Comprehensive Bioinformatics Identifies Key microRNA Players in ATG7-Deficient Lung Fibroblasts


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
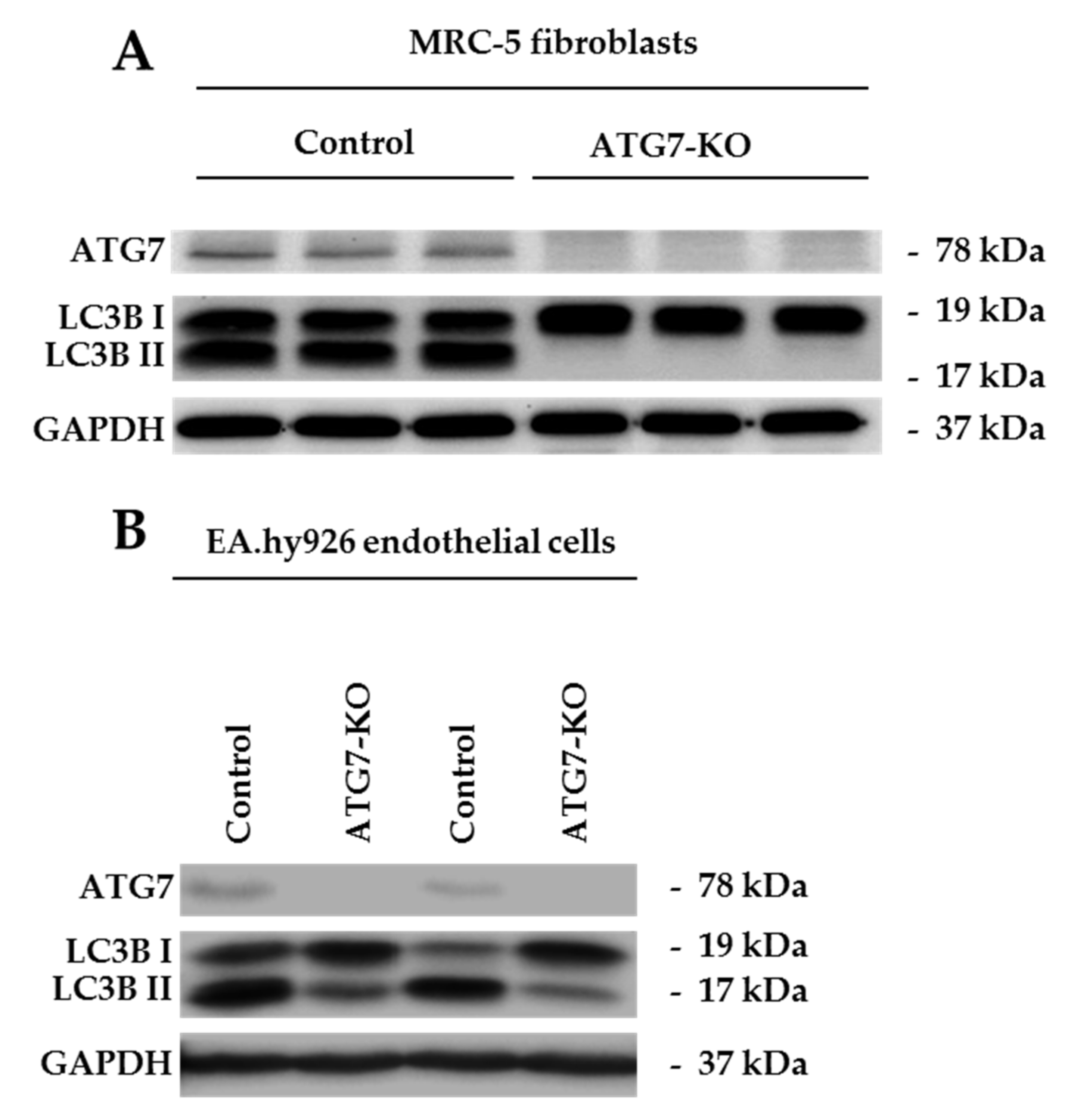
