Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses

 and
and 
Abstract
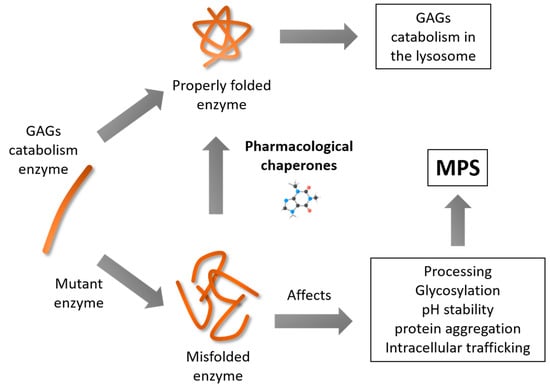
1. Introduction
2. Use of Pharmacological Chaperones in LSDs
3. Pharmacological Chaperones for MPS
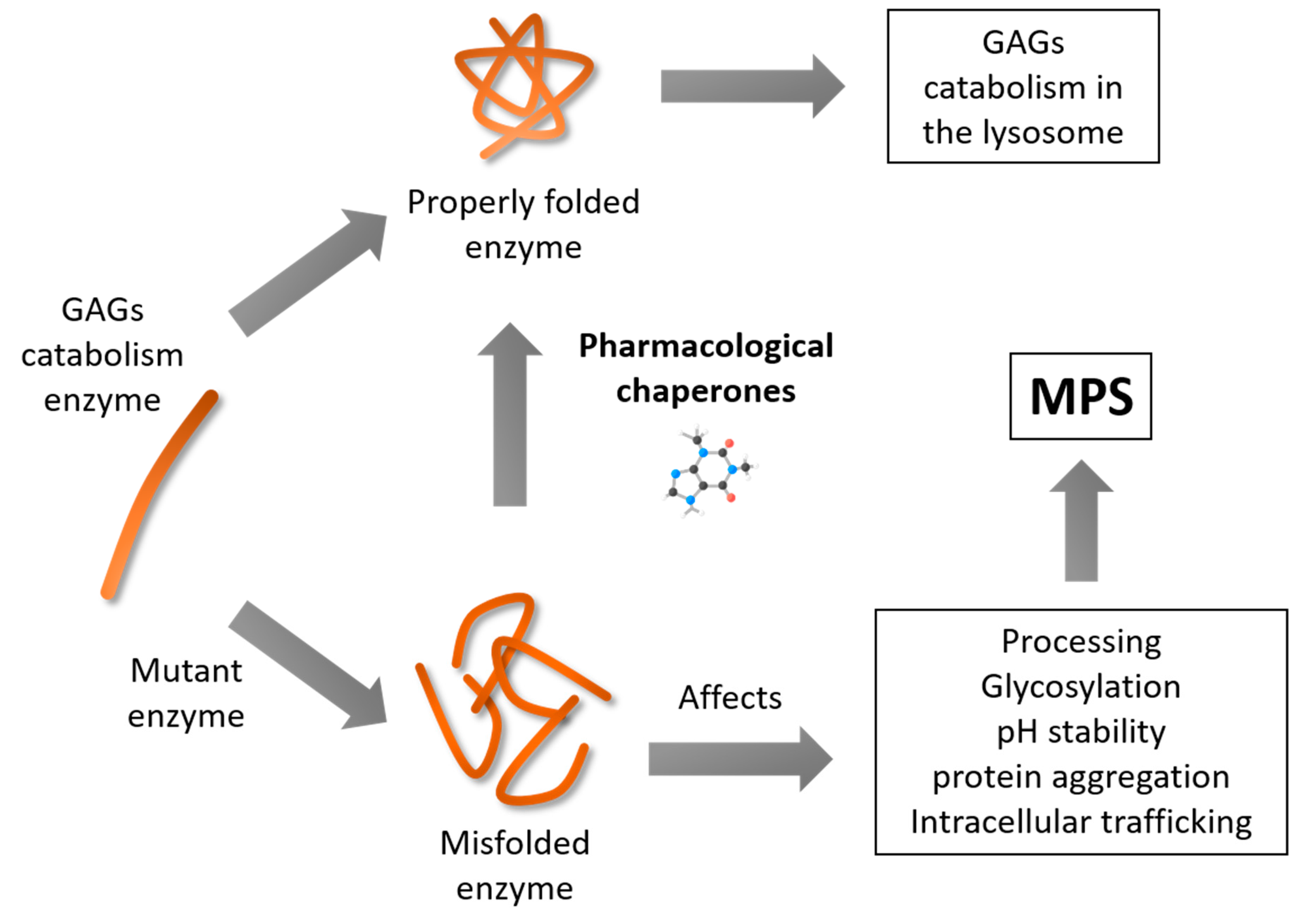
3.1. Mucopolysaccharidosis Type II
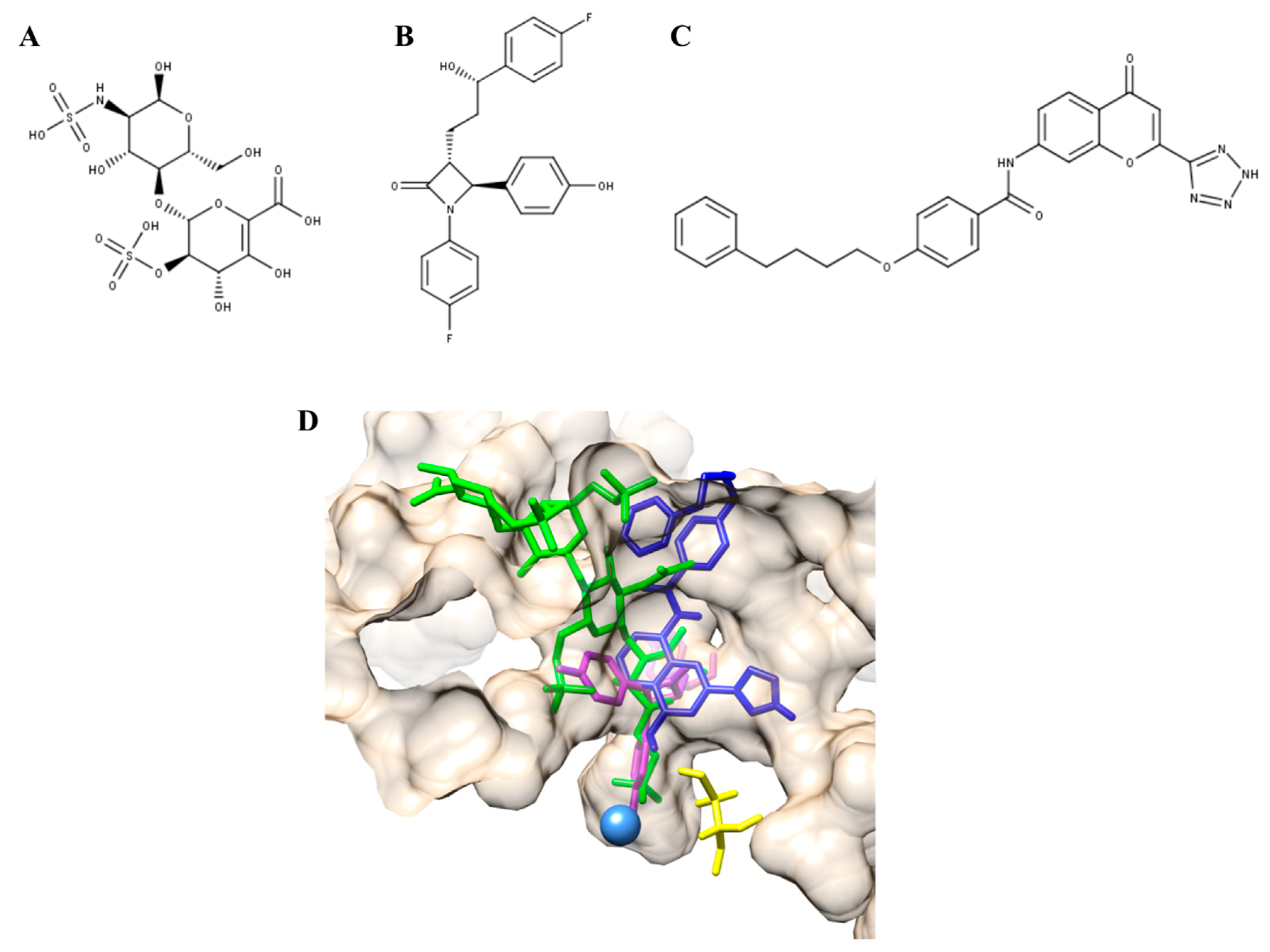
3.2. Mucopolysaccharidosis Type IVA
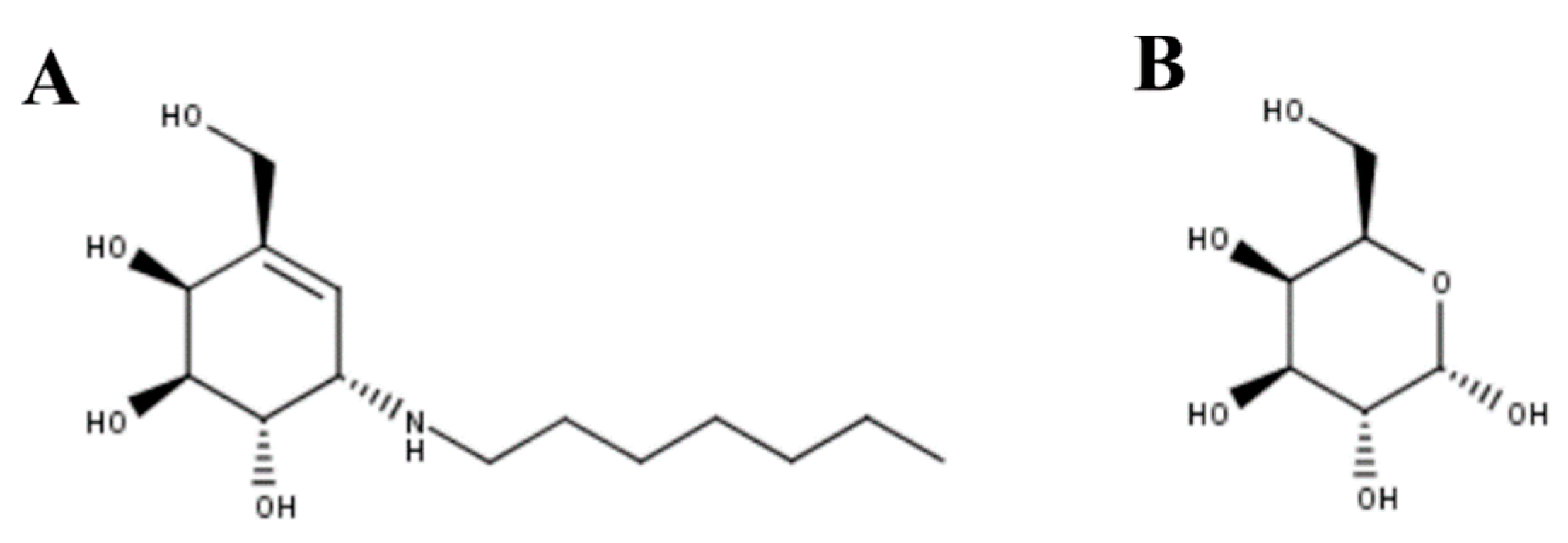
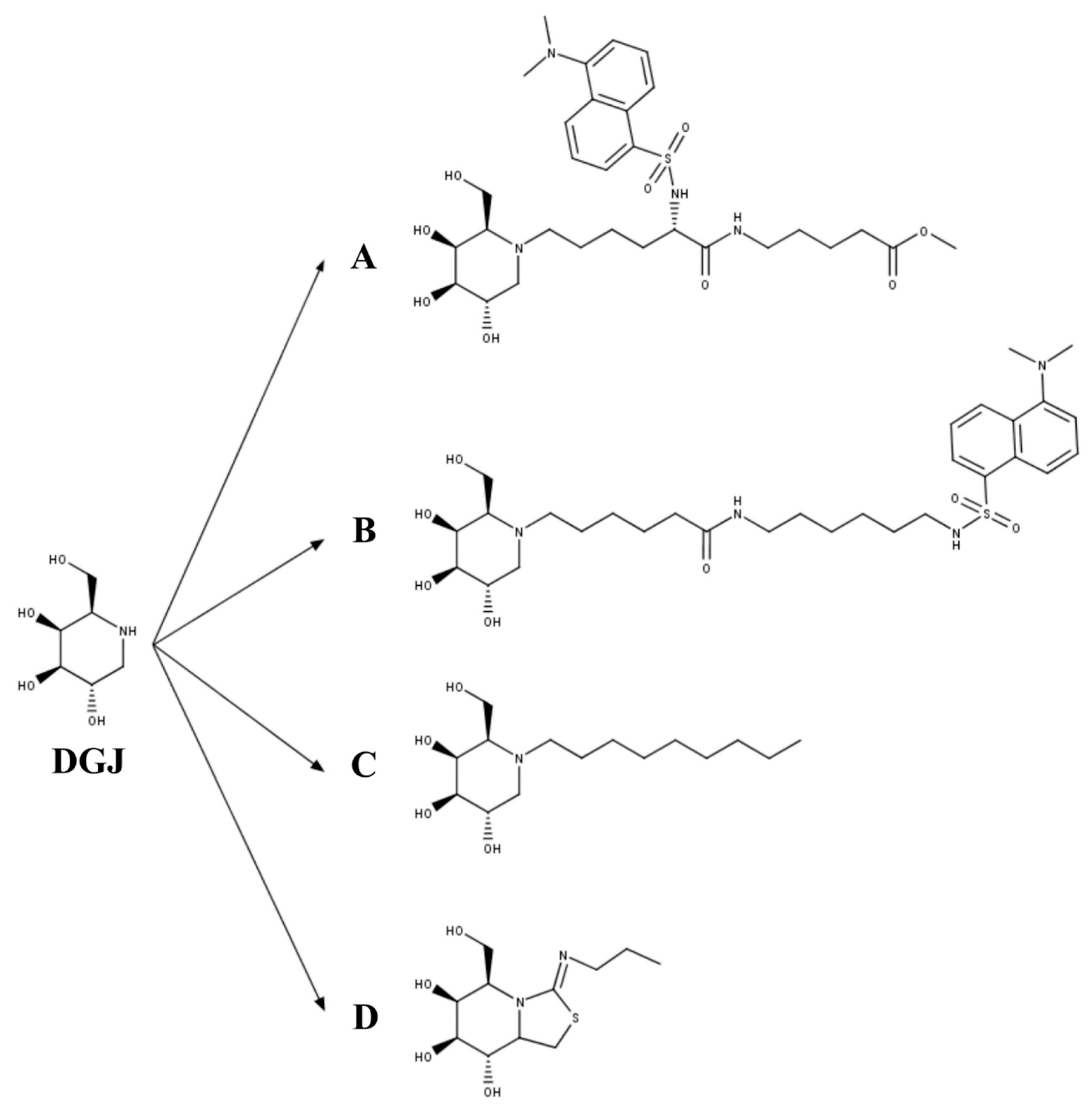
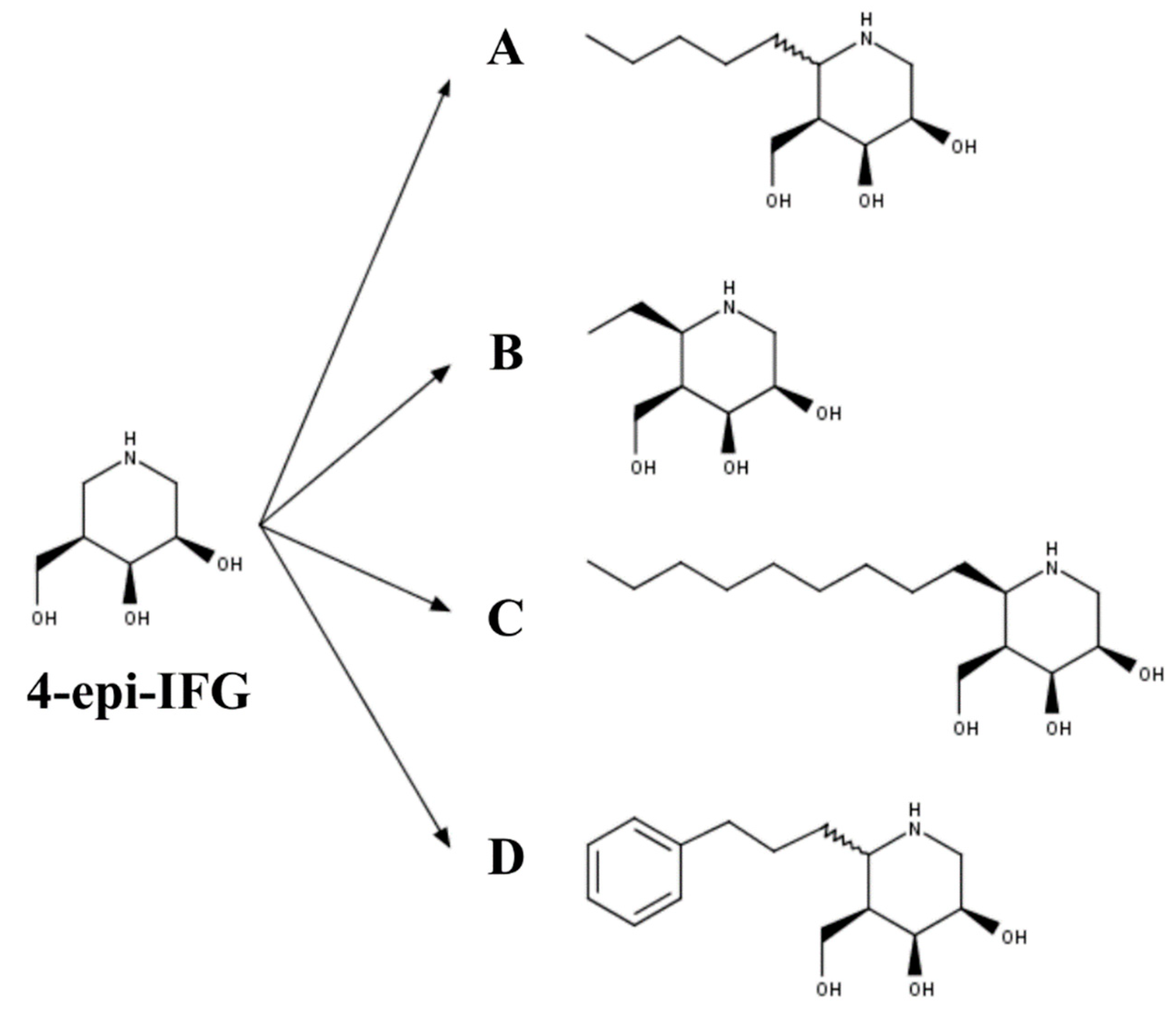
3.3. Mucopolysaccharidosis Type IVB
4. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.X.; Conn, P.M. Pharmacoperones as Novel Therapeutics for Diverse Protein Conformational Diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- Auluck, P.K.; Chan, H.Y.; Trojanowski, J.Q.; Lee, V.M.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef]
- Chen, W.; Helenius, J.; Braakman, I.; Helenius, A. Cotranslational folding and calnexin binding during glycoprotein synthesis. Proc. Natl. Acad. Sci. USA 1995, 92, 6229–6233. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Young, J.C. Mechanisms of the Hsp70 chaperone system. Biochem. Cell Biol. 2010, 88, 291–300. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, C.; Yuan, Y.; Zhu, J.; Li, N.; Chen, C.; Wu, S.; Yu, L.; Lei, J.; Gao, N. Structural basis for interaction of a cotranslational chaperone with the eukaryotic ribosome. Nat. Struct. Mol. Biol. 2014, 21, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Morgner, N.; Schmidt, C.; Beilsten-Edmands, V.; Ebong, I.O.; Patel, N.A.; Clerico, E.M.; Kirschke, E.; Daturpalli, S.; Jackson, S.E.; Agard, D.; et al. Hsp70 forms antiparallel dimers stabilized by post-translational modifications to position clients for transfer to Hsp90. Cell Rep. 2015, 11, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Ejima, D.; Kita, Y.; Tsumoto, K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim. Biophys. Acta 2006, 1764, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.E.; Kelly, J.W. Therapeutic approaches to protein-misfolding diseases. Nature 2003, 426, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Janovick, J.A.; Brothers, S.P.; Cornea, A.; Bush, E.; Goulet, M.T.; Ashton, W.T.; Sauer, D.R.; Haviv, F.; Greer, J.; Conn, P.M. Refolding of misfolded mutant GPCR: Post-translational pharmacoperone action in vitro. Mol. Cell. Endocrinol. 2007, 272, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T.P. Ligand detection in the allosteric world. J. Biomol. Screen. 2010, 15, 119–130. [Google Scholar] [CrossRef]
- Wagner, J.R.; Lee, C.T.; Durrant, J.D.; Malmstrom, R.D.; Feher, V.A.; Amaro, R.E. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016, 116, 6370–6390. [Google Scholar] [CrossRef]
- Boyd, R.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.; Valenzano, K. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef]
- Valenzano, K.J.; Khanna, R.; Powe, A.C.; Boyd, R.; Lee, G.; Flanagan, J.J.; Benjamin, E.R. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Dev. Technol. 2011, 9, 213–235. [Google Scholar] [CrossRef]
- Pereira, D.M.; Valentao, P.; Andrade, P.B. Tuning protein folding in lysosomal storage diseases: The chemistry behind pharmacological chaperones. Chem. Sci. 2018, 9, 1740–1752. [Google Scholar] [CrossRef]
