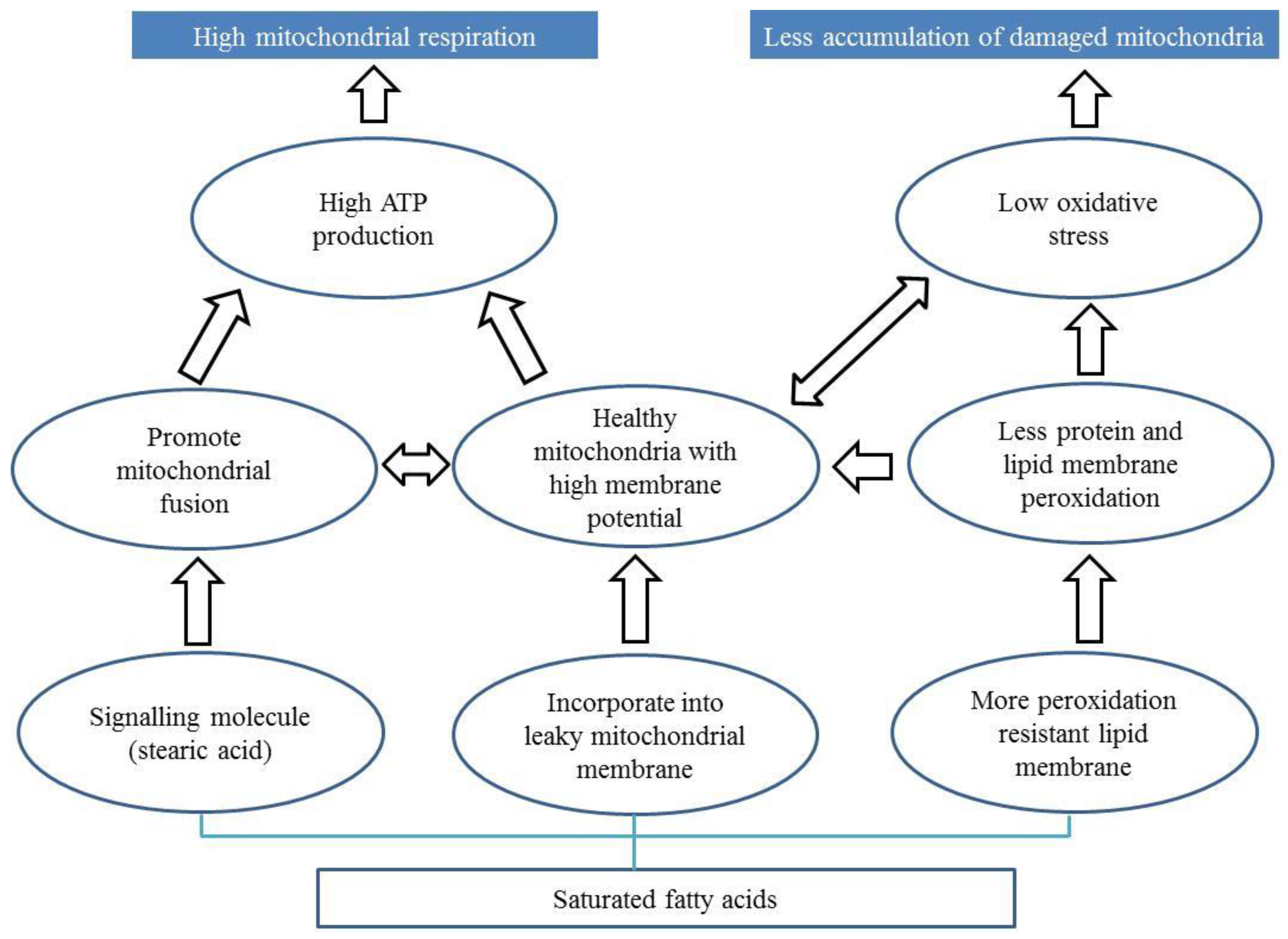
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bajracharya, R.; Youngson, N.A.; Ballard, J.W.O. Dietary Macronutrient Management to Treat Mitochondrial Dysfunction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 1850. https://doi.org/10.3390/ijms20081850
AMA Style
Bajracharya R, Youngson NA, Ballard JWO. Dietary Macronutrient Management to Treat Mitochondrial Dysfunction in Parkinson’s Disease. International Journal of Molecular Sciences. 2019; 20(8):1850. https://doi.org/10.3390/ijms20081850
Chicago/Turabian StyleBajracharya, Rijan, Neil A. Youngson, and J. William O. Ballard. 2019. "Dietary Macronutrient Management to Treat Mitochondrial Dysfunction in Parkinson’s Disease" International Journal of Molecular Sciences 20, no. 8: 1850. https://doi.org/10.3390/ijms20081850
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.