Dissection of Dynamic Transcriptome Landscape of Leaf, Bract, and Lupulin Gland in Hop (Humulus lupulus L.)


 , , , ,
, , , ,  ,
,
Abstract
1. Introduction
2. Results
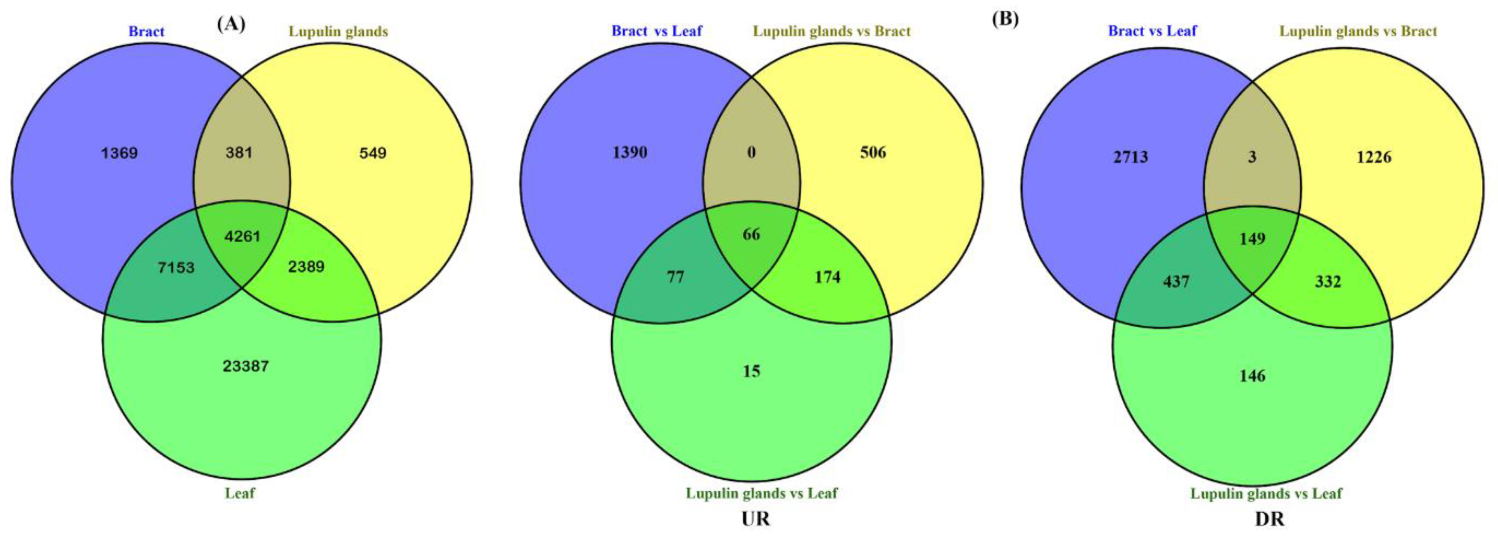
2.1. Transcriptome Sequence Analysis and De Novo Assembly
2.2. Functional Annotation and Classification of Assembled Transcriptome
2.3. Analysis of Expression Profiles of the Unigenes in Different Tissues
2.4. Validation of the Candidate Gene Expression Patterns by qRT-PCR Analysis
2.5. Metabolite Profiling of Important Secondary Metabolite Content
3. Discussion
3.1. Building a Transcriptome Resource and Gene Annotation
3.2. The Differential Expression Patterns of Unigenes in Different Tissues
4. Material and Methods
4.1. Plant Materials
4.2. RNA Extraction, Illumina Sequencing, and Data Processing
4.3. Functional Annotation of Hop Assembled Unigenes
4.4. Differential Gene Expression and Biological Pathway Analysis
4.5. Real-Time PCR Analysis of Gene Expression
4.6. Extraction and Quantitative Analysis of Important Secondary Metabolites
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Yoshimaru, T.; Komatsu, M.; Tashiro, E.; Imoto, M.; Osada, H.; Miyoshi, Y.; Honda, J.; Sasa, M.; Katagiri, T. Xanthohumol suppresses oestrogen-signalling in breast cancer through the inhibition of BIG3-PHB2 interactions. Sci. Rep. 2014, 4, 7355. [Google Scholar] [CrossRef] [PubMed]
- Quiñones, M.; Miguel, M.; Aleixandre, A. Beneficial effects of polyphenols on cardiovascular disease. Pharmacol. Res. 2013, 68, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.M. Phenolic Compounds in Botanical Extracts Used in Foods, Flavors, Cosmetics, and Pharmaceuticals. In Phenolic Compounds in Food and Their Effects on Health I; ACS Symposium Series; American Chemical Society: Washington, WA, USA, 1992; Volume 506, pp. 154–168. ISBN 9780841224759. [Google Scholar]
- Hartkorn, A.; Hoffmann, F.; Ajamieh, H.; Vogel, S.; Heilmann, J.; Gerbes, A.L.; Vollmar, A.M.; Zahler, S. Antioxidant Effects of Xanthohumol and Functional Impact on Hepatic Ischemia−Reperfusion Injury. J. Nat. Prod. 2009, 72, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Mizutani, M.; Hitomi, Y.; Yajima, H.; Kondo, K. Isohumulones, the bitter component of beer, improve hyperglycemia and decrease body fat in Japanese subjects with prediabetes. Clin. Nutr. 2009, 28, 278–284. [Google Scholar] [CrossRef]
- Raza, S.S.; Khan, M.M.; Ahmad, A.; Ashafaq, M.; Islam, F.; Wagner, A.P.; Safhi, M.M.; Islam, F. Neuroprotective effect of naringenin is mediated through suppression of NF-κB signaling pathway in experimental stroke. Neuroscience 2013, 230, 157–171. [Google Scholar] [CrossRef]
- Franco, L.; Sánchez, C.; Bravo, R.; Rodriguez, A.; Barriga, C.; Juánez, J. The sedative effects of hops (Humulus lupulus), a component of beer, on the activity/rest rhythm. Acta Physiol. Hung. 2012, 99, 133–139. [Google Scholar] [CrossRef]