- Shen, J.S.; Edwards, N.J.; Hong, Y.B.; Murray, G.J. Isofagomine increases lysosomal delivery of exogenous glucocerebrosidase. Biochem. Biophys. Res. Commun. 2008, 369, 1071–1075. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kato, A.; Nakagome, I.; Nakagawa, S.; Kinami, K.; Adachi, I.; Jenkinson, S.F.; Desire, J.; Bleriot, Y.; Nash, R.J.; Fleet, G.W.J.; et al. In silico analyses of essential interactions of iminosugars with the Hex A active site and evaluation of their pharmacological chaperone effects for Tay-Sachs disease. Org. Biomol. Chem. 2017, 15, 9297–9304. [Google Scholar] [CrossRef] [PubMed]
- Marugan, J.; Huang, W.; Motabar, O.; Zheng, W.; Xiao, J.; Patnaik, S.; Southall, N.; Westbroek, W.; Lea, W.A.; Simeonov, A.; et al. Non-iminosugar glucocerebrosidase small molecule chaperones. MedChemComm 2012, 3, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Padia, J.; Urban, D.J.; Jadhav, A.; Goker-Alpan, O.; Simeonov, A.; Goldin, E.; Auld, D.; LaMarca, M.E.; Inglese, J.; et al. Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 13192–13197. [Google Scholar] [CrossRef]
- Berardi, A.; Pannuzzo, G.; Graziano, A.; Costantino-Ceccarini, E.; Piomboni, P.; Luddi, A. Pharmacological chaperones increase residual beta-galactocerebrosidase activity in fibroblasts from Krabbe patients. Mol. Genet. Metab. 2014, 112, 294–301. [Google Scholar] [CrossRef]
- Lavinder, J.J.; Hari, S.B.; Sullivan, B.J.; Magliery, T.J. High-throughput thermal scanning: A general, rapid dye-binding thermal shift screen for protein engineering. J. Am. Chem. Soc. 2009, 131, 3794–3795. [Google Scholar] [CrossRef]
- Conn, P.M.; Smith, E.; Hodder, P.; Janovick, J.A.; Smithson, D.C. High-throughput screen for pharmacoperones of the vasopressin type 2 receptor. J. Biomol. Screen. 2013, 18, 930–937. [Google Scholar] [CrossRef]
- Mecozzi, V.J.; Berman, D.E.; Simoes, S.; Vetanovetz, C.; Awal, M.R.; Patel, V.M.; Schneider, R.T.; Petsko, G.A.; Ringe, D.; Small, S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. [Google Scholar] [CrossRef]
- Shin, M.H.; Lim, H.S. Screening methods for identifying pharmacological chaperones. Mol. Biosyst. 2017, 13, 638–647. [Google Scholar] [CrossRef]
- Fecarotta, S.; Gasperini, S.; Parenti, G. New treatments for the mucopolysaccharidoses: From pathophysiology to therapy. Ital. J. Pediatrics 2018, 44, 124. [Google Scholar] [CrossRef]
- Grayson, M. Lysosomal storage disorders. Nature 2016, 537, S145. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.; Muro, S. Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives. Adv. Drug Deliv. Rev. 2017, 118, 109–134. [Google Scholar] [CrossRef]
- McCafferty, E.H.; Scott, L.J. Vestronidase Alfa: A Review in Mucopolysaccharidosis VII. BioDrugs 2019, 33, 233–240. [Google Scholar] [CrossRef]
- Markham, A. Cerliponase Alfa: First Global Approval. Drugs 2017, 77, 1247–1249. [Google Scholar] [CrossRef]
- Harmatz, P.; Cattaneo, F.; Ardigo, D.; Geraci, S.; Hennermann, J.B.; Guffon, N.; Lund, A.; Hendriksz, C.J.; Borgwardt, L. Enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase): Novel global treatment response model and outcomes in patients with alpha-mannosidosis. Mol. Genet. Metab. 2018, 124, 152–160. [Google Scholar] [CrossRef]
- Khan, F.I.; Shahbaaz, M.; Bisetty, K.; Waheed, A.; Sly, W.S.; Ahmad, F.; Hassan, M.I. Large scale analysis of the mutational landscape in β-glucuronidase: A major player of mucopolysaccharidosis type VII. Gene 2016, 576, 36–44. [Google Scholar] [CrossRef]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the structural biology of Gaucher disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef]
