Differences in Stability of Viral and Viral-Cellular Fusion Transcripts in HPV-Induced Cervical Cancers

 , ,
, ,
Abstract
1. Introduction
2. Results
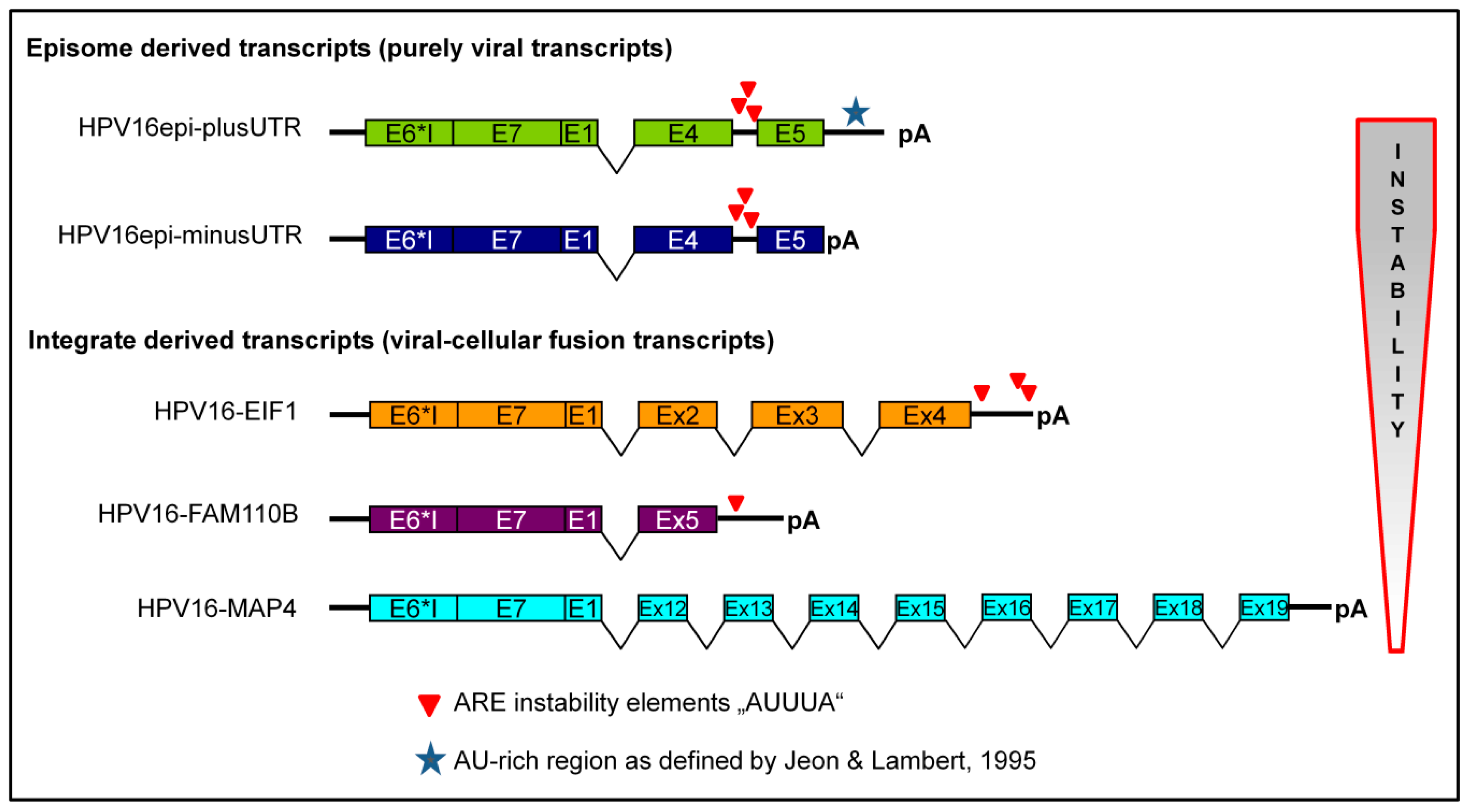
2.1. Recombinant Adenoviruses for Transduction of cDNAs Derived from Integrated and Episomal HPV16 DNA
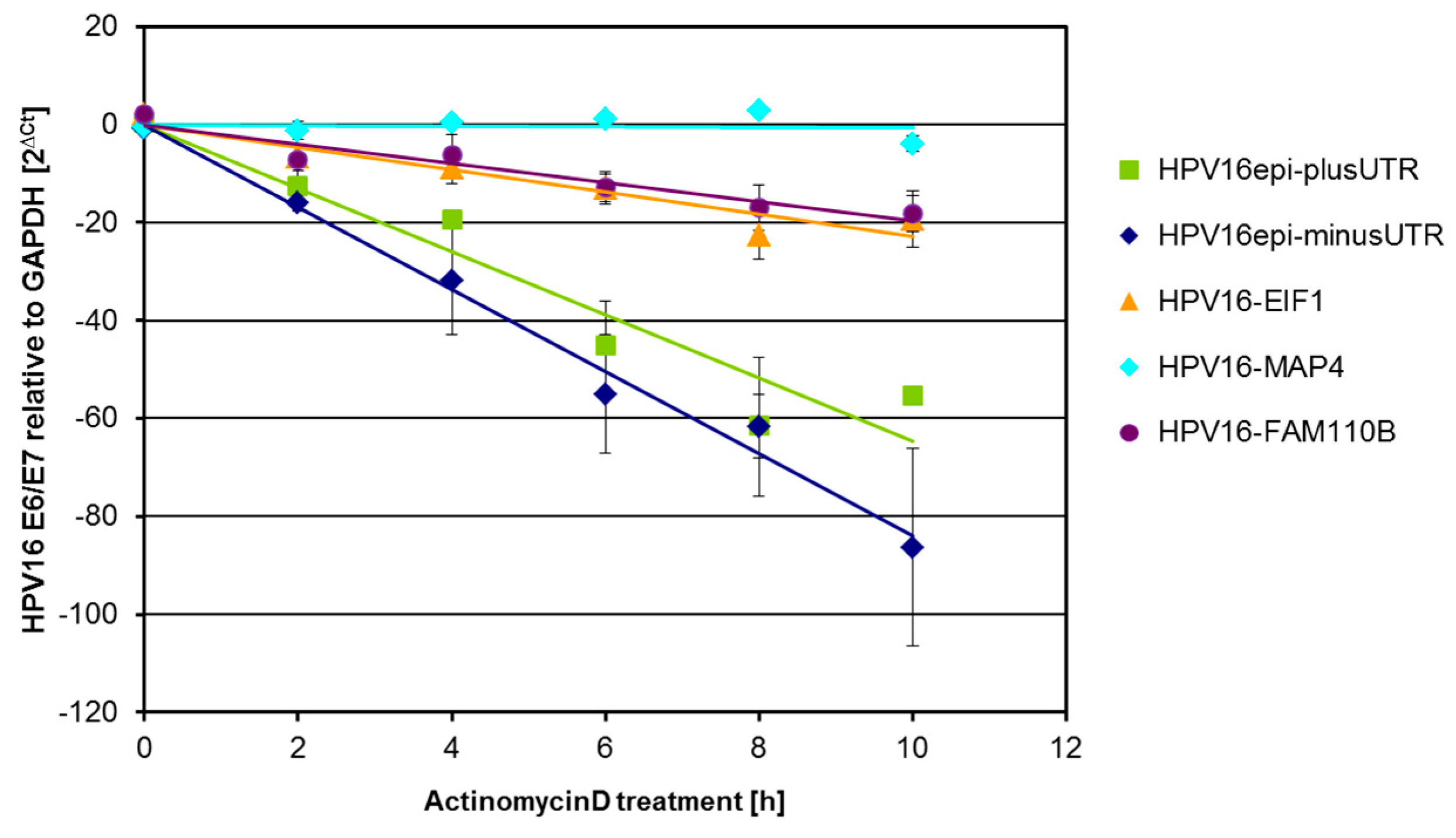
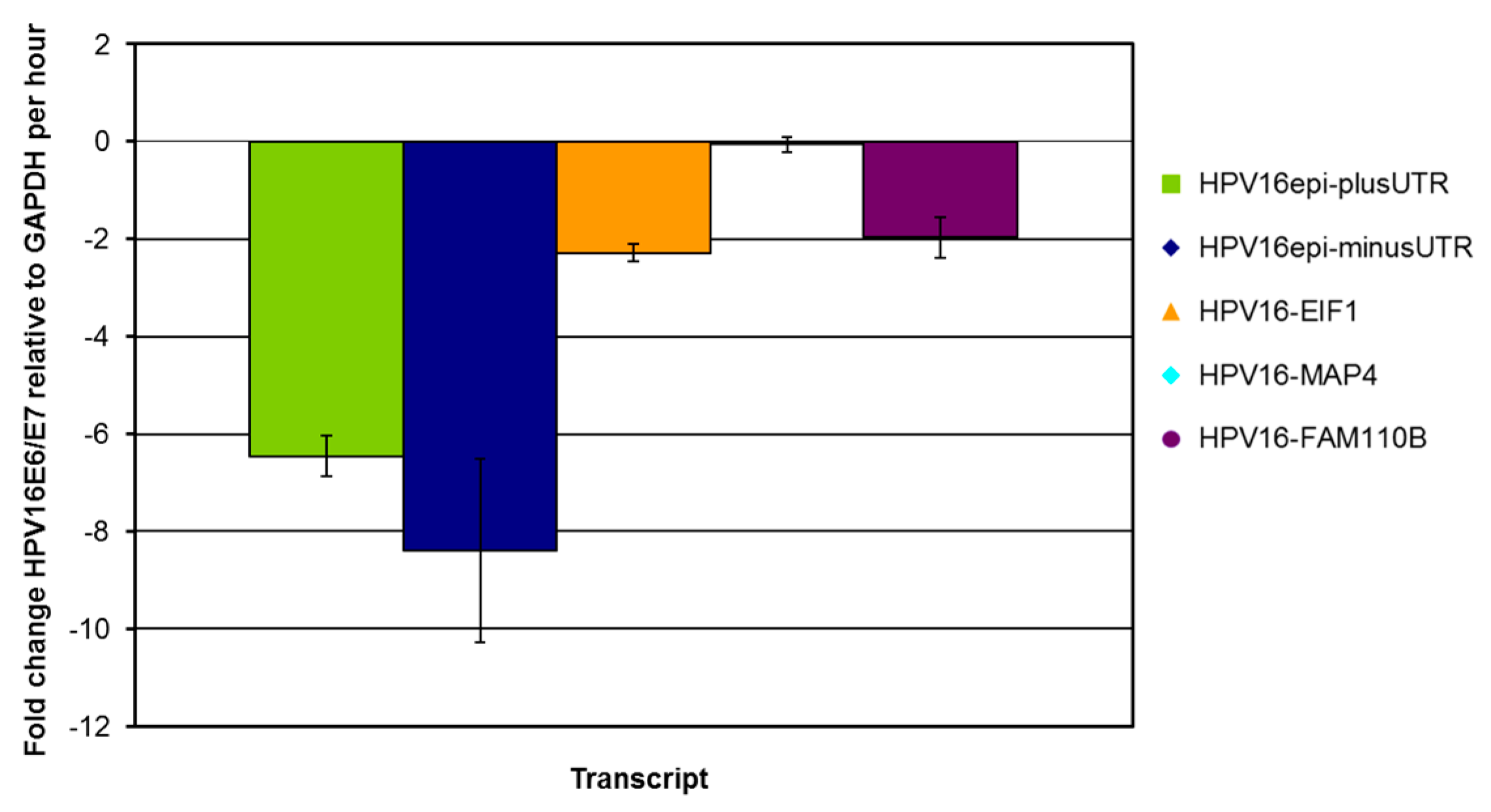
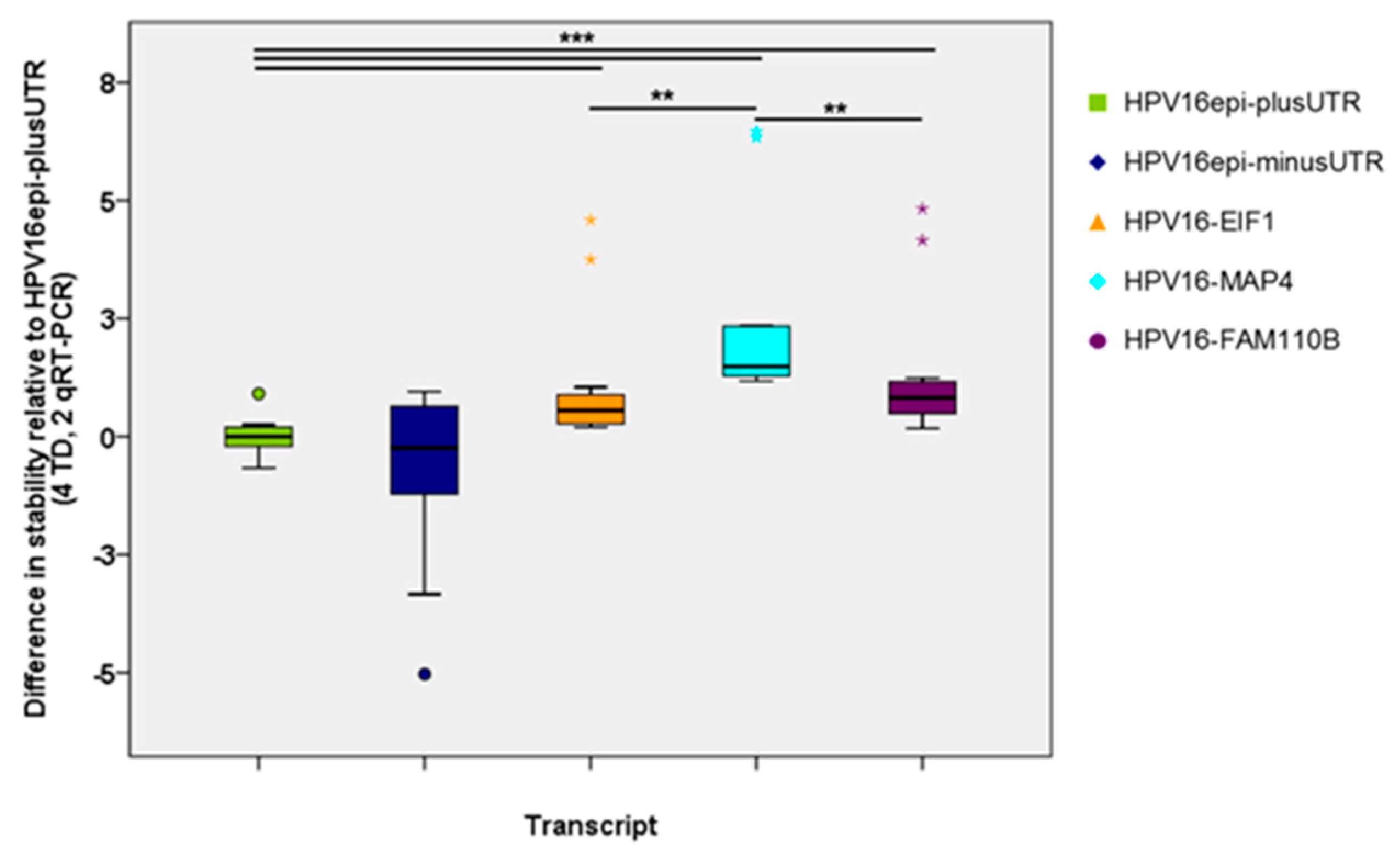
2.2. HPV16 Transcript Stability after Transduction of Human Primary Keratinocytes
2.3. Expression at the Protein Level
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of Adenovirus Constructs
4.3. Quantification of RNA Stability
4.4. HPV16-E7 Immunocytochemistry
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APOT | Amplification of Papillomavirus Oncogene Transcripts |
| ARE | Adenine and uridine rich element |
| CIN | Cervical epithelial neoplasia |
| Ct | Cycle threshold |
| moi | Multiplicity of infection |
| ncr | Non-coding region |
| qPCR | Quantitative PCR |
| RACE | Rapid amplification of cDNA ends |
| UTR | Untranslated region |
References
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. (Lond) 2006, 110, 525–541. [Google Scholar] [CrossRef]