- Sugiyama, R.; Oda, H.; Kurosaki, F. Two distinct phases of glandular trichome development in hop (Humulus lupulus L.). Plant Biotechnol. 2006, 23, 493–496. [Google Scholar] [CrossRef][Green Version]
- Nagel, J.; Culley, L.K.; Lu, Y.; Liu, E.; Matthews, P.D.; Stevens, J.F.; Page, J.E. EST analysis of hop glandular trichomes identifies an O-methyltransferase that catalyzes the biosynthesis of xanthohumol. Plant Cell 2008, 20, 186–200. [Google Scholar] [CrossRef]
- Larkin, J.C.; Brown, M.L.; Schiefelbein, J. How Do Cells Know What They Want to Be When They Grow Up? Lessons from Epidermal Patterning in Arabidopsis. Annu. Rev. Plant Biol. 2003, 54, 403–430. [Google Scholar] [CrossRef]
- Turner, G.W.; Gershenzon, J.; Croteau, R.B. Development of Peltate Glandular Trichomes of Peppermint. Plant Physiol. 2000, 124, 665–680. [Google Scholar] [CrossRef]
- Bergau, N.; Bennewitz, S.; Syrowatka, F.; Hause, G.; Tissier, A. The development of type VI glandular trichomes in the cultivated tomato Solanum lycopersicum and a related wild species S. habrochaites. BMC Plant Biol. 2015, 15, 289. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bartholomew, E.; Cai, Y.; Ren, H. Trichome-Related Mutants Provide a New Perspective on Multicellular Trichome Initiation and Development in Cucumber (Cucumis sativus L). Front. Plant Sci. 2016, 7, 1187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Morohashi, K.; Hatlestad, G.; Grotewold, E.; Lloyd, A. The TTG1-bHLH-MYB complex controls trichome cell fate and patterning through direct targeting of regulatory loci. Development 2008, 135, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Glover, B.J.; Perez-Rodriguez, M.; Martin, C. Development of several epidermal cell types can be specified by the same MYB-related plant transcription factor. Development 1998, 125, 3497–3508. [Google Scholar]
- Yang, S.; Miao, H.; Zhang, S.; Cheng, Z.; Zhou, J.; Dong, S.; Wehner, T.C.; Gu, X. Genetic Analysis and Mapping of gl-2 Gene in Cucumber(Cucumis sativus L.). Acta Hortic. Sin. 2011, 38, 1685–1692. [Google Scholar]
- Li, Q.; Cao, C.; Zhang, C.; Zheng, S.; Wang, Z.; Wang, L.; Ren, Z. The identification of Cucumis sativus Glabrous 1 (CsGL1) required for the formation of trichomes uncovers a novel function for the homeodomain-leucine zipper I gene. J. Exp. Bot. 2015, 66, 2515–2526. [Google Scholar] [CrossRef]
- Cui, J.-Y.; Miao, H.; Ding, L.-H.; Wehner, T.C.; Liu, P.-N.; Wang, Y.; Zhang, S.-P.; Gu, X.-F. A New Glabrous Gene (csgl3) Identified in Trichome Development in Cucumber (Cucumis sativus L.). PLoS ONE 2016, 11, e0148422. [Google Scholar] [CrossRef]