- Ugrinov, K.G.; Freed, S.D.; Thomas, C.L.; Lee, S.W. A Multiparametric Computational Algorithm for Comprehensive Assessment of Genetic Mutations in Mucopolysaccharidosis Type IIIA (Sanfilippo Syndrome). PLoS ONE 2015, 10, e0121511. [Google Scholar] [CrossRef]
- Yogalingam, G.; Hopwood, J.J. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: Diagnostic, clinical, and biological implications. Hum. Mutat. 2001, 18, 264–281. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Nijmeijer, S.C.M.; IJlst, L.; te Brinke, H.; van Vlies, N.; Wijburg, F.A. Prediction of phenotypic severity in mucopolysaccharidosis type IIIA. Ann. Neurol. 2017, 82, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Meijer, O.L.M.; te Brinke, H.; Ofman, R.; Ijlst, L.; Wijburg, F.A.; van Vlies, N. Processing of mutant N-acetyl-α-glucosaminidase in mucopolysaccharidosis type IIIB fibroblasts cultured at low temperature. Mol. Genet. Metab. 2017, 122, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Giese, A.-K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional Characterisation of Alpha-Galactosidase A Mutations as a Basis for a New Classification System in Fabry Disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A.; Yoshida, K.; Itoh, K.; Kase, R.; Sakuraba, H.; Suzuki, Y. Intracellular processing and maturation of mutant gene products in hereditary β-galactosidase deficiency (β-galactosidosis). Hum. Genet. 1994, 93, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Chang, H.-H.; Kawasaki, K.; Yasuda, K.; Wu, H.-L.; Garman Scott, C.; Fan, J.-Q. Mutant α-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Guce, A.I.; Clark, N.E.; Rogich, J.J.; Garman, S.C. The Molecular Basis of Pharmacological Chaperoning in Human α-Galactosidase. Chem. Biol. 2011, 18, 1521–1526. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Montaño, A.M.; Dung, V.C.; Grubb, J.H.; Sly, W.S. Mutations and polymorphisms in GUSB gene in mucopolysaccharidosis VII (Sly Syndrome). Hum. Mutat. 2009, 30, 511–519. [Google Scholar] [CrossRef]
- Olarte-Avellaneda, S.; Rodríguez-López, A.; Alméciga-Díaz, C.J.; Barrera, L.A. Computational analysis of human N-acetylgalactosamine-6-sulfate sulfatase enzyme: An update in genotype–phenotype correlation for Morquio A. Mol. Biol. Rep. 2014, 41, 7073–7088. [Google Scholar] [CrossRef]
- Parenti, G.; Moracci, M.; Fecarotta, S.; Andria, G. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med. Chem. 2014, 6, 1031–1045. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological chaperone therapy: Preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef]
- Leidenheimer, N.J. Pharmacological chaperones: Beyond conformational disorders. Handb. Exp. Pharm. 2018, 245, 135–153. [Google Scholar] [CrossRef]
- Fan, J.-Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α–galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Yam, G.H.-F.; Bosshard, N.; Zuber, C.; Steinmann, B.; Roth, J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. Am. J. Physiol. Cell Physiol. 2006, 290, C1076–C1082. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.E.; Metcalf, M.C.; Best, D.; Fleet, G.W.J.; Garman, S.C. Pharmacological chaperones for human α-N-acetylgalactosaminidase. Proc. Natl. Acad. Sci. USA 2012, 109, 17400–17405. [Google Scholar] [CrossRef] [PubMed]