- Woodman, C.B.; Collins, S.I.; Young, L.S. The natural history of cervical HPV infection: Unresolved issues. Nat. Rev. 2007, 7, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Vinokurova, S.; Wentzensen, N.; Kraus, I.; Klaes, R.; Driesch, C.; Melsheimer, P.; Kisseljov, F.; Durst, M.; Schneider, A.; von Knebel Doeberitz, M. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 2008, 68, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Hafner, N.; Driesch, C.; Gajda, M.; Jansen, L.; Kirchmayr, R.; Runnebaum, I.B.; Durst, M. Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts. Oncogene 2008, 27, 1610–1617. [Google Scholar] [CrossRef]
- Xu, B.; Chotewutmontri, S.; Wolf, S.; Klos, U.; Schmitz, M.; Durst, M.; Schwarz, E. Multiplex Identification of Human Papillomavirus 16 DNA Integration Sites in Cervical Carcinomas. PLoS ONE 2013, 8, e66693. [Google Scholar] [CrossRef]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef]
- Zhao, J.W.; Fang, F.; Guo, Y.; Zhu, T.L.; Yu, Y.Y.; Kong, F.F.; Han, L.F.; Chen, D.S.; Li, F. HPV16 integration probably contributes to cervical oncogenesis through interrupting tumor suppressor genes and inducing chromosome instability. J. Exp. Clin. Cancer Res. 2016, 35, 180. [Google Scholar] [CrossRef]