- Ma, D.; Hu, Y.; Yang, C.; Liu, B.; Fang, L.; Wan, Q.; Liang, W.; Mei, G.; Wang, L.; Wang, H.; et al. Genetic basis for glandular trichome formation in cotton. Nat. Commun. 2016, 7, 10456. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, D.; Hu, R.; Hua, C.; Ali, I.; Zhang, A.; Liu, B.; Wu, M.; Huang, L.; Gan, Y. AtGIS, a C2H2 zinc-finger transcription factor from Arabidopsis regulates glandular trichome development through GA signaling in tobacco. Biochem. Biophys. Res. Commun. 2017, 483, 209–215. [Google Scholar] [CrossRef]
- Li, C.-F.; Zhu, Y.; Yu, Y.; Zhao, Q.-Y.; Wang, S.-J.; Wang, X.-C.; Yao, M.-Z.; Luo, D.; Li, X.; Chen, L.; et al. Global transcriptome and gene regulation network for secondary metabolite biosynthesis of tea plant (Camellia sinensis). BMC Genom. 2015, 16, 560. [Google Scholar] [CrossRef]
- MATOUSEK, J.; VRBA, L.; SKOPEK, J.; ORCTOVA, L.; PESINA, K.; Heyerick, A.; BAULCOMBE, D.; De Keukeleire, D. Sequence analysis of a “true” chalcone synthase (chs_H1) oligofamily from hop (Humulus lupulus L.) and PAP1 activation of chs_H1 in heterologous systems. J. Agric. Food Chem. 2006, 54, 7606–7615. [Google Scholar] [CrossRef] [PubMed]
- Tsurumaru, Y.; Sasaki, K.; Miyawaki, T.; Uto, Y.; Momma, T.; Umemoto, N.; Momose, M.; Yazaki, K. HlPT-1, a membrane-bound prenyltransferase responsible for the biosynthesis of bitter acids in hops. Biochem. Biophys. Res. Commun. 2012, 417, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Kocábek, T.; Patzak, J.; Stehlík, J.; Füssy, Z.; Krofta, K.; Heyerick, A.; Roldán-Ruiz, I.; Maloukh, L.; Keukeleire, D. De Cloning and Molecular Analysis of HlbZip1 and HlbZip2 Transcription Factors Putatively Involved in the Regulation of the Lupulin Metabolome in Hop (Humulus lupulus L.). J. Agric. Food Chem. 2010, 58, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Kocábek, T.; Patzak, J.; Füssy, Z.; Procházková, J.; Heyerick, A. Combinatorial analysis of lupulin gland transcription factors from R2R3Myb, bHLH and WDR families indicates a complex regulation of chs_H1 genes essential for prenylflavonoid biosynthesis in hop (Humulus Lupulus L.). BMC Plant Biol. 2012, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Kocábek, T.; Patzak, J.; Bříza, J.; Siglová, K.; Mishra, A.K.; Duraisamy, G.S.; Týcová, A.; Ono, E.; Krofta, K. The “putative” role of transcription factors from HlWRKY family in the regulation of the final steps of prenylflavonid and bitter acids biosynthesis in hop (Humulus lupulus L.). Plant Mol. Biol. 2016, 92, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Duraisamy, G.S.; Khare, M.; Kocábek, T.; Jakse, J.; Bříza, J.; Patzak, J.; Sano, T.; Matoušek, J. Genome-wide transcriptome profiling of transgenic hop (Humulus lupulus L.) constitutively overexpressing HlWRKY1 and HlWDR1 transcription factors. BMC Genom. 2018, 19, 739. [Google Scholar] [CrossRef] [PubMed]
- Kocábek, T.; Mishra, A.K.; Matoušek, J.; Patzak, J.; Lomnická, A.; Khare, M.; Krofta, K. The R2R3 transcription factor HlMYB8 and its role in flavonoid biosynthesis in hop (Humulus lupulus L.). Plant Sci. 2018, 269, 32–46. [Google Scholar] [CrossRef]