- Gary, S.E.; Ryan, E.; Steward, A.M.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Narita, A.; Shirai, K.; Itamura, S.; Matsuda, A.; Ishihara, A.; Matsushita, K.; Fukuda, C.; Kubota, N.; Takayama, R.; Shigematsu, H.; et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: A pilot study. Ann. Clin. Transl. Neurol. 2016, 3, 200–215. [Google Scholar] [CrossRef]
- Charkhand, B.; Scantlebury, M.H.; Narita, A.; Zimran, A.; Al-Hertani, W. Effect of Ambroxol chaperone therapy on Glucosylsphingosine (Lyso-Gb1) levels in two Canadian patients with type 3 Gaucher disease. Mol. Genet. Metab. Rep. 2019, 20, 100476. [Google Scholar] [CrossRef]
- Aflaki, E.; Stubblefield, B.K.; Maniwang, E.; Lopez, G.; Moaven, N.; Goldin, E.; Marugan, J.; Patnaik, S.; Dutra, A.; Southall, N.; et al. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci. Transl. Med. 2014, 6, 240ra73. [Google Scholar] [CrossRef]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Jung, O.; Patnaik, S.; Marugan, J.; Sidransky, E.; Westbroek, W. Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev. Proteom. 2016, 13, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.H.; Viuff, A.H.; Spratley, S.J.; Salamone, S.; Christensen, S.H.; Read, R.J.; Moriarty, N.W.; Jensen, H.H.; Deane, J.E. Azasugar inhibitors as pharmacological chaperones for Krabbe disease. Chem. Sci. 2015, 6, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Spratley, S.J.; Deane, J.E. New therapeutic approaches for Krabbe disease: The potential of pharmacological chaperones. J. Neurosci. Res. 2016, 94, 1203–1219. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Higaki, K.; Saito, S.; Ohno, K.; Sakuraba, H.; Nanba, E.; Suzuki, Y.; Ozono, K.; Sakai, N. Chaperone therapy for Krabbe disease: Potential for late-onset GALC mutations. J. Hum. Genet. 2015, 60, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Kang, D.; Causevic, E.; Herdt, A.R.; Eckman, E.A.; Eckman, C.B. Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones. J. Neurosci. 2010, 30, 5489–5497. [Google Scholar] [CrossRef] [PubMed]
- Porto, C.; Ferrara, M.C.; Meli, M.; Acampora, E.; Avolio, V.; Rosa, M.; Cobucci-Ponzano, B.; Colombo, G.; Moracci, M.; Andria, G.; et al. Pharmacological Enhancement of α-Glucosidase by the Allosteric Chaperone N-acetylcysteine. Mol. Ther. 2012, 20, 2201–2211. [Google Scholar] [CrossRef]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef]
- Porto, C.; Cardone, M.; Fontana, F.; Rossi, B.; Tuzzi, M.R.; Tarallo, A.; Barone, M.V.; Andria, G.; Parenti, G. The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol. Ther. 2009, 17, 964–971. [Google Scholar] [CrossRef]
- Parenti, G.; Fecarotta, S.; la Marca, G.; Rossi, B.; Ascione, S.; Donati, M.A.; Morandi, L.O.; Ravaglia, S.; Pichiecchio, A.; Ombrone, D.; et al. A chaperone enhances blood α-glucosidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol. Ther. 2014, 22, 2004–2012. [Google Scholar] [CrossRef]
- Hossain, M.A.; Higaki, K.; Shinpo, M.; Nanba, E.; Suzuki, Y.; Ozono, K.; Sakai, N. Chemical chaperone treatment for galactosialidosis: Effect of NOEV on β-galactosidase activities in fibroblasts. Brain Dev. 2016, 38, 175–180. [Google Scholar] [CrossRef]
- Maegawa, G.H.B.; Tropak, M.; Buttner, J.; Stockley, T.; Kok, F.; Clarke, J.T.R.; Mahuran, D.J. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161. [Google Scholar] [CrossRef]
