Pathogenic Puppetry: Manipulation of the Host Actin Cytoskeleton by Chlamydia trachomatis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. A Multilayered Assault: Chlamydia Redistributes the Actin Cytoskeleton to Invade Host Cells
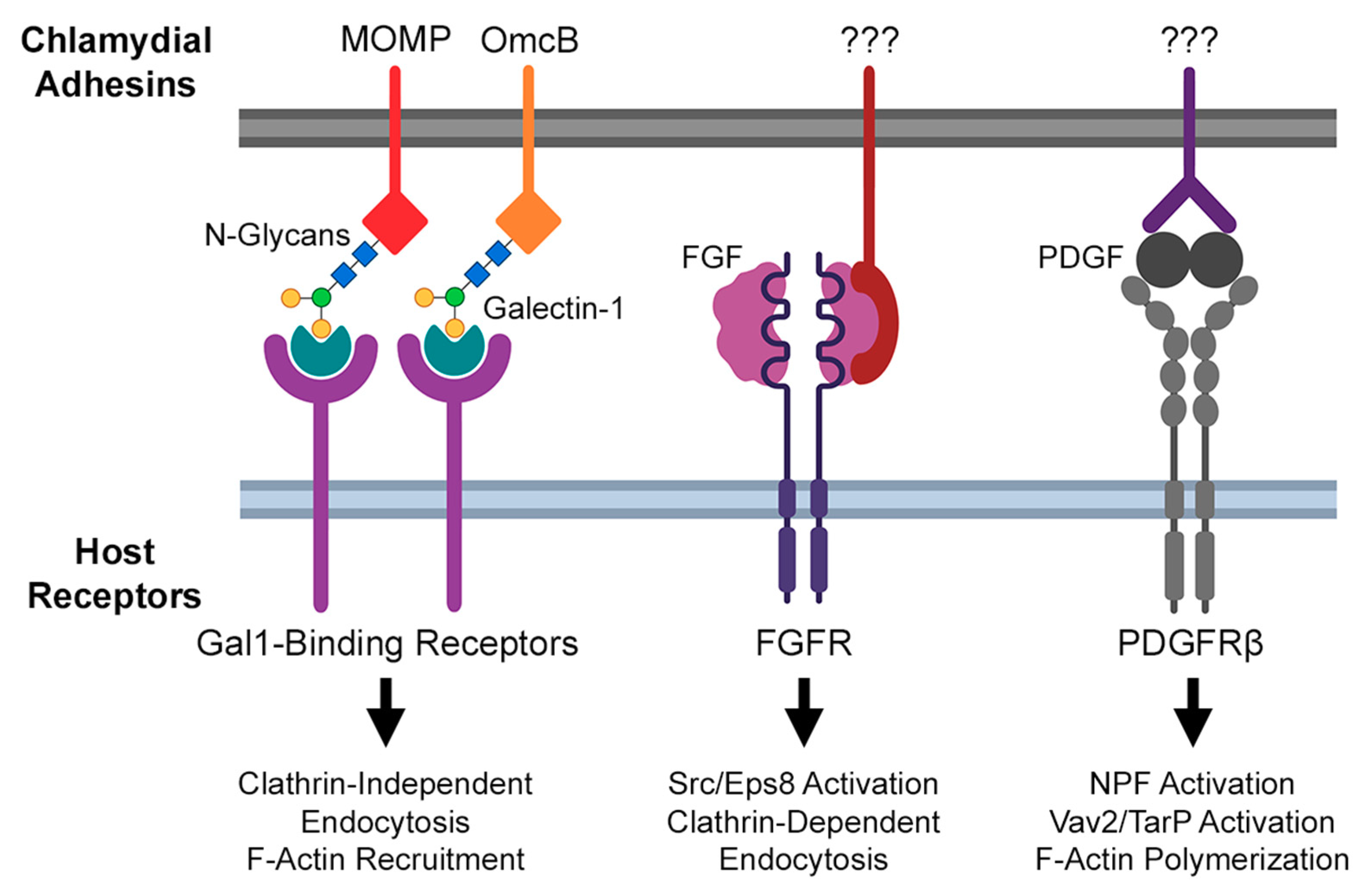
2.1. Actin Modulation during Transient Chlamydial Adhesion
2.2. Actin Modulation during Irreversible Chlamydial Attachment
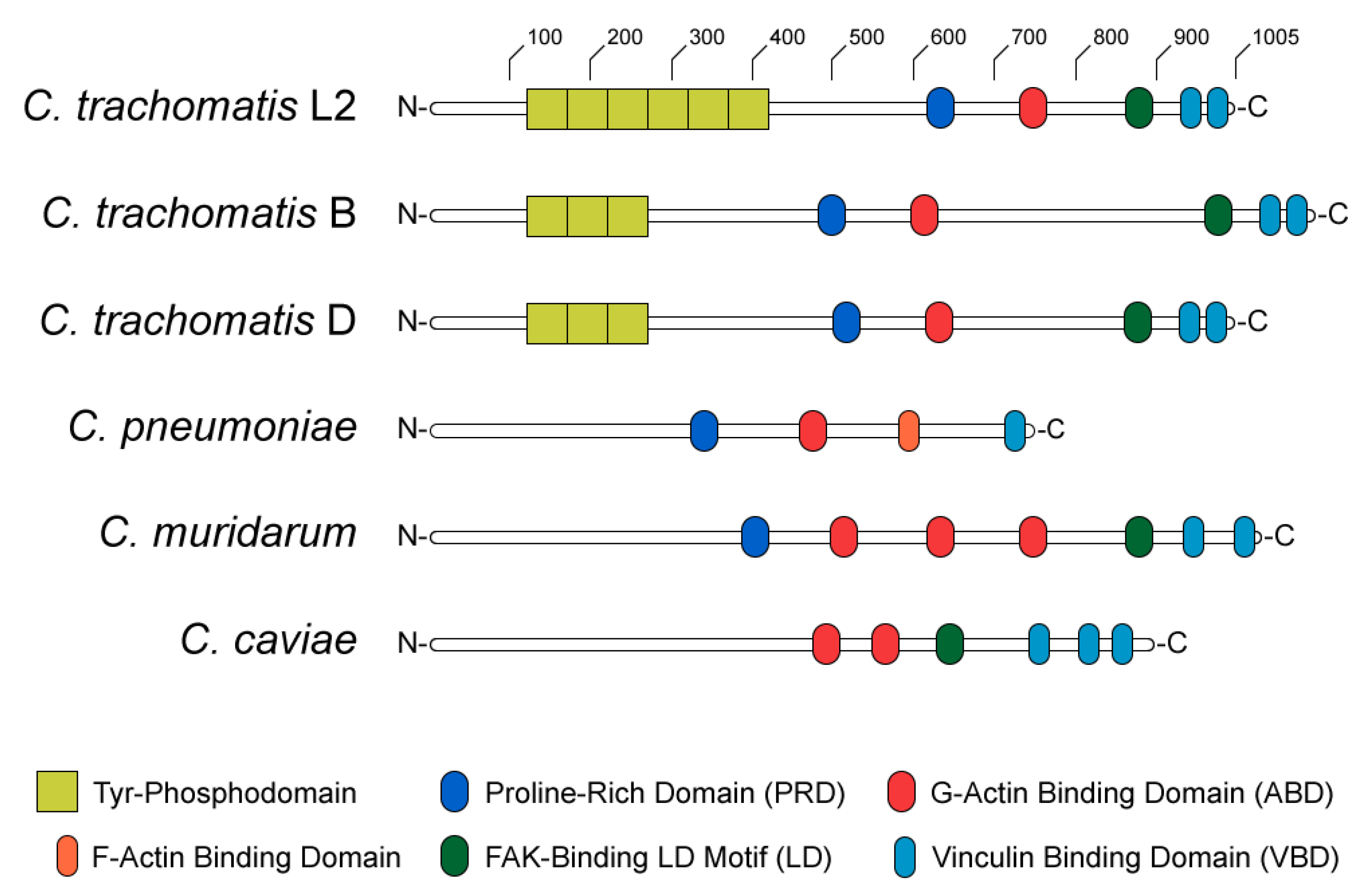
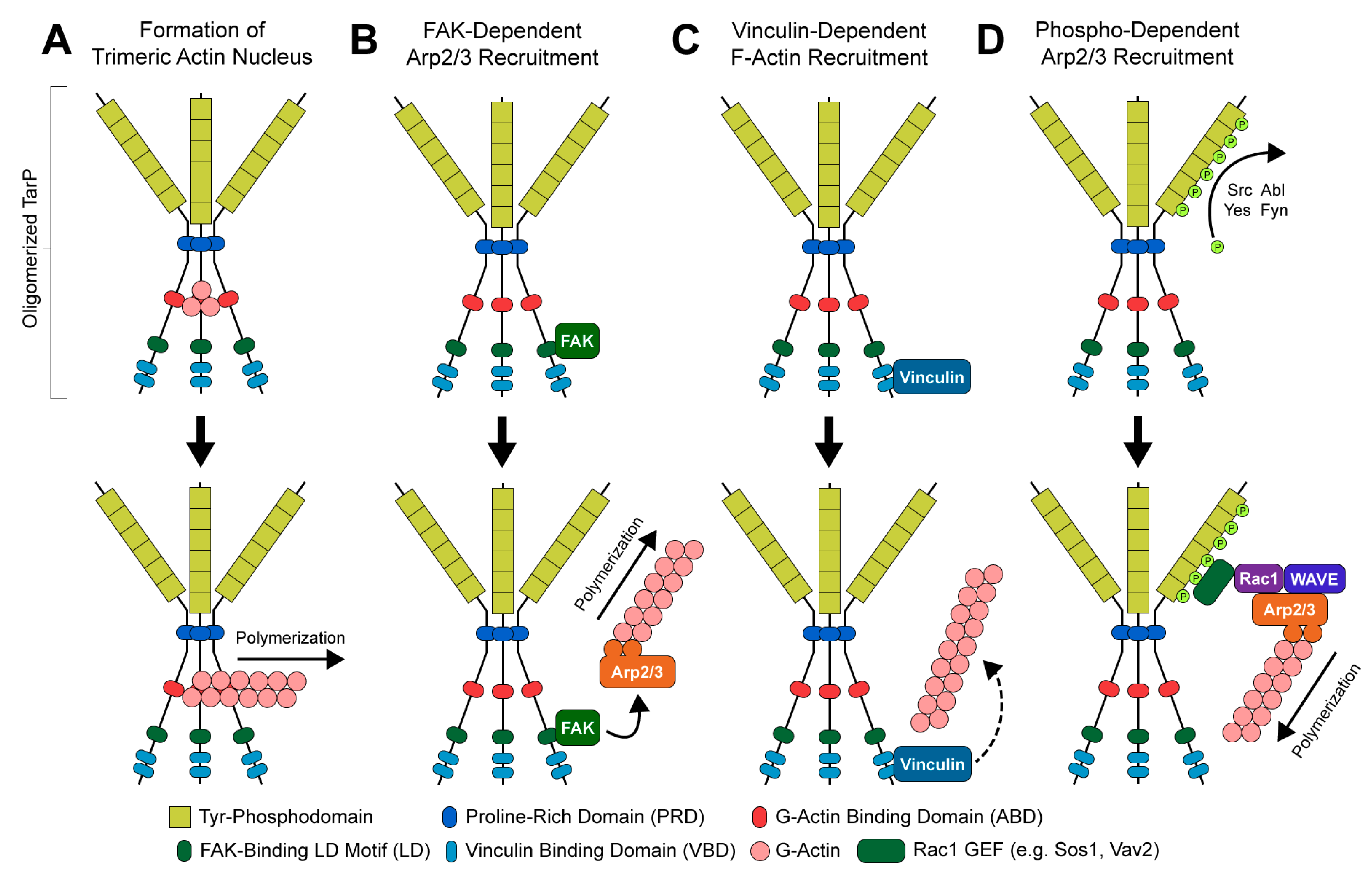
2.3. TarP, a Multifunctional Actin-Recruiting Effector
2.4. Actin-Depolymerizing Chlamydial Effectors
3. Life after Invasion: Chlamydia Modulates Actin for Inclusion Stability and Host Egress
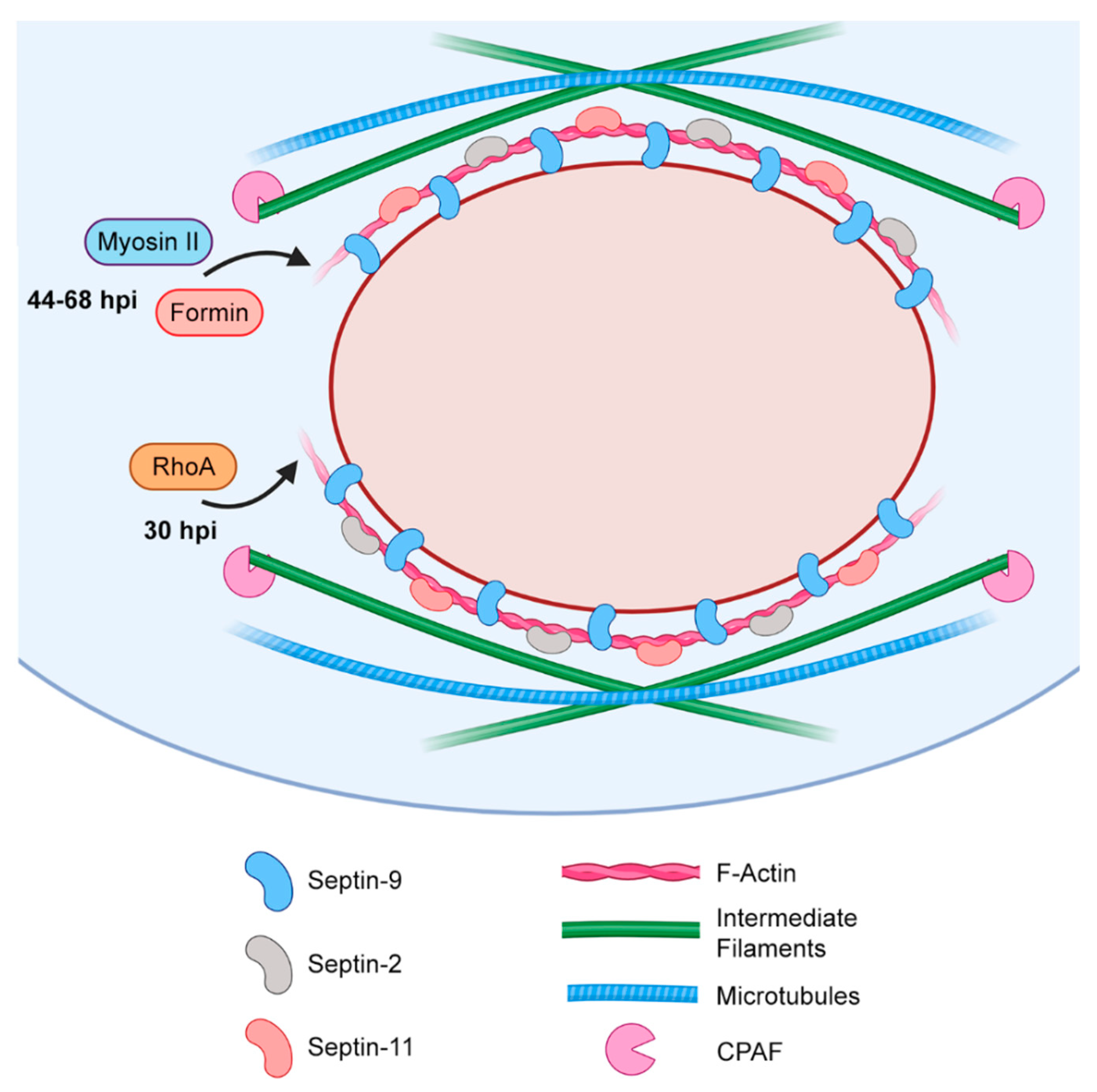
3.1. Actin-Mediated Reinforcement of the Inclusion
3.2. The Role of Actin in Chlamydial Extrusion
4. Conclusions and Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| G-actin | Globular actin |
| F-actin | Filamentous actin |
| Arp2/3 | Actin-related protein 2/3 |
| NPF | Nucleation promotion factor |
| N-WASP | Neuronal Wiskott–Aldrich Syndrome protein |
| WAVE | WASP-family verprolin-homologous family protein |
| WASH | Wiskott–Aldrich syndrome and SCAR homolog |
| GTP | Guanosine-5’-triphosphate |
| EPEC | Enteropathogenic Escherichia coli |
| EHEC | Enterohemorrhagic Escherichia coli |
| T3SS | Type III Secretion System |
| EB | Elementary body |
| GAG | Glycosaminoglycan |
| MOMP | Major outer membrane protein |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| PDGFRβ | Platelet-derived growth factor receptor beta |
| Abl | Abelson-family kinase |
| TarP | Translocated actin recruitment protein |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| PRD | Proline-rich domain |
| ABD | Actin binding domain |
| FAK | Focal adhesion kinase |
| LD | Leucine-aspartatic acid repeat domain |
| VBD | Vinculin binding domain |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| hpi | Hours post-infection |
| MEF | Mouse embryonic fibroblast |
| MTOC | Microtubule organizing center |
| ER | Endoplasmic reticulum |
| MVB | Multivesicular body |
| ROCK | Rho-associated protein kinase |
| CPAF | Chlamydial protease/proteasome-like activity factor |
| MLC2 | Myosin light chain 2 |
| MLCK | Myosin light chain kinase |
| MYPT1 | Myosin phosphatase target subunit 1 |
| PP2 | Protein phosphatase 2 |
| IPTR3 | Inositol-1,4,5-triphosphate receptor, type 3 |
| SRF | Serum response factor |
| MRTF | Myocardin-related transcription factor |
| INM | Inner nuclear membrane |
| LINC | Linker of nucleoskeleton and cytoskeleton |
| ILK | Integrin-linked kinase |
References
- Goley, E.D.; Welch, M.D. The ARP2/3 complex: An actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006, 7, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 451–477. [Google Scholar] [CrossRef] [PubMed]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M.; Mullins, R.D.; Higgs, H.N.; Kaiser, D.A.; Blanchoin, L.; May, R.C.; Hall, M.E.; Pollard, T.D. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc. Natl. Acad. Sci. USA 1999, 96, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Linardopoulou, E.V.; Parghi, S.S.; Friedman, C.; Osborn, G.E.; Parkhurst, S.M.; Trask, B.J. Human Subtelomeric WASH Genes Encode a New Subclass of the WASP Family. PLoS Genet. 2007, 3, e237. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. Bacterial factors exploit eukaryotic Rho GTPase signaling cascades to promote invasion and proliferation within their host. Small GTPases 2014, 5, e983863. [Google Scholar] [CrossRef]
- Zhou, D.; Chen, L.M.; Hernandez, L.; Shears, S.B.; Galán, J.E. A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol. Microbiol. 2001, 39, 248–259. [Google Scholar] [CrossRef]
- Stender, S.; Friebel, A.; Linder, S.; Rohde, M.; Mirold, S.; Hardt, W.-D. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol. Microbiol. 2000, 36, 1206–1221. [Google Scholar] [CrossRef]
- Hardt, W.D.; Chen, L.M.; Schuebel, K.E.; Bustelo, X.R.; Galán, J.E.S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 1998, 93, 815–826. [Google Scholar] [CrossRef]
- Friebel, A.; Ilchmann, H.; Aepfelbacher, M.; Ehrbar, K.; Machleidt, W.; Hardt, W.-D. SopE and SopE2 from Salmonella typhimurium Activate Different Sets of RhoGTPases of the Host Cell. J. Biol. Chem. 2001, 276, 34035–34040. [Google Scholar] [CrossRef] [PubMed]