- Cripe, T.P.; Haugen, T.H.; Turk, J.P.; Tabatabai, F.; Schmid, P.G., 3rd; Durst, M.; Gissmann, L.; Roman, A.; Turek, L.P. Transcriptional regulation of the human papillomavirus-16 E6-E7 promoter by a keratinocyte-dependent enhancer, and by viral E2 trans-activator and repressor gene products: Implications for cervical carcinogenesis. EMBO J. 1987, 6, 3745–3753. [Google Scholar] [CrossRef]
- Bechtold, V.; Beard, P.; Raj, K. Human papillomavirus type 16 E2 protein has no effect on transcription from episomal viral DNA. J. Virol. 2003, 77, 2021–2028. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high-and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Warburton, A.; Redmond, C.J.; Dooley, K.E.; Fu, H.; Gillison, M.L.; Akagi, K.; Symer, D.E.; Aladjem, M.I.; McBride, A.A. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLoS Genet. 2018, 14, e1007179. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.; Lameiras, S.; Jeannot, E.; Marie, Y.; Castera, L.; Sastre-Garau, X.; Nicolas, A. Mechanistic signatures of HPV insertions in cervical carcinomas. NPJ Genom. Med. 2016, 1, 16004. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Lambert, P.F. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 1654–1658. [Google Scholar] [CrossRef]
- Ziegert, C.; Wentzensen, N.; Vinokurova, S.; Kisseljov, F.; Einenkel, J.; Hoeckel, M.; von Knebel Doeberitz, M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 2003, 22, 3977–3984. [Google Scholar] [CrossRef]