2.1. ATG7-Knockout Disables LC3B Traffic in Endothelial Cells and Fibroblasts
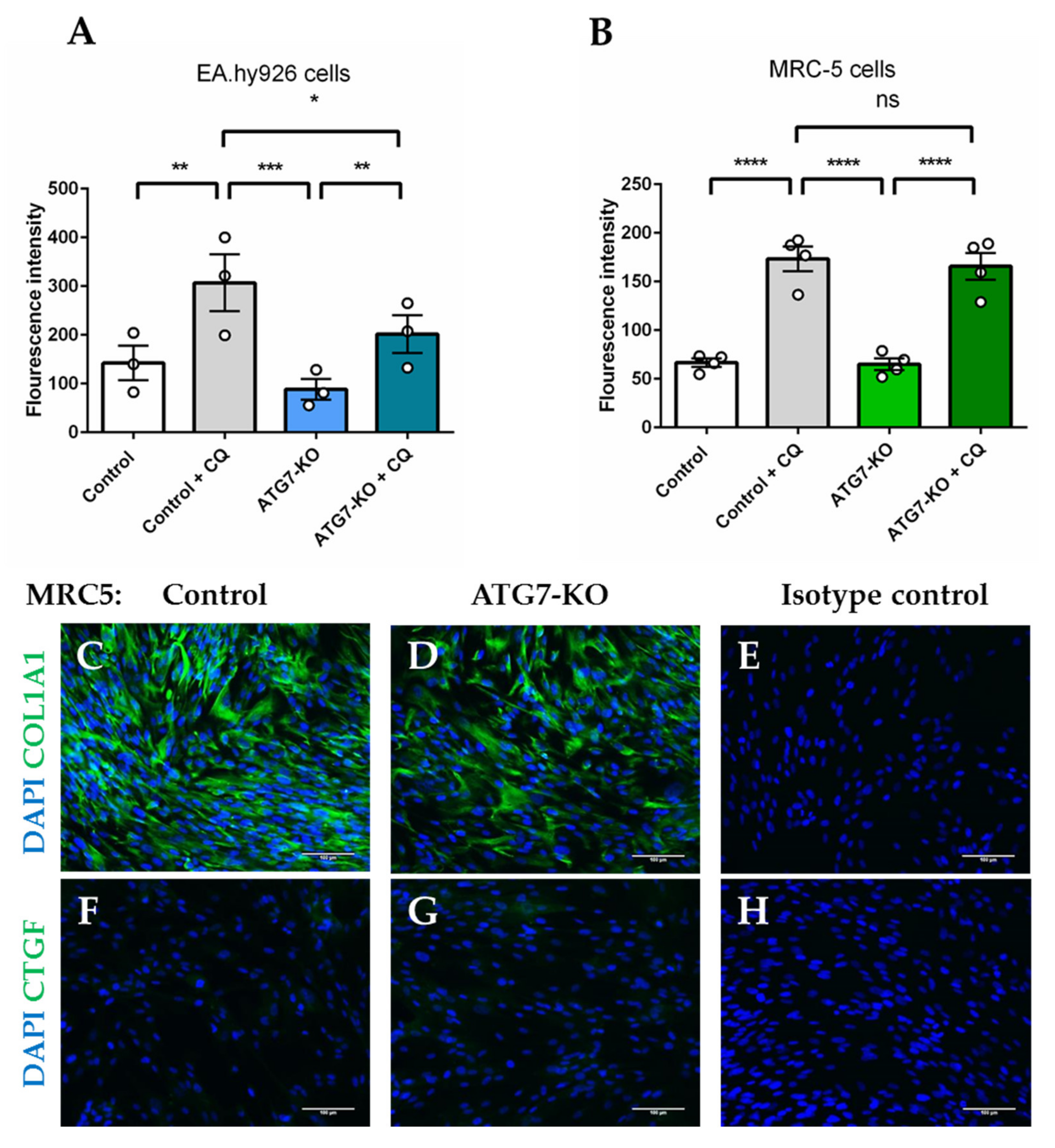
2.2. Autophagosomal Generation is Hampered in ATG7-Knockout Endothelial Cells, but not in Lung Fibroblasts
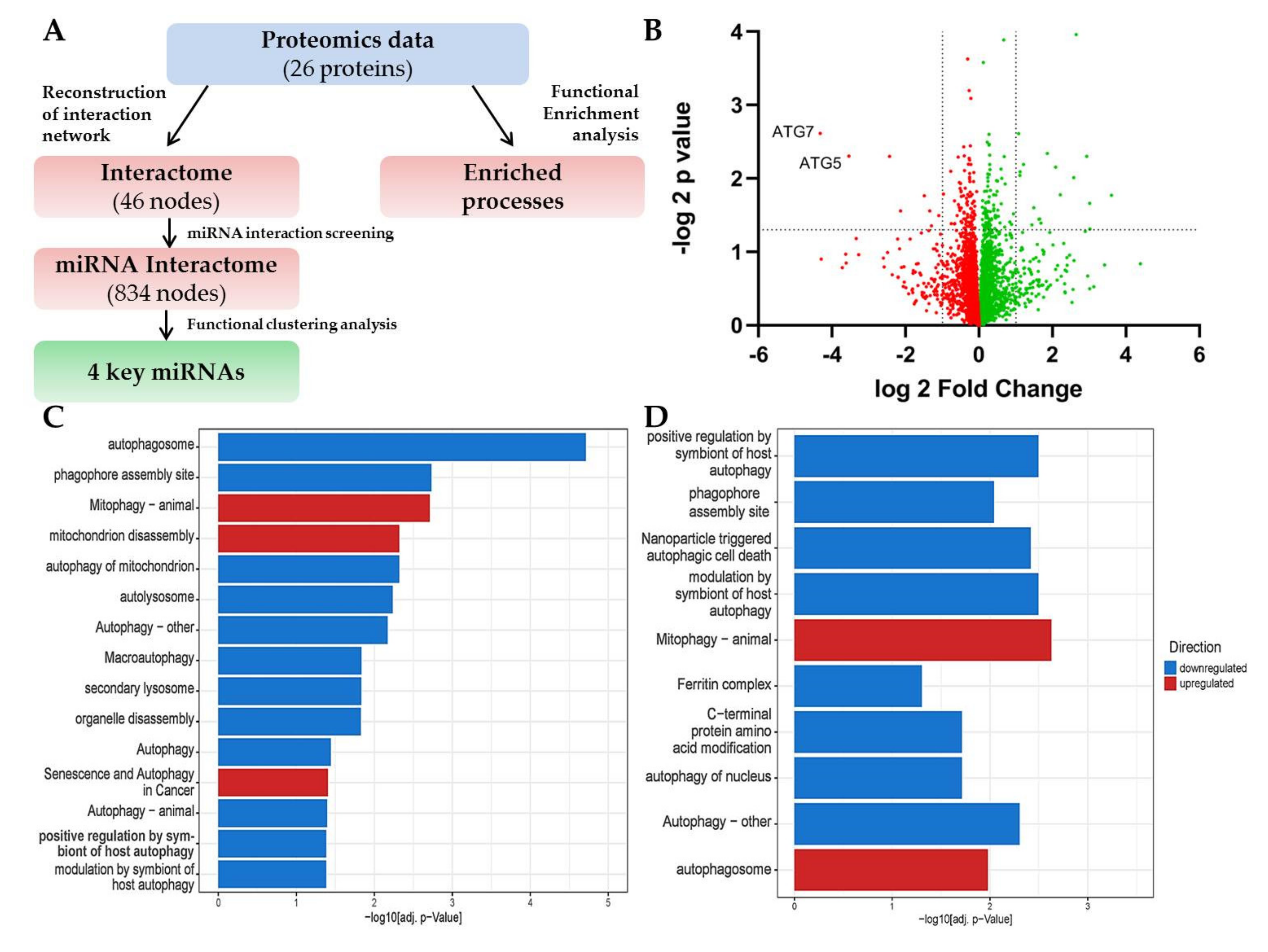
2.3. LC-MS Shows a Signature of ATG5/7-Independent Autophagy in ATG7-Knockout Lung Fibroblasts