- Clarke, J.T.R.; Mahuran, D.J.; Sathe, S.; Kolodny, E.H.; Rigat, B.A.; Raiman, J.A.; Tropak, M.B. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol. Genet. Metab. 2011, 102, 6–12. [Google Scholar] [CrossRef]
- Banning, A.; Gülec, C.; Rouvinen, J.; Gray, S.J.; Tikkanen, R. Identification of Small Molecule Compounds for Pharmacological Chaperone Therapy of Aspartylglucosaminuria. Sci. Rep. 2016, 6, 37583. [Google Scholar] [CrossRef]
- Rísquez-Cuadro, R.; Matsumoto, R.; Ortega-Caballero, F.; Nanba, E.; Higaki, K.; García Fernández, J.M.; Ortiz Mellet, C. Pharmacological Chaperones for the Treatment of α-Mannosidosis. J. Med. Chem. 2019, 62, 5832–5843. [Google Scholar] [CrossRef]
- Clarke, L.; Ellaway, C.; Foster, H.E.; Giugliani, R.; Goizet, C.; Goring, S.; Hawley, S.; Jurecki, E.; Khan, Z.; Lampe, C.; et al. Understanding the Early Presentation of Mucopolysaccharidoses Disorders: Results of a Systematic Literature Review and Physician Survey. J. Inborn Errors Metab. Screen. 2018, 6. [Google Scholar] [CrossRef]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef]
- Tomatsu, S.; Kubasky, F.; Stapleton, M.; Suzuki, Y.; Orii, K.; Vairo, F.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Graeff, M.; Fischinger, C.; et al. Mucopolysaccharidosis Type II: Clinical Features, Biochemistry, Diagnosis, Genetics, and Treatment. In Mucopolysaccharidoses Update (2 Volume Set); Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Węgrzyn, G., Orii, T., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2018; Volume I, pp. 165–209. [Google Scholar]
- Hoshina, H.; Shimada, Y.; Higuchi, T.; Kobayashi, H.; Ida, H.; Ohashi, T. Chaperone effect of sulfated disaccharide from heparin on mutant iduronate-2-sulfatase in mucopolysaccharidosis type II. Mol. Genet. Metab. 2017, 123, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Demydchuk, M.; Hill, C.H.; Zhou, A.; Bunkoczi, G.; Stein, P.E.; Marchesan, D.; Deane, J.E.; Read, R.J. Insights into Hunter syndrome from the structure of iduronate-2-sulfatase. Nat. Commun. 2017, 8, 15786. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Alméciga-Díaz, C.J.; Mason, R.W.; Orii, T.; Tomatsu, S. Mucopolysaccharidosis Type IVA: Clinical Features, Biochemistry, Diagnosis, Genetics, and Treatment. In Mucopolysaccharidoses Update (2 Volume Set); Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Węgrzyn, G., Orii, T., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2018; Volume I, pp. 235–272. [Google Scholar]
- Khan, S.; Almeciga-Diaz, C.J.; Sawamoto, K.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Mason, R.W.; Orii, T.; Tomatsu, S. Mucopolysaccharidosis IVA and glycosaminoglycans. Mol. Genet. Metab. 2017, 120, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Montaño, A.M.; Oikawa, H.; Smith, M.; Barrera, L.; Chinen, Y.; Thacker, M.M.; Mackenzie, W.G.; Suzuki, Y.; Orii, T. Mucopolysaccharidosis type IVA (Morquio A disease): Clinical review and current treatment. Curr. Pharm. Biotechnol. 2011, 12, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses: Past, present, and future. Biol. Blood Marrow Transplant. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Burton, B.; Fleming, T.R.; Harmatz, P.; Hughes, D.; Jones, S.A.; Lin, S.P.; Mengel, E.; Scarpa, M.; Valayannopoulos, V.; et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): A phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 2014, 37, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Giugliani, R.; Harmatz, P.; Mengel, E.; Guffon, N.; Valayannopoulos, V.; Parini, R.; Hughes, D.; Pastores, G.M.; Lau, H.A.; et al. Multi-domain impact of elosufase alfa in Morquio A syndrome in the pivotal phase III trial. Mol. Genet. Metab. 2015, 114, 178–185. [Google Scholar] [CrossRef]