- Kalra, S.; Puniya, B.L.; Kulshreshtha, D.; Kumar, S.; Kaur, J.; Ramachandran, S.; Singh, K. De Novo Transcriptome Sequencing Reveals Important Molecular Networks and Metabolic Pathways of the Plant, Chlorophytum borivilianum. PLoS ONE 2013, 8, e83336. [Google Scholar] [CrossRef]
- Devi, K.; Mishra, S.K.; Sahu, J.; Panda, D.; Modi, M.K.; Sen, P. Genome wide transcriptome profiling reveals differential gene expression in secondary metabolite pathway of Cymbopogon winterianus. Sci. Rep. 2016, 6, 21026. [Google Scholar] [CrossRef]
- Loke, K.-K.; Rahnamaie-Tajadod, R.; Yeoh, C.-C.; Goh, H.-H.; Mohamed-Hussein, Z.-A.; Mohd Noor, N.; Zainal, Z.; Ismail, I. RNA-seq analysis for secondary metabolite pathway gene discovery in Polygonum minus. Genom. Data 2015, 7, 12–13. [Google Scholar] [CrossRef]
- Pokorn, T.; Radišek, S.; Javornik, B.; Štajner, N.; Jakše, J. Development of hop transcriptome to support research into host-viroid interactions. PLoS ONE 2017, 12, e0184528. [Google Scholar] [CrossRef] [PubMed]
- Considine, M.J.; Foyer, C.H. Redox regulation of plant development. Antioxid. Redox Signal. 2014, 21, 1305–1326. [Google Scholar] [CrossRef] [PubMed]
- Camara, B.; Bouvier, F. Oxidative remodeling of plastid carotenoids. Arch. Biochem. Biophys. 2004, 430, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.M.; Vaitheeswaran, V.; Ambrose, S.J.; Purves, R.W.; Page, J.E. Transcriptome analysis of bitter acid biosynthesis and precursor pathways in hop (Humulus lupulus). BMC Plant Biol. 2013, 13, 12. [Google Scholar] [CrossRef]
- Hölzer, M.; Marz, M. De novo transcriptome assembly: A comprehensive cross-species comparison of short-read RNA-Seq assemblers. GigaScience 2019, 8, giz039. [Google Scholar] [CrossRef]
- OKADA, Y.; ITO, K. Cloning and Analysis of Valerophenone Synthase Gene Expressed Specifically in Lupulin Gland of Hop (Humulus lupulus L.). Biosci. Biotechnol. Biochem. 2001, 65, 150–155. [Google Scholar] [CrossRef]
- Binder, S.; Knill, T.; Schuster, J. Branched-chain amino acid metabolism in higher plants. Physiol. Plant. 2007, 129, 68–78. [Google Scholar] [CrossRef]
- Zheng, X.; Xu, H.; Ma, X.; Zhan, R.; Chen, W. Triterpenoid saponin biosynthetic pathway profiling and candidate gene mining of the Ilex asprella root using RNA-Seq. Int. J. Mol. Sci. 2014, 15, 5970–5987. [Google Scholar] [CrossRef]
- Weitzel, C.; Simonsen, H.T. Cytochrome P450-enzymes involved in the biosynthesis of mono-and sesquiterpenes. Phytochem. Rev. 2015, 14, 7–24. [Google Scholar] [CrossRef]
- Lynch, J.H.; Orlova, I.; Zhao, C.; Guo, L.; Jaini, R.; Maeda, H.; Akhtar, T.; Cruz-Lebron, J.; Rhodes, D.; Morgan, J.; et al. Multifaceted plant responses to circumvent Phe hyperaccumulation by downregulation of flux through the shikimate pathway and by vacuolar Phe sequestration. Plant J. 2017, 92, 939–950. [Google Scholar] [CrossRef]