- Kraus, I.; Driesch, C.; Vinokurova, S.; Hovig, E.; Schneider, A.; von Knebel Doeberitz, M.; Durst, M. The majority of viral-cellular fusion transcripts in cervical carcinomas cotranscribe cellular sequences of known or predicted genes. Cancer Res. 2008, 68, 2514–2522. [Google Scholar] [CrossRef]
- Khabar, K.S. The AU-rich transcriptome: More than interferons and cytokines, and its role in disease. J. Interferon Cytokine Res. 2005, 25, 1–10. [Google Scholar] [CrossRef]
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2005, 33, 7138–7150. [Google Scholar] [CrossRef]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Schmitz, M.; Driesch, C.; Jansen, L.; Runnebaum, I.B.; Dürst, M. Non-Random Integration of the HPV Genome in Cervical Cancer. PLoS ONE 2012, 7, e39632. [Google Scholar] [CrossRef] [PubMed]
- Durst, M.; Dzarlieva-Petrusevska, R.T.; Boukamp, P.; Fusenig, N.E.; Gissmann, L. Molecular and cytogenetic analysis of immortalized human primary keratinocytes obtained after transfection with human papillomavirus type 16 DNA. Oncogene 1987, 1, 251–256. [Google Scholar] [PubMed]
- Klaes, R.; Woerner, S.M.; Ridder, R.; Wentzensen, N.; Duerst, M.; Schneider, A.; Lotz, B.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999, 59, 6132–6136. [Google Scholar] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef]