- Tomatsu, S.; Sawamoto, K.; Shimada, T.; Bober, M.B.; Kubaski, F.; Yasuda, E.; Mason, R.W.; Khan, S.; Alméciga-Díaz, C.J.; Barrera, L.A.; et al. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): Effect and limitations. Expert Opin. Orphan Drugs 2015, 3, 1279–1290. [Google Scholar] [CrossRef]
- Doherty, C.; Stapleton, M.; Piechnik, M.; Mason, R.W.; Mackenzie, W.G.; Yamaguchi, S.; Kobayashi, H.; Suzuki, Y.; Tomatsu, S. Effect of enzyme replacement therapy on the growth of patients with Morquio A. J. Hum. Genet. 2019, 64, 625–635. [Google Scholar] [CrossRef]
- Schweighardt, B.; Tompkins, T.; Lau, K.; Jesaitis, L.; Qi, Y.; Musson, D.G.; Farmer, P.; Haller, C.; Shaywitz, A.J.; Yang, K.; et al. Immunogenicity of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome: Results from MOR-004, a phase III trial. Clin. Ther. 2015, 37, 1012–1021. [Google Scholar] [CrossRef]
- Chinen, Y.; Higa, T.; Tomatsu, S.; Suzuki, Y.; Orii, T.; Hyakuna, N. Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol. Genet. Metab. Rep. 2014, 1, 31–41. [Google Scholar] [CrossRef]
- Yabe, H.; Tanaka, A.; Chinen, Y.; Kato, S.; Sawamoto, K.; Yasuda, E.; Shintaku, H.; Suzuki, Y.; Orii, T.; Tomatsu, S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 2016, 117, 84–94. [Google Scholar] [CrossRef]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis-A ten-year report from the China children transplant group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef]
- Almeciga-Diaz, C.J.; Hidalgo, O.A.; Olarte-Avellaneda, S.; Rodriguez-Lopez, A.; Guzman, E.; Garzon, R.; Pimentel-Vera, L.N.; Puentes-Tellez, M.A.; Rojas-Rodriguez, A.F.; Gorshkov, K.; et al. Identification of Ezetimibe and Pranlukast as Pharmacological Chaperones for the Treatment of the Rare Disease Mucopolysaccharidosis Type IVA. J. Med. Chem. 2019, 62, 6175–6189. [Google Scholar] [CrossRef] [PubMed]
- Phan, B.A.; Dayspring, T.D.; Toth, P.P. Ezetimibe therapy: Mechanism of action and clinical update. Vasc. Health Risk Manag. 2012, 8, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J.; Lyseng-Williamson, K.A.; Goa, K.L. Pranlukast: A review of its use in the management of asthma. Drugs 2003, 63, 991–1019. [Google Scholar] [CrossRef]
- Delic, M.; Gongrich, R.; Mattanovich, D.; Gasser, B. Engineering of protein folding and secretion-strategies to overcome bottlenecks for efficient production of recombinant proteins. Antioxid. Redox Signal. 2014, 21, 414–437. [Google Scholar] [CrossRef]
- Gasser, B.; Prielhofer, R.; Marx, H.; Maurer, M.; Nocon, J.; Steiger, M.; Puxbaum, V.; Sauer, M.; Mattanovich, D. Pichia pastoris: Protein production host and model organism for biomedical research. Future Microbiol. 2013, 8, 191–208. [Google Scholar] [CrossRef]
- Maas, C.; Hermeling, S.; Bouma, B.; Jiskoot, W.; Gebbink, M.F. A role for protein misfolding in immunogenicity of biopharmaceuticals. J. Biol. Chem. 2007, 282, 2229–2236. [Google Scholar] [CrossRef]
- Regier, D.S.; Proia, R.L.; D’Azzo, A.; Tifft, C.J. The GM1 and GM2 Gangliosidoses: Natural History and Progress toward Therapy. Pediatr. Endocrinol. Rev. 2016, 13, 663–673. [Google Scholar]