- Gatica-Arias, A.; Farag, M.A.; Stanke, M.; Matoušek, J.; Wessjohann, L.; Weber, G. Flavonoid production in transgenic hop (Humulus lupulus L.) altered by PAP1/MYB75 from Arabidopsis thaliana L. Plant Cell Rep. 2012, 31, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Watanabe, M.; Hoefgen, R.; Fernie, A.R. Shikimate and phenylalanine biosynthesis in the green lineage. Front. Plant Sci. 2013, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Schilmiller, A.L.; Last, R.L.; Pichersky, E. Harnessing plant trichome biochemistry for the production of useful compounds. Plant J. 2008, 54, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Jirschitzka, J.; Schmidt, G.W.; Reichelt, M.; Schneider, B.; Gershenzon, J.; D’Auria, J.C. Plant tropane alkaloid biosynthesis evolved independently in the Solanaceae and Erythroxylaceae. Proc. Natl. Acad. Sci. USA 2012, 109, 10304–10309. [Google Scholar] [CrossRef]
- Negri, G.; di Santi, D.; Tabach, R. Bitter acids from hydroethanolic extracts of Humulus lupulus L., Cannabaceae, used as anxiolytic. Rev. Bras. Farmacogn. 2010, 20, 850–859. [Google Scholar] [CrossRef]
- Jing, F.; Cantu, D.C.; Tvaruzkova, J.; Chipman, J.P.; Nikolau, B.J.; Yandeau-Nelson, M.D.; Reilly, P.J. Phylogenetic and experimental characterization of an acyl-ACP thioesterase family reveals significant diversity in enzymatic specificity and activity. BMC Biochem. 2011, 12, 44. [Google Scholar] [CrossRef]
- Huchelmann, A.; Boutry, M.; Hachez, C. Plant Glandular Trichomes: Natural Cell Factories of High Biotechnological Interest. Plant Physiol. 2017, 175, 6–22. [Google Scholar] [CrossRef]
- Wang, G.; Tian, L.; Aziz, N.; Broun, P.; Dai, X.; He, J.; King, A.; Zhao, P.X.; Dixon, R.A. Terpene Biosynthesis in Glandular Trichomes of Hop. Plant Physiol. 2008, 148, 1254–1266. [Google Scholar] [CrossRef]
- Schellmann, S.; Schnittger, A.; Kirik, V.; Wada, T.; Okada, K.; Beermann, A.; Thumfahrt, J.; Jürgens, G.; Hülskamp, M. TRIPTYCHON and CAPRICE mediate lateral inhibition during trichome and root hair patterning in Arabidopsis. EMBO J. 2002, 21, 5036–5046. [Google Scholar] [CrossRef]
- Saddic, L.A.; Huvermann, B.; Bezhani, S.; Su, Y.; Winter, C.M.; Kwon, C.S.; Collum, R.P.; Wagner, D. The LEAFY target LMI1 is a meristem identity regulator and acts together with LEAFY to regulate expression of CAULIFLOWER. Development 2006, 133, 1673–1682. [Google Scholar] [CrossRef]
- Zhao, J.-L.; Pan, J.-S.; Guan, Y.; Nie, J.-T.; Yang, J.-J.; Qu, M.-L.; He, H.-L.; Cai, R. Transcriptome analysis in Cucumis sativus identifies genes involved in multicellular trichome development. Genomics 2015, 105, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, Z.; Li, F. Updates on molecular mechanisms in the development of branched trichome in Arabidopsis and nonbranched in cotton. Plant Biotechnol. J. 2019, 17, 1706–1722. [Google Scholar] [CrossRef] [PubMed]