- Hardt, W.D.; Urlaub, H.; Galán, J.E. A substrate of the centisome 63 type III protein secretion system of Salmonella typhimurium is encoded by a cryptic bacteriophage. Proc. Natl. Acad. Sci. USA 1998, 95, 2574–2579. [Google Scholar] [CrossRef] [PubMed]
- Garcia-del Portillo, F.; Finlay, B.B. Salmonella invasion of nonphagocytic cells induces formation of macropinosomes in the host cell. Infect. Immun. 1994, 62, 4641–4645. [Google Scholar] [PubMed]
- Humphreys, D.; Davidson, A.; Hume, P.J.; Koronakis, V. Salmonella virulence effector SopE and Host GEF ARNO cooperate to recruit and activate WAVE to trigger bacterial invasion. Cell Host Microbe 2012, 11, 129–139. [Google Scholar] [CrossRef]
- Boujemaa-Paterski, R.; Gouin, E.; Hansen, G.; Samarin, S.; Le Clainche, C.; Didry, D.; Dehoux, P.; Cossart, P.; Kocks, C.; Carlier, M.F.; et al. Listeria protein ActA mimics WASp family proteins: It activates filament barbed end branching by Arp2/3 complex. Biochemistry 2001, 40, 11390–11404. [Google Scholar] [CrossRef]
- Welch, M.D.; Rosenblatt, J.; Skoble, J.; Portnoy, D.A.; Mitchison, T.J. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science 1998, 281, 105–108. [Google Scholar] [CrossRef]
- Skoble, J.; Portnoy, D.A.; Welch, M.D. Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J. Cell Biol. 2000, 150, 527–538. [Google Scholar] [CrossRef]
- Ireton, K. Molecular mechanisms of cell-cell spread of intracellular bacterial pathogens. Open Biol. 2013, 3, 130079. [Google Scholar] [CrossRef]
- Welch, M.D.; Way, M. Arp2/3-mediated actin-based motility: A tail of pathogen abuse. Cell Host Microbe 2013, 14, 242–255. [Google Scholar] [CrossRef]
- Lamason, R.L.; Welch, M.D. Actin-based motility and cell-to-cell spread of bacterial pathogens. Curr. Opin. Microbiol. 2017, 35, 48–57. [Google Scholar] [CrossRef]
- Kenny, B.; DeVinney, R.; Stein, M.; Reinscheid, D.J.; Frey, E.A.; Finlay, B.B. Enteropathogenic E. coli (EPEC) Transfers Its Receptor for Intimate Adherence into Mammalian Cells. Cell 1997, 91, 511–520. [Google Scholar] [CrossRef]
- Deibel, C.; Krämer, S.; Chakraborty, T.; Ebel, F. EspE, a novel secreted protein of attaching and effacing bacteria, is directly translocated into infected host cells, where it appears as a tyrosine-phosphorylated 90 kDa protein. Mol. Microbiol. 1998, 28, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Warawa, J.; Kenny, B. Phosphoserine modification of the enteropathogenic Escherichia coli Tir molecule is required to trigger conformational changes in Tir and efficient pedestal elongation. Mol. Microbiol. 2001, 42, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Rankin, S.; Pawson, T.; Kirschner, M.W.; Tipper, D.J.; Leong, J.M. Clustering of Nck by a 12-residue Tir phosphopeptide is sufficient to trigger localized actin assembly. J. Cell Biol. 2004, 164, 407–416. [Google Scholar] [CrossRef]
- Gruenheid, S.; DeVinney, R.; Bladt, F.; Goosney, D.; Gelkop, S.; Gish, G.D.; Pawson, T.; Finlay, B.B. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat. Cell Biol. 2001, 3, 856–859. [Google Scholar] [CrossRef]
- Frankel, G.; Phillips, A.D. Attaching effacing Escherichia coli and paradigms of Tir-triggered actin polymerization: Getting off the pedestal. Cell. Microbiol. 2008, 10, 549–556. [Google Scholar] [CrossRef]
- Garmendia, J.; Frankel, G.; Crepin, V.F. Enteropathogenic and Enterohemorrhagic Escherichia coli Infections: Translocation, Translocation, Translocation. Infect. Immun. 2005, 73, 2573–2585. [Google Scholar] [CrossRef]
- Elwell, C.; Mirrashidi, K.; Engel, J. Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 2016, 14, 385–400. [Google Scholar] [CrossRef]
- Malhotra, M.; Sood, S.; Mukherjee, A.; Muralidhar, S.; Bala, M. Genital Chlamydia trachomatis: An update. Indian J. Med. Res. 2013, 138, 303–316. [Google Scholar]
- Braun, C.; Alcázar-Román, A.R.; Laska, A.; Mölleken, K.; Fleig, U.; Hegemann, J.H. CPn0572, the C. pneumoniae ortholog of TarP, reorganizes the actin cytoskeleton via a newly identified F-actin binding domain and recruitment of vinculin. PLoS ONE 2019, 14, e0210403. [Google Scholar] [CrossRef]