- Lidqvist, M.; Nilsson, O.; Holmgren, J.; Holters, S.; Roijer, E.; Durst, M.; Fermer, C. Detection of human papillomavirus oncoprotein E7 in liquid-based cytology. J. Gen. Virol. 2012, 93, 356–363. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lab Code | cDNA for Transduction | Insert Size | Additional Information |
|---|---|---|---|
| HPV16epi-plusUTR (Ref) | E6*I-E7-E1VE4-E5 plus 3’UTR | 1477 bp | RefSeq: NC_001526.2 nt 83-4211 |
| HPV16epi-minusUTR | E6*I-E7-E1VE4-E5 minus 3’UTR | 1358 bp | RefSeq: NC_001526.2 nt 83-4101 |
| HPV16-EIF1 | E6*I-E7-E1VEIF1 exons 2V3V4 plus 3’UTR | 1757 bp | Integration site 17q21.2 RefSeq: NM_005801 nt 186-1327 |
| HPV16-MAP4 | E6*I-E7-E1VMAP4 exons 12v13v14v15v16v17v18v19 plus 3’UTR | 2732 bp | Integration site 3p21 RefSeq: NM_002375 nt 2602-4718 |
| HPV16-FAM110B | E6*I-E7-E1VFAM110B exon 5 plus 3’UTR | 3492 bp | Integration site 8q12.1 RefSeq: NM_147189 nt 588-3463 |
| Primer | Sequence | Product Size and Annealing Temperature |
|---|---|---|
| GAPDH-F | 5′-GCGACACCCACTCCTCCACC-3′ | 119 bp, 58 °C |
| GAPDH-R | 5′-GAGGTCCACCACCCTGTTGC-3′ | |
| HPV16-F | 5′-AATGTTTCAGGACCCACAGG-3′ | 124 bp, 58 °C |