- Traw, M.B.; Bergelson, J. Interactive effects of jasmonic acid, salicylic acid, and gibberellin on induction of trichomes in Arabidopsis. Plant Physiol. 2003, 133, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Kazama, H.; Dan, H.; Imaseki, H.; Wasteneys, G.O. Transient Exposure to Ethylene Stimulates Cell Division and Alters the Fate and Polarity of Hypocotyl Epidermal Cells. Plant Physiol. 2004, 134, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Sun, L.; Zhao, Y.; An, L.; Yan, A.; Meng, X.; Gan, Y. Zinc Finger Protein 6 (ZFP6) regulates trichome initiation by integrating gibberellin and cytokinin signaling in Arabidopsis thaliana. New Phytol. 2013, 198, 699–708. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Natsume, S.; Takagi, H.; Shiraishi, A.; Murata, J.; Toyonaga, H.; Patzak, J.; Takagi, M.; Yaegashi, H.; Uemura, A.; Mitsuoka, C.; et al. The Draft Genome of Hop (Humulus lupulus), an Essence for Brewing. Plant Cell Physiol. 2014, 56, 428–441. [Google Scholar] [CrossRef]
- Iwata, H.; Gotoh, O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 2012, 40, e161. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 138–148. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Guo, Y.; Sheng, Q.; Shyr, Y. Advanced Heat Map and Clustering Analysis Using Heatmap3. Biomed Res. Int. 2014, 986048. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant. Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. mapman: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leaf | Bracts | Lupulin Glands | Combined | |
|---|---|---|---|---|
| Raw data | ||||
| Reads [M] | 66.02 | 53.15 | 22.98 | 127.37 |
| Amount of data [Gb] | 7.3 | 5.9 | 2.6 | 12.94 |
| Clean data | ||||
| Reads [M] | 61.13 | 50.01 | 20.18 | 118.32 |
| Average length [bp] | 75.14 | 108.55 | 112.25 | 100.10 |
| Amount of data [Gb] | 6.06 | 5.4 | 2.2 | 11.53 |
| Unigenes | ||||
| No. of Unigenes (n) | 37,690 | 13,164 | 7580 | 43,550 |
| Maximum Length (bp) | 19,887 | 7298 | 3257 | 21,367 |
| Average Length (bp) | 650 | 385 | 396 | 653 |
| Minimum Length (bp) | 105 | 127 | 121 | 144 |
| N25 | 1743 | 550 | 577 | 1740 |
| N50 | 1008 | 398 | 402 | 959 |
| GC% | 41.5 | 38.9 | 39.7 | 40.9 |
| Number of Unigenes in Pathway | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| KEGG Categories | Count of Pathways | UG | Bracts | Lupulin Glands | Leaf | Bract vs. Leaf | Lupulin Gland vs. Bract | Lupulin Gland vs. Leaf | |||
| Metabolism | UR | DR | UR | DR | UR | DR | |||||
| Carbohydrate Metabolism | 15 | 1000 | 330 | 222 | 511 | 50 | 160 | 67 | 65 | 23 | 65 |
| Energy metabolism | 07 | 379 | 211 | 152 | 240 | 27 | 79 | 23 | 56 | 4 | 52 |
| Lipid metabolism | 15 | 581 | 143 | 99 | 310 | 24 | 79 | 33 | 28 | 14 | 27 |