- Zrieq, R.; Braun, C.; Hegemann, J.H. The Chlamydia pneumoniae Tarp Ortholog CPn0572 Stabilizes Host F-Actin by Displacement of Cofilin. Front. Cell. Infect. Microbiol. 2017, 7, 511. [Google Scholar] [CrossRef] [PubMed]
- Jiwani, S.; Alvarado, S.; Ohr, R.J.; Romero, A.; Nguyen, B.; Jewett, T.J. Chlamydia trachomatis Tarp Harbors Distinct G and F Actin Binding Domains That Bundle Actin Filaments. J. Bacteriol. 2013, 195, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Elwell, C.A.; Ceesay, A.; Kim, J.H.; Kalman, D.; Engel, J.N. RNA Interference Screen Identifies Abl Kinase and PDGFR Signaling in Chlamydia trachomatis Entry. PLoS Pathog. 2008, 4, e1000021. [Google Scholar] [CrossRef] [PubMed]
- Clifton, D.R.; Dooley, C.A.; Grieshaber, S.S.; Carabeo, R.A.; Fields, K.A.; Hackstadt, T. Tyrosine Phosphorylation of the Chlamydial Effector Protein Tarp Is Species Specific and Not Required for Recruitment of Actin. Infect. Immun. 2005, 73, 3860–3868. [Google Scholar] [CrossRef] [PubMed]
- Carabeo, R. Bacterial subversion of host actin dynamics at the plasma membrane. Cell. Microbiol. 2011, 13, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Carabeo, R.A.; Grieshaber, S.S.; Fischer, E.; Hackstadt, T. Chlamydia trachomatis Induces Remodeling of the Actin Cytoskeleton during Attachment and Entry into HeLa Cells. Infect. Immun. 2002, 70, 3793–3803. [Google Scholar] [CrossRef]
- Ford, C.; Nans, A.; Boucrot, E.; Hayward, R.D. Chlamydia exploits filopodial capture and a macropinocytosis-like pathway for host cell entry. PLoS Pathog. 2018, 14, e1007051. [Google Scholar] [CrossRef]
- Kuo, C.C.; Grayston, T. Interaction of Chlamydia trachomatis organisms and HeLa 229 cells. Infect. Immun. 1976, 13, 1103–1109. [Google Scholar]
- Byrne, G.I. Kinetics of phagocytosis of Chlamydia psittaci by mouse fibroblasts (L cells): Separation of the attachment and ingestion stages. Infect. Immun. 1978, 19, 607–612. [Google Scholar]
- Carabeo, R.A.; Hackstadt, T. Isolation and Characterization of a Mutant Chinese Hamster Ovary Cell Line That Is Resistant to Chlamydia trachomatis Infection at a Novel Step in the Attachment Process. Infect. Immun. 2001, 69, 5899–5904. [Google Scholar] [CrossRef]
- Moulder, J.W. Interaction of chlamydiae and host cells in vitro. Microbiol. Mol. Biol. Rev. 1991, 55, 143–190. [Google Scholar]
- Tiwari, V.; Maus, E.; Sigar, I.M.; Ramsey, K.H.; Shukla, D. Role of heparan sulfate in sexually transmitted infections. Glycobiology 2012, 22, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Taraktchoglou, M.; Pacey, A.A.; Turnbull, J.E.; Eley, A. Infectivity of Chlamydia trachomatis serovar LGV but not E is dependent on host cell heparan sulfate. Infect. Immun. 2001, 69, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Kitagawa, H. Heparin and heparan sulfate biosynthesis. IUBMB Life 2002, 54, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen-Lathrop, S.J.; Koshiyama, K.; Phillips, N.; Stephens, R.S. Chlamydia-dependent biosynthesis of a heparan sulphate-like compound in eukaryotic cells. Cell. Microbiol. 2000, 2, 137–144. [Google Scholar] [CrossRef]
- Stephens, R.S. Molecular mimicry and Chlamydia trachomatis infection of eukaryotic cells. Trends Microbiol. 1994, 2, 99–101. [Google Scholar] [CrossRef]
- Zhang, J.P.; Stephens, R.S. Mechanism of C. trachomatis attachment to eukaryotic host cells. Cell 1992, 69, 861–869. [Google Scholar] [CrossRef]
- Fadel, S.; Eley, A. Chlamydia trachomatis OmcB protein is a surface-exposed glycosaminoglycan-dependent adhesin. J. Med. Microbiol. 2007, 56, 15–22. [Google Scholar] [CrossRef]
- Moelleken, K.; Hegemann, J.H. The Chlamydia outer membrane protein OmcB is required for adhesion and exhibits biovar-specific differences in glycosaminoglycan binding. Mol. Microbiol. 2008, 67, 403–419. [Google Scholar] [CrossRef]
- Fechtner, T.; Stallmann, S.; Moelleken, K.; Meyer, K.L.; Hegemann, J.H. Characterization of the interaction between the chlamydial adhesin OmcB and the human host cell. J. Bacteriol. 2013, 195, 5323–5333. [Google Scholar] [CrossRef]