- Higaki, K.; Ninomiya, H. Mucopolysaccharidosis Type IVB: Clinical Features, Biochemistry, Diagnosis, Genetics, and Treatment. In Mucopolysaccharidoses Update (2 Volume Set); Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P., Scarpa, M., Węgrzyn, G., Orii, T., Eds.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2018; Volume I, pp. 273–283. [Google Scholar]
- Regier, D.S.; Tifft, C.J. GLB1-Related Disorders. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA, 1993. [Google Scholar]
- Matsuda, J.; Suzuki, O.; Oshima, A.; Yamamoto, Y.; Noguchi, A.; Takimoto, K.; Itoh, M.; Matsuzaki, Y.; Yasuda, Y.; Ogawa, S.; et al. Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15912–15917. [Google Scholar] [CrossRef]
- Caciotti, A.; Donati, M.A.; d’Azzo, A.; Salvioli, R.; Guerrini, R.; Zammarchi, E.; Morrone, A. The potential action of galactose as a “chemical chaperone”: Increase of beta galactosidase activity in fibroblasts from an adult GM1-gangliosidosis patient. Eur. J. Paediatr. Neurol. 2009, 13, 160–164. [Google Scholar] [CrossRef]
- Fantur, K.; Hofer, D.; Schitter, G.; Steiner, A.J.; Pabst, B.M.; Wrodnigg, T.M.; Stutz, A.E.; Paschke, E. DLHex-DGJ, a novel derivative of 1-deoxygalactonojirimycin with pharmacological chaperone activity in human G(M1)-gangliosidosis fibroblasts. Mol. Genet. Metab. 2010, 100, 262–268. [Google Scholar] [CrossRef]
- Frohlich, R.F.; Fantur, K.; Furneaux, R.H.; Paschke, E.; Stutz, A.E.; Wicki, J.; Withers, S.G.; Wrodnigg, T.M. A fluorescent probe for GM1 gangliosidosis related beta-galactosidase: N-( dansylamino)hexylaminocarbonylpentyl-1,5-dideoxy-1,5-imino-d-galactitol. Bioorg. Med. Chem. Lett. 2011, 21, 6872–6875. [Google Scholar] [CrossRef] [PubMed]
- Rigat, B.A.; Tropak, M.B.; Buttner, J.; Crushell, E.; Benedict, D.; Callahan, J.W.; Martin, D.R.; Mahuran, D.J. Evaluation of N-nonyl-deoxygalactonojirimycin as a pharmacological chaperone for human GM1 gangliosidosis leads to identification of a feline model suitable for testing enzyme enhancement therapy. Mol. Genet. Metab. 2012, 107, 203–212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takai, T.; Higaki, K.; Aguilar-Moncayo, M.; Mena-Barragan, T.; Hirano, Y.; Yura, K.; Yu, L.; Ninomiya, H.; Garcia-Moreno, M.I.; Sakakibara, Y.; et al. A bicyclic 1-deoxygalactonojirimycin derivative as a novel pharmacological chaperone for GM1 gangliosidosis. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Front, S.; Biela-Banas, A.; Burda, P.; Ballhausen, D.; Higaki, K.; Caciotti, A.; Morrone, A.; Charollais-Thoenig, J.; Gallienne, E.; Demotz, S.; et al. (5aR)-5a-C-Pentyl-4-epi-isofagomine: A powerful inhibitor of lysosomal beta-galactosidase and a remarkable chaperone for mutations associated with GM1-gangliosidosis and Morquio disease type B. Eur. J. Med. Chem. 2017, 126, 160–170. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Gene | Enzyme Deficiency | OMIM |
|---|---|---|---|
| MPS I (Hurler, Hurler–Scheie, and Scheie syndrome) | IDUA | alpha-l-iduronidase | 607014 607015 607016 |
| MPS II (Hunter syndrome) | IDS | Iduronate-2-sulfatase | 309900 |
| MPS IIIA (Sanfilippo syndrome) | SGSH | Heparan-N-sulfatase | 252900 |
| MPS IIIB (Sanfilippo syndrome) | NAGLU | N-acetylglucosaminidase | 252920 |
| MPS IIIC (Sanfilippo syndrome) | HGSNAT | Acetyl CoA glucosamine N-acetyltransferase | 252930 |
| MPS IIID (Sanfilippo syndrome) | GNS | N-acetyl-glucosamine-6-sulfatase | 252940 |
| MPS IVA (Morquio A syndrome) | GALNS | N-acetylgalactosamine-6-sulfate sulfatase | 253000 |
| MPS IVB (Morquio B syndrome) | GLB1 | β-galactosidase | 253010 |
| MPS VI (Maroteaux–Lamy syndrome) | ARSB | Arylsulfatase B | 253200 |
| MPS VII (Sly syndrome) | GUSB | β-glucuronidase | 253220 |
| MPS IX | HYAL1 | Hyaluronidase | 601492 |
| Pharmacological Chaperone | IC50/Ki (µM) | GLB1 Mutations | Maximum Activity Enhancement (Fold Change) | Reference |
|---|---|---|---|---|
| NOEV | IC50: 0.2 | Patient Fibroblasts p.R201C p.R201H p.R457Q p.W273L * p.Y83H * | 5.1 4.5 2.4 2.2 2.0 | [102] |
| Galactose | N.D. | Patient Fibroblasts p.S54N/p.R59C p.R201/p.G579D p.T329A/p.R442Q | 1.0 1.4 2.6 | [103] |
| DLHex-DGJ | IC50: 6.0 Ki: 0.6 | Patient Fibroblasts p.I181K p.R201C p.R201H/p.R457X p.R201H/p.S149F p.R201H/p.H281Y p.R208C/p.W161X p.C230R p.Y270D p.W273L p.A301V p.Y333H p.G438E p.P549L | 2.1 9.4 11.1 12.3 12.5 2.5 9.0 1.7 1.3 1.3 1.8 2.3 1.4 | [104] |
| N-(dansylamino)hexylaminocarbonylpentyl-DGJ | Ki: 0.7 | Patient Fibroblasts p.R201C | ~7.0 | [105] |
| NN-DGJ | IC50: 0.12 | Patient Fibroblasts p.R351X/p.R351X p.R148S/p.D332N p.R148S/p.R482H p.R201H/IVS14-2A>G p.R201H/p.W509C p.W273L/p.R482H * p.G438E/p.G438E * | 1.4 4.1 4.9 7.3 0.9 1.0 1.0 | |
| 6S-NBI-DGJ | IC50: 32 | Patient Fibroblasts p.I51T p.I51T/p.Y316C p.I51T/p.R457Q p.R59H p.G190D p.R201C p.G438E p.R457Q COS7 cells p.I51T p.R148T p.L155R p.G190D p.R201C p.R201H p.R208C p.D214Y p.V216A p.C230Y p.L264S p.N266S p.W273R p.D332N p.K346N p.S434L p.G438E p.Y444C p.R457Q p.R482H p.D491Y p.R590H p.E632G p.D640E | 2.6 5.5 4.9 1.1 4.1 4.9 2.7 3.0 1,9 1,8 1,6 1,5 1,9 2,5 1,8 1,4 1,7 2,3 1,9 2,0 1,9 2,2 1,4 2,2 2,2 2,8 2,4 1,5 1,7 2,0 1,6 1,7 | [107] |
| (5aR)-5a-C-pentyl-4-epi-IFG | IC50: 0.008 | Patient Fibroblasts 733 + 2T > C p.I51T p.R59H p.R59H p.C127Y p.R148S/p.S532G/p.L305F p.S191N/R351X p.R201C p.R201C p.R201C/p.H281Y p.R201H/c.247dup1 p.R201H/p.G76E p.R201H/ p.H281Y p.R208C/IVS10 + 1G > A p.Q255H/p.K578R p.H281Y/splicing p.R351Ter/p.R351X p.G438E p.R442Q/p.W92X p.R457Q p.P549L p.W273L/p.R482H * p.W273L/ p.W509C * | 1.1 1.6 1.4 1.1 1.1 1.0 11.0 15.0 5.4 18.0 3.5 6.7 4.7 4.4 20.0 35.0 1.0 2.0 1.5 7.3 1.3 1.5 1.5 | [107] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losada Díaz, J.C.; Cepeda del Castillo, J.; Rodriguez-López, E.A.; Alméciga-Díaz, C.J. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 232. https://doi.org/10.3390/ijms21010232
Losada Díaz JC, Cepeda del Castillo J, Rodriguez-López EA, Alméciga-Díaz CJ. Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. International Journal of Molecular Sciences. 2020; 21(1):232. https://doi.org/10.3390/ijms21010232
Chicago/Turabian StyleLosada Díaz, Juan Camilo, Jacobo Cepeda del Castillo, Edwin Alexander Rodriguez-López, and Carlos J. Alméciga-Díaz. 2020. "Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses" International Journal of Molecular Sciences 21, no. 1: 232. https://doi.org/10.3390/ijms21010232
APA StyleLosada Díaz, J. C., Cepeda del Castillo, J., Rodriguez-López, E. A., & Alméciga-Díaz, C. J. (2020). Advances in the Development of Pharmacological Chaperones for the Mucopolysaccharidoses. International Journal of Molecular Sciences, 21(1), 232. https://doi.org/10.3390/ijms21010232