| HPV16-R | 5′-CTCACGTCGCAGTAACTGTTG-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ehrig, F.; Häfner, N.; Driesch, C.; Kraus Christiansen, I.; Beer, K.; Schmitz, M.; Runnebaum, I.B.; Dürst, M. Differences in Stability of Viral and Viral-Cellular Fusion Transcripts in HPV-Induced Cervical Cancers. Int. J. Mol. Sci. 2020, 21, 112. https://doi.org/10.3390/ijms21010112
Ehrig F, Häfner N, Driesch C, Kraus Christiansen I, Beer K, Schmitz M, Runnebaum IB, Dürst M. Differences in Stability of Viral and Viral-Cellular Fusion Transcripts in HPV-Induced Cervical Cancers. International Journal of Molecular Sciences. 2020; 21(1):112. https://doi.org/10.3390/ijms21010112
Chicago/Turabian StyleEhrig, Franziska, Norman Häfner, Corina Driesch, Irene Kraus Christiansen, Katrin Beer, Martina Schmitz, Ingo B. Runnebaum, and Matthias Dürst. 2020. "Differences in Stability of Viral and Viral-Cellular Fusion Transcripts in HPV-Induced Cervical Cancers" International Journal of Molecular Sciences 21, no. 1: 112. https://doi.org/10.3390/ijms21010112
APA StyleEhrig, F., Häfner, N., Driesch, C., Kraus Christiansen, I., Beer, K., Schmitz, M., Runnebaum, I. B., & Dürst, M. (2020). Differences in Stability of Viral and Viral-Cellular Fusion Transcripts in HPV-Induced Cervical Cancers. International Journal of Molecular Sciences, 21(1), 112. https://doi.org/10.3390/ijms21010112