- Su, H.; Raymond, L.; Rockey, D.D.; Fischer, E.; Hackstadt, T.; Caldwell, H.D. A recombinant Chlamydia trachomatis major outer membrane protein binds to heparan sulfate receptors on epithelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 11143–11148. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.A.; Lee, A.; Kuo, C. Cleavage of the N-linked oligosaccharide from the surfaces of Chlamydia species affects infectivity in the mouse model of lung infection. Infect. Immun. 2006, 74, 3027–3029. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Swanson, A.F.; Kuo, C.C. Evidence that the major outer membrane protein of Chlamydia trachomatis is glycosylated. Infect. Immun. 1991, 59, 2120–2125. [Google Scholar] [PubMed]
- Lujan, A.L.; Croci, D.O.; Gambarte Tudela, J.A.; Losinno, A.D.; Cagnoni, A.J.; Mariño, K.V.; Damiani, M.T.; Rabinovich, G.A. Glycosylation-dependent galectin-receptor interactions promote Chlamydia trachomatis infection. Proc. Natl. Acad. Sci. USA 2018, 115, E6000–E6009. [Google Scholar] [CrossRef]
- Quintá, H.R.; Wilson, C.; Blidner, A.G.; González-Billault, C.; Pasquini, L.A.; Rabinovich, G.A.; Pasquini, J.M. Ligand-mediated Galectin-1 endocytosis prevents intraneural H2O2 production promoting F-actin dynamics reactivation and axonal re-growth. Exp. Neurol. 2016, 283, 165–178. [Google Scholar] [CrossRef]
- Boleti, H.; Benmerah, A.; Ojcius, D.M.; Cerf-Bensussan, N.; Dautry-Varsat, A. Chlamydia infection of epithelial cells expressing dynamin and Eps15 mutants: Clathrin-independent entry into cells and dynamin-dependent productive growth. J. Cell Sci. 1999, 112, 1487–1496. [Google Scholar]
- Benmerah, A.; Gagnon, J.; Bègue, B.; Mégarbané, B.; Dautry-Varsat, A.; Cerf-Bensussan, N. The tyrosine kinase substrate eps15 is constitutively associated with the plasma membrane adaptor AP-2. J. Cell Biol. 1995, 131, 1831–1838. [Google Scholar] [CrossRef]
- Tebar, F.; Sorkina, T.; Sorkin, A.; Ericsson, M.; Kirchhausen, T. Eps15 is a component of clathrin-coated pits and vesicles and is located at the rim of coated pits. J. Biol. Chem. 1996, 271, 28727–28730. [Google Scholar] [CrossRef]
- Benmerah, A.; Lamaze, C.; Bègue, B.; Schmid, S.L.; Dautry-Varsat, A.; Cerf-Bensussan, N. AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J. Cell Biol. 1998, 140, 1055–1062. [Google Scholar] [CrossRef]
- Benmerah, A.; Bayrou, M.; Cerf-Bensussan, N.; Dautry-Varsat, A. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J. Cell Sci. 1999, 112, 1303–1311. [Google Scholar]
- Hybiske, K.; Stephens, R.S. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. USA 2007, 104, 11430–11435. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.D.; Mölleken, K.; Hegemann, J.H.; Carabeo, R.A. Chlamydia Adhesion and Invasion. In Chlamydia Biology: From Genome to Disease; Caister Academic Press: Poole, UK, 2020; ISBN 978-1-912530-28-1. [Google Scholar]
- Kim, J.H.; Jiang, S.; Elwell, C.A.; Engel, J.N. Chlamydia trachomatis co-opts the FGF2 signaling pathway to enhance infection. PLoS Pathog. 2011, 7, e1002285. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, N. Regulation of growth factor signaling by FRS2 family docking/scaffold adaptor proteins. Cancer Sci. 2008, 99, 1319–1325. [Google Scholar] [CrossRef]
- Birkelund, S.; Johnsen, H.; Christiansen, G. Chlamydia trachomatis serovar L2 induces protein tyrosine phosphorylation during uptake by HeLa cells. Infect. Immun. 1994, 62, 4900–4908. [Google Scholar] [PubMed]
- Fawaz, F.S.; van Ooij, C.H.; Homola, E.L.; Mutka, S.C.; Engel, J.N. Infection with Chlamydia trachomatis alters the tyrosine phosphorylation and/or localization of several host cell proteins including cortactin. Infect. Immun. 1997, 65, 5301–5308. [Google Scholar]
- Clifton, D.R.; Fields, K.A.; Grieshaber, S.S.; Dooley, C.A.; Fischer, E.R.; Mead, D.J.; Carabeo, R.A.; Hackstadt, T. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc. Natl. Acad. Sci. USA 2004, 101, 10166–10171. [Google Scholar] [CrossRef]
- Lutter, E.I.; Bonner, C.; Holland, M.J.; Suchland, R.J.; Stamm, W.E.; Jewett, T.J.; McClarty, G.; Hackstadt, T. Phylogenetic Analysis of Chlamydia trachomatis Tarp and Correlation with Clinical Phenotype. Infect. Immun. 2010, 78, 3678–3688. [Google Scholar] [CrossRef]
- Jewett, T.J.; Miller, N.J.; Dooley, C.A.; Hackstadt, T. The Conserved Tarp Actin Binding Domain Is Important for Chlamydial Invasion. PLoS Pathog. 2010, 6, e1000997. [Google Scholar] [CrossRef]