| Nucleotide metabolism | 02 | 205 | 44 | 23 | 106 | 5 | 33 | 5 | 7 | 1 | 13 |
| Amino acid metabolism | 14 | 711 | 230 | 155 | 409 | 31 | 115 | 55 | 30 | 18 | 31 |
| Metabolism of other amino acids | 09 | 202 | 59 | 36 | 112 | 12 | 33 | 9 | 11 | 3 | 12 |
| Glycan biosynthesis and metabolism | 11 | 331 | 63 | 23 | 193 | 10 | 18 | 6 | 8 | 0 | 5 |
| Metabolism of cofactors and vitamins | 12 | 382 | 105 | 67 | 209 | 15 | 62 | 19 | 22 | 6 | 26 |
| Metabolism of terpenoids and polyketides | 09 | 209 | 56 | 35 | 119 | 15 | 30 | 19 | 23 | 10 | 16 |
| Biosynthesis of other secondary metabolites | 17 | 321 | 95 | 56 | 165 | 40 | 30 | 32 | 20 | 27 | 12 |
| Xenobiotics biodegradation and metabolism | 18 | 156 | 52 | 35 | 75 | 5 | 19 | 17 | 13 | 4 | 12 |
| Genetic information processing | |||||||||||
| Transcription | 03 | 388 | 95 | 71 | 173 | 16 | 23 | 3 | 10 | 0 | 2 |
| Translation | 05 | 835 | 228 | 173 | 505 | 64 | 90 | 12 | 44 | 2 | 18 |
| Folding, sorting and degradation | 07 | 657 | 182 | 141 | 363 | 35 | 73 | 12 | 29 | 5 | 16 |
| Replication and repair | 07 | 469 | 65 | 26 | 281 | 14 | 32 | 5 | 19 | 1 | 8 |
| Cellular Process | |||||||||||
| Transport and catabolism | 09 | 815 | 192 | 129 | 456 | 35 | 100 | 20 | 36 | 4 | 30 |
| Cell growth and death | 11 | 724 | 137 | 79 | 423 | 33 | 68 | 7 | 20 | 2 | 19 |
| Cellular community—eukaryotes | 05 | 108 | 23 | 18 | 49 | 7 | 17 | 4 | 4 | 0 | 3 |
| Cellular community—prokaryotes | 03 | 60 | 18 | 9 | 30 | 4 | 10 | 2 | 5 | 1 | 4 |
| α Acids | β Acids | Cohumulone | Colupulone | XN | DMX | Total Oils | |
|---|---|---|---|---|---|---|---|
| hop sample | % w/w | % w/w | % rel | % rel | % w/w | % w/w | % w/w |
| lupulin glands | 26.06 ± 14.0 | 17.44 ± 7.8 | 26.1 ± 4.63 | 51.4 ± 7.29 | 1.63 ± 0.78 | 0.76 ± 0.06 | 3.24 ± 8.3 |
| bracts/bracteoles * | 5.39 ± 0.85 | 3.44 ± 0.26 | 23.2 ± 3.87 | 42.9 ± 5.18 | 0.39 ± 0.08 | 0.11 ± 0.03 | 1.24 ± 0.41 |
| leaves | 0.22 ± 0.08 | 0.16 ± 0.07 | ND | ND | 0.01 ± 0.00 | ND | 0.14 ± 0.03 |
| HopBase ID | Gene/Transcription Factors | Bract vs. Leaf | Lupulin Gland vs. Bract | Lupulin Gland vs. Leaf | Function |
|---|---|---|---|---|---|
| HL.SW.v1.0.G039151 | HlMIXTA1 | NDE | −22.76 | NDE | initiation of glandular trichomes |
| HL.SW.v1.0.G009685 | non-specific lipid-transfer protein 1-like | 38.87 | −3.82 | 37.49 | expansion of epidermal cells and certain secretory tissues. |
| HL.SW.v1.0.G021650 | protein ABIL2 | NDE | 43.57 | 27.20 | part of a WAVE complex that activates the Arp2/3 complex |
| HL.SW.v1.0.G041748 | signaling peptide TAXIMIN | NDE | 53.18 | NDE | organ boundary specification between lateral organs and the meristem |
| HL.SW.v1.0.G010965 | xyloglucanase inhibitor 1 | NDE | NDE | 15.12 | involved in the trichome cell wall composition |