- Thwaites, T.; Nogueira, A.T.; Campeotto, I.; Silva, A.P.; Grieshaber, S.S.; Carabeo, R.A. The Chlamydia Effector TarP Mimics the Mammalian Leucine-Aspartic Acid Motif of Paxillin to Subvert the Focal Adhesion Kinase during Invasion. J. Biol. Chem. 2014, 289, 30426–30442. [Google Scholar] [CrossRef]
- Thwaites, T.R.; Pedrosa, A.T.; Peacock, T.P.; Carabeo, R.A. Vinculin Interacts with the Chlamydia Effector TarP Via a Tripartite Vinculin Binding Domain to Mediate Actin Recruitment and Assembly at the Plasma Membrane. Front. Cell. Infect. Microbiol. 2015, 5, 88. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Schaller, M.D.; Hildebrand, J.D.; Shannon, J.D.; Fox, J.W.; Vines, R.R.; Parsons, J.T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 1994, 14, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Calalb, M.B.; Polte, T.R.; Hanks, S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for Src family kinases. Mol. Cell. Biol. 1995, 15, 954–963. [Google Scholar] [CrossRef]
- Schlaepfer, D.D.; Hunter, T. Signal transduction from the extracellular matrix--a role for the focal adhesion protein-tyrosine kinase FAK. Cell Struct. Funct. 1996, 21, 445–450. [Google Scholar] [CrossRef]
- Huveneers, S.; Danen, E.H.J. Adhesion signaling–crosstalk between integrins, Src and Rho. J. Cell Sci. 2009, 122, 1059–1069. [Google Scholar] [CrossRef]
- Stallmann, S.; Hegemann, J.H. The Chlamydia trachomatis Ctad1 invasin exploits the human integrin β1 receptor for host cell entry. Cell. Microbiol. 2016, 18, 761–775. [Google Scholar] [CrossRef]
- Igietseme, J.U.; Omosun, Y.; Stuchlik, O.; Reed, M.S.; Partin, J.; He, Q.; Joseph, K.; Ellerson, D.; Bollweg, B.; George, Z.; et al. Role of Epithelial-Mesenchyme Transition in Chlamydia Pathogenesis. PLoS ONE 2015, 10, e0145198. [Google Scholar] [CrossRef]
- Belland, R.J.; Nelson, D.E.; Virok, D.; Crane, D.D.; Hogan, D.; Sturdevant, D.; Beatty, W.L.; Caldwell, H.D. Transcriptome analysis of chlamydial growth during IFN-γ-mediated persistence and reactivation. Proc. Natl. Acad. Sci. USA 2003, 100, 15971–15976. [Google Scholar] [CrossRef]
- Carlson, J.H.; Porcella, S.F.; McClarty, G.; Caldwell, H.D. Comparative Genomic Analysis of Chlamydia trachomatis Oculotropic and Genitotropic Strains. Infect. Immun. 2005, 73, 6407–6418. [Google Scholar] [CrossRef]
- Thomson, N.R.; Holden, M.T.G.; Carder, C.; Lennard, N.; Lockey, S.J.; Marsh, P.; Skipp, P.; O’Connor, C.D.; Goodhead, I.; Norbertzcak, H.; et al. Chlamydia trachomatis: Genome sequence analysis of lymphogranuloma venereum isolates. Genome Res. 2008, 18, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Jewett, T.J.; Dooley, C.A.; Mead, D.J.; Hackstadt, T. Chlamydia trachomatis TarP is phosphorylated by src family tyrosine kinases. Biochem. Biophys. Res. Commun. 2008, 371, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.J.; Mutchler, C.; Al Khodor, S.; Grieshaber, S.S.; Carabeo, R.A. Chlamydial Entry Involves TARP Binding of Guanine Nucleotide Exchange Factors. PLoS Pathog. 2008, 4, e1000014. [Google Scholar] [CrossRef] [PubMed]
- Carabeo, R.A.; Dooley, C.A.; Grieshaber, S.S.; Hackstadt, T. Rac interacts with Abi-1 and WAVE2 to promote an Arp2/3-dependent actin recruitment during chlamydial invasion. Cell. Microbiol. 2007, 9, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Park, J.; Thomas, M.; Cruz, E.; Cardona, O.; Kang, H.; Jewett, T. Biophysical characterization of actin bundles generated by the Chlamydia trachomatis Tarp effector. Biochem. Biophys. Res. Commun. 2018, 500, 423–428. [Google Scholar] [CrossRef] [PubMed]
- McGhie, E.J.; Hayward, R.D.; Koronakis, V. Control of actin turnover by a salmonella invasion protein. Mol. Cell 2004, 13, 497–510. [Google Scholar] [CrossRef]
- Bierne, H.; Gouin, E.; Roux, P.; Caroni, P.; Yin, H.L.; Cossart, P. A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J. Cell Biol. 2001, 155, 101–112. [Google Scholar] [CrossRef]
- Bastidas, R.J.; Valdivia, R.H. Emancipating Chlamydia: Advances in the Genetic Manipulation of a Recalcitrant Intracellular Pathogen. Microbiol. Mol. Biol. Rev. 2016, 80, 411–427. [Google Scholar] [CrossRef]
- Wickstrum, J.; Sammons, L.R.; Restivo, K.N.; Hefty, P.S. Conditional Gene Expression in Chlamydia trachomatis using the Tet System. PLoS ONE 2013, 8, e76743. [Google Scholar] [CrossRef]
- Ouellette, S.P. Feasibility of a Conditional Knockout System for Chlamydia Based on CRISPR Interference. Front. Cell. Infect. Microbiol. 2018, 8, 59. [Google Scholar] [CrossRef]
- Yother, J.; Goguen, J.D. Isolation and characterization of Ca2+-blind mutants of Yersinia pestis. J. Bacteriol. 1985, 164, 704–711. [Google Scholar] [PubMed]
- Fields, K.A.; Hackstadt, T. Evidence for the secretion of Chlamydia trachomatis CopN by a type III secretion mechanism. Mol. Microbiol. 2000, 38, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Jamison, W.P.; Hackstadt, T. Induction of type III secretion by cell-free Chlamydia trachomatis elementary bodies. Microb. Pathog. 2008, 45, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Betts-Hampikian, H.J.; Fields, K.A. The Chlamydial Type III Secretion Mechanism: Revealing Cracks in a Tough Nut. Front. Microbiol. 2010, 1, 114. [Google Scholar] [CrossRef]
- Ferrell, J.C.; Fields, K.A. A working model for the type III secretion mechanism in Chlamydia. Microbes Infect. 2016, 18, 84–92. [Google Scholar] [CrossRef]
- Lee, J.J.; Kim, D.G.; Kim, D.H.; Simborio, H.L.; Min, W.; Lee, H.J.; Her, M.; Jung, S.C.; Watarai, M.; Kim, S. Interplay between Clathrin and Rab5 Controls the Early Phagocytic Trafficking and Intracellular Survival of Brucella abortus within HeLa cells. J. Biol. Chem. 2013, 288, 28049–28057. [Google Scholar] [CrossRef]
- Hower, S.; Wolf, K.; Fields, K.A. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol. Microbiol. 2009, 72, 1423–1437. [Google Scholar] [CrossRef]
- Chen, Y.-S.; Bastidas, R.J.; Saka, H.A.; Carpenter, V.K.; Richards, K.L.; Plano, G.V.; Valdivia, R.H. The Chlamydia trachomatis Type III Secretion Chaperone Slc1 Engages Multiple Early Effectors, Including TepP, a Tyrosine-phosphorylated Protein required for the Recruitment of CrkI-II to Nascent Inclusions and Innate Immune Signaling. PLoS Pathog. 2014, 10, e1003954. [Google Scholar] [CrossRef]
- Benaud, C.; Gentil, B.J.; Assard, N.; Court, M.; Garin, J.; Delphin, C.; Baudier, J. AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J. Cell Biol. 2004, 164, 133–144. [Google Scholar] [CrossRef]
- Haase, H.; Pagel, I.; Khalina, Y.; Zacharzowsky, U.; Person, V.; Lutsch, G.; Petzhold, D.; Kott, M.; Schaper, J.; Morano, I. The carboxyl-terminal ahnak domain induces actin bundling and stabilizes muscle contraction. FASEB J. 2004, 18, 839–841. [Google Scholar] [CrossRef]
- McKuen, M.J.; Mueller, K.E.; Bae, Y.S.; Fields, K.A. Fluorescence-Reported Allelic Exchange Mutagenesis Reveals a Role for Chlamydia trachomatis TmeA in Invasion That Is Independent of Host AHNAK. Infect. Immun. 2017, 85, e00640-17. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Lim, H.J.; Yoon, S.; Seong, J.K.; Bae, D.S.; Rhee, S.G.; Bae, Y.S. Ahnak Protein Activates Protein Kinase C (PKC) through Dissociation of the PKC-Protein Phosphatase 2A Complex. J. Biol. Chem. 2008, 283, 6312–6320. [Google Scholar] [CrossRef] [PubMed]
- Belland, R.J.; Scidmore, M.A.; Crane, D.D.; Hogan, D.M.; Whitmire, W.; McClarty, G.; Caldwell, H.D. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proc. Natl. Acad. Sci. USA 2001, 98, 13984–13989. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium difficile Toxin Biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, R.; Lacy, D.B. The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 2017, 41, 723–750. [Google Scholar] [CrossRef]
- Thalmann, J.; Janik, K.; May, M.; Sommer, K.; Ebeling, J.; Hofmann, F.; Genth, H.; Klos, A. Actin Re-Organization Induced by Chlamydia trachomatis Serovar D—Evidence for a Critical Role of the Effector Protein CT166 Targeting Rac. PLoS ONE 2010, 5, e9887. [Google Scholar] [CrossRef]
- Bothe, M.; Dutow, P.; Pich, A.; Genth, H.; Klos, A. DXD Motif-Dependent and -Independent Effects of the Chlamydia trachomatis Cytotoxin CT166. Toxins 2015, 7, 621–637. [Google Scholar] [CrossRef]
- Dennis, M.W.; Storz, J. Infectivity of Chlamydia psittaci of bovine and ovine origins for cultured cells. Am. J. Vet. Res. 1982, 43, 1897–1902. [Google Scholar]
- Campbell, S.; Richmond, S.J.; Yates, P.S. The effect of Chlamydia trachomatis infection on the host cell cytoskeleton and membrane compartments. J. Gen. Microbiol. 1989, 135, 2379–2386. [Google Scholar] [CrossRef]