| HL.SW.v1.0.G020081 | adaptor protein GIR1-like | 4.53 | 8.59 | 38.59 | involved in the initiation of trichome development; interacts with GLABRA2 |
| HL.SW.v1.0.G008259 | R3 Myb ETC1 | 76.34 | NDE | 32.51 | trichome development repressor |
| HL.SW.v1.0.G009660 | R3 Myb CPC-like | −6.61 | NDE | NDE | trichome development repressor |
| HL.SW.v1.0.G001811 | HlMIXTA1 Myb93-like | NDE | 62.26 | NDE | lateral root development |
| HL.SW.v1.0.G046443 | rax2; Myb 36 | −25.8 | 108.74 | NDE | cell differentiation |
| HL.SW.v1.0.G001378 | WRKY4-like | NDE | 8.53 | NDE | defense response; epidermal cell fate specification |
| HL.SW.v1.0.G044612 | homeobox-leucine zipper protein hox32 | 18.92 | −25.39 | NDE | polarity specification |
| HL.SW.v1.0.G026259 | homeobox-leucine zipper protein athb-51 | NDE | 90.91 | 42.03 | meristem regulator to control leaf or bract morphogenesis |
| HL.SW.v1.0.G021599 | Glabra2 | −3.75 | NDE | NDE | trichome initiation, trichome spacing |
| HL.SW.v1.0.G005787 | glabrous 11 | NDE | 48.69 | NDE | trichome branching |
| HL.SW.v1.0.G015009 | G2-like family protein; putative transcription factor KAN1 | NDE | 17.23 | NDE | lateral organ polarity |
| HL.SW.v1.0.G025215 | ap2 erf and b3 domain-containing transcription factor rav1-like | 4.61 | −8.84 | NDE | negative regulator of plant growth and development |
| HL.SW.v1.0.G041557 | yabby 1 | NDE | −88.54 | −16.89 | abaxial cell fate determination |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.K.; Kocábek, T.; Sukumari Nath, V.; Awasthi, P.; Shrestha, A.; Kumar Killi, U.; Jakse, J.; Patzak, J.; Krofta, K.; Matoušek, J. Dissection of Dynamic Transcriptome Landscape of Leaf, Bract, and Lupulin Gland in Hop (Humulus lupulus L.). Int. J. Mol. Sci. 2020, 21, 233. https://doi.org/10.3390/ijms21010233
Mishra AK, Kocábek T, Sukumari Nath V, Awasthi P, Shrestha A, Kumar Killi U, Jakse J, Patzak J, Krofta K, Matoušek J. Dissection of Dynamic Transcriptome Landscape of Leaf, Bract, and Lupulin Gland in Hop (Humulus lupulus L.). International Journal of Molecular Sciences. 2020; 21(1):233. https://doi.org/10.3390/ijms21010233
Chicago/Turabian StyleMishra, Ajay Kumar, Tomáš Kocábek, Vishnu Sukumari Nath, Praveen Awasthi, Ankita Shrestha, Uday Kumar Killi, Jernej Jakse, Josef Patzak, Karel Krofta, and Jaroslav Matoušek. 2020. "Dissection of Dynamic Transcriptome Landscape of Leaf, Bract, and Lupulin Gland in Hop (Humulus lupulus L.)" International Journal of Molecular Sciences 21, no. 1: 233. https://doi.org/10.3390/ijms21010233
APA StyleMishra, A. K., Kocábek, T., Sukumari Nath, V., Awasthi, P., Shrestha, A., Kumar Killi, U., Jakse, J., Patzak, J., Krofta, K., & Matoušek, J. (2020). Dissection of Dynamic Transcriptome Landscape of Leaf, Bract, and Lupulin Gland in Hop (Humulus lupulus L.). International Journal of Molecular Sciences, 21(1), 233. https://doi.org/10.3390/ijms21010233