- Grieshaber, S.S.; Grieshaber, N.A.; Hackstadt, T. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J. Cell Sci. 2003, 116, 3793–3802. [Google Scholar] [CrossRef]
- Campbell, S.; Richmond, S.J.; Yates, P. The development of Chlamydia trachomatis inclusions within the host eukaryotic cell during interphase and mitosis. J. Gen. Microbiol. 1989, 135, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Derré, I.; Swiss, R.; Agaisse, H. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog. 2011, 7, e1002092. [Google Scholar] [CrossRef] [PubMed]
- Dumoux, M.; Clare, D.K.; Saibil, H.R.; Hayward, R.D. Chlamydiae assemble a pathogen synapse to hijack the host endoplasmic reticulum. Traffic 2012, 13, 1612–1627. [Google Scholar] [CrossRef] [PubMed]
- Beatty, W.L. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J. Cell Sci. 2006, 119, 350–359. [Google Scholar] [CrossRef]
- Beatty, W.L. Late endocytic multivesicular bodies intersect the chlamydial inclusion in the absence of CD63. Infect. Immun. 2008, 76, 2872–2881. [Google Scholar] [CrossRef]
- Robertson, D.K.; Gu, L.; Rowe, R.K.; Beatty, W.L. Inclusion biogenesis and reactivation of persistent Chlamydia trachomatis requires host cell sphingolipid biosynthesis. PLoS Pathog. 2009, 5, e1000664. [Google Scholar] [CrossRef]
- Kumar, Y.; Valdivia, R.H. Actin and Intermediate Filaments Stabilize the Chlamydia trachomatis Vacuole by Forming Dynamic Structural Scaffolds. Cell Host Microbe 2008, 4, 159–169. [Google Scholar] [CrossRef]
- Chin, E.; Kirker, K.; Zuck, M.; James, G.; Hybiske, K. Actin Recruitment to the Chlamydia Inclusion Is Spatiotemporally Regulated by a Mechanism That Requires Host and Bacterial Factors. PLoS ONE 2012, 7, e46949. [Google Scholar] [CrossRef]
- Dong, F.; Su, H.; Huang, Y.; Zhong, Y.; Zhong, G. Cleavage of host keratin 8 by a Chlamydia-secreted protease. Infect. Immun. 2004, 72, 3863–3868. [Google Scholar] [CrossRef]
- Savijoki, K.; Alvesalo, J.; Vuorela, P.; Leinonen, M.; Kalkkinen, N. Proteomic analysis of Chlamydia pneumoniae-infected HL cells reveals extensive degradation of cytoskeletal proteins. FEMS Immunol. Med. Microbiol. 2008, 54, 375–384. [Google Scholar] [CrossRef]
- Nogueira, A.T.; Pedrosa, A.T.; Carabeo, R.A. Manipulation of the Host Cell Cytoskeleton by Chlamydia. Curr. Top. Microbiol. Immunol. 2018, 412, 59–80. [Google Scholar] [PubMed]
- Todd, W.J.; Caldwell, H.D. The interaction of Chlamydia trachomatis with host cells: Ultrastructural studies of the mechanism of release of a biovar II strain from HeLa 229 cells. J. Infect. Dis. 1985, 151, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Lutter, E.I.; Barger, A.C.; Nair, V.; Hackstadt, T. Chlamydia trachomatis inclusion membrane protein CT228 recruits elements of the myosin phosphatase pathway to regulate release mechanisms. Cell Rep. 2013, 3, 1921–1931. [Google Scholar] [CrossRef] [PubMed]
- Mital, J.; Miller, N.J.; Fischer, E.R.; Hackstadt, T. Specific chlamydial inclusion membrane proteins associate with active Src family kinases in microdomains that interact with the host microtubule network. Cell. Microbiol. 2010, 12, 1235–1249. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, F.; Hartshorne, D.J. Myosin phosphatase target subunit: Many roles in cell function. Biochem. Biophys. Res. Commun. 2008, 369, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.H.; Key, C.E.; Snider, T.A.; Sah, P.; Shaw, E.I.; Fisher, D.J.; Lutter, E.I. Genetic Inactivation of Chlamydia trachomatis Inclusion Membrane Protein CT228 Alters MYPT1 Recruitment, Extrusion Production, and Longevity of Infection. Front. Cell. Infect. Microbiol. 2018, 8, 415. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Lutter, E.I.; Hackstadt, T. Chlamydia trachomatis inclusion membrane protein MrcA interacts with the inositol 1,4,5-trisphosphate receptor type 3 (ITPR3) to regulate extrusion formation. PLoS Pathog. 2018, 14, e1006911. [Google Scholar] [CrossRef]
- Volceanov, L.; Herbst, K.; Biniossek, M.; Schilling, O.; Haller, D.; Nölke, T.; Subbarayal, P.; Rudel, T.; Zieger, B.; Häcker, G. Septins Arrange F-Actin-Containing Fibers on the Chlamydia trachomatis Inclusion and Are Required for Normal Release of the Inclusion by Extrusion. MBio 2014, 5, e01802-14. [Google Scholar] [CrossRef]
- Mostowy, S.; Cossart, P. Septins: The fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 183–194. [Google Scholar] [CrossRef]
- Geneste, O.; Copeland, J.W.; Treisman, R. LIM kinase and Diaphanous cooperate to regulate serum response factor and actin dynamics. J. Cell Biol. 2002, 157, 831–838. [Google Scholar] [CrossRef]
- Zhao, X.-H.; Laschinger, C.; Arora, P.; Szászi, K.; Kapus, A.; McCulloch, C.A. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J. Cell Sci. 2007, 120, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chi, L.; Zhao, J.; Wang, X.; Chen, Z.; Meng, L.; Liu, G.; Guan, G.; Wang, F. Serum response factor provokes epithelial-mesenchymal transition in renal tubular epithelial cells of diabetic nephropathy. Physiol. Genom. 2016, 48, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; He, L.; Li, T.; Lu, Y.; Miao, Y.; Liang, S.; Guo, H.; Bai, M.; Xie, H.; Luo, G.; et al. SRF expedites metastasis and modulates the epithelial to mesenchymal transition by regulating miR-199a-5p expression in human gastric cancer. Cell Death Differ. 2014, 21, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Zadora, P.K.; Chumduri, C.; Imami, K.; Berger, H.; Mi, Y.; Selbach, M.; Meyer, T.F.; Gurumurthy, R.K. Integrated Phosphoproteome and Transcriptome Analysis Reveals Chlamydia-Induced Epithelial-to-Mesenchymal Transition in Host Cells. Cell Rep. 2019, 26, 1286–1302. [Google Scholar] [CrossRef] [PubMed]
- Igietseme, J.U.; Omosun, Y.; Nagy, T.; Stuchlik, O.; Reed, M.S.; He, Q.; Partin, J.; Joseph, K.; Ellerson, D.; George, Z.; et al. Molecular Pathogenesis of Chlamydia Disease Complications: Epithelial-Mesenchymal Transition and Fibrosis. Infect. Immun. 2018, 86, e00585-17. [Google Scholar] [CrossRef] [PubMed]
- Rajić, J.; Inic-Kanada, A.; Stein, E.; Dinić, S.; Schuerer, N.; Uskoković, A.; Ghasemian, E.; Mihailović, M.; Vidaković, M.; Grdović, N.; et al. Chlamydia trachomatis Infection Is Associated with E-Cadherin Promoter Methylation, Downregulation of E-Cadherin Expression, and Increased Expression of Fibronectin and α-SMA—Implications for Epithelial-Mesenchymal Transition. Front. Cell. Infect. Microbiol. 2017, 7, 253. [Google Scholar] [CrossRef]
- Thorpe, S.D.; Lee, D.A. Dynamic regulation of nuclear architecture and mechanics—A rheostatic role for the nucleus in tailoring cellular mechanosensitivity. Nucleus 2017, 8, 287–300. [Google Scholar] [CrossRef]
- Lee, K.K.; Haraguchi, T.; Lee, R.S.; Koujin, T.; Hiraoka, Y.; Wilson, K.L. Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J. Cell Sci. 2001, 114, 4567–4573. [Google Scholar]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef]
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511. [Google Scholar] [CrossRef]
- Plessner, M.; Melak, M.; Chinchilla, P.; Baarlink, C.; Grosse, R. Nuclear F-actin formation and reorganization upon cell spreading. J. Biol. Chem. 2015, 290, 11209–11216. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ogawa, M.; Mimuro, H.; Sasakawa, C. Reinforcement of epithelial cell adhesion to basement membrane by a bacterial pathogen as a new infectious stratagem. Virulence 2010, 1, 52–55. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, M.; Ogawa, M.; Fujita, Y.; Yoshikawa, Y.; Nagai, T.; Koyama, T.; Nagai, S.; Lange, A.; Fässler, R.; Sasakawa, C. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature 2009, 459, 578–582. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caven, L.; Carabeo, R.A. Pathogenic Puppetry: Manipulation of the Host Actin Cytoskeleton by Chlamydia trachomatis. Int. J. Mol. Sci. 2020, 21, 90. https://doi.org/10.3390/ijms21010090
Caven L, Carabeo RA. Pathogenic Puppetry: Manipulation of the Host Actin Cytoskeleton by Chlamydia trachomatis. International Journal of Molecular Sciences. 2020; 21(1):90. https://doi.org/10.3390/ijms21010090
Chicago/Turabian StyleCaven, Liam, and Rey A. Carabeo. 2020. "Pathogenic Puppetry: Manipulation of the Host Actin Cytoskeleton by Chlamydia trachomatis" International Journal of Molecular Sciences 21, no. 1: 90. https://doi.org/10.3390/ijms21010090
APA StyleCaven, L., & Carabeo, R. A. (2020). Pathogenic Puppetry: Manipulation of the Host Actin Cytoskeleton by Chlamydia trachomatis. International Journal of Molecular Sciences, 21(1), 90. https://doi.org/10.3390/ijms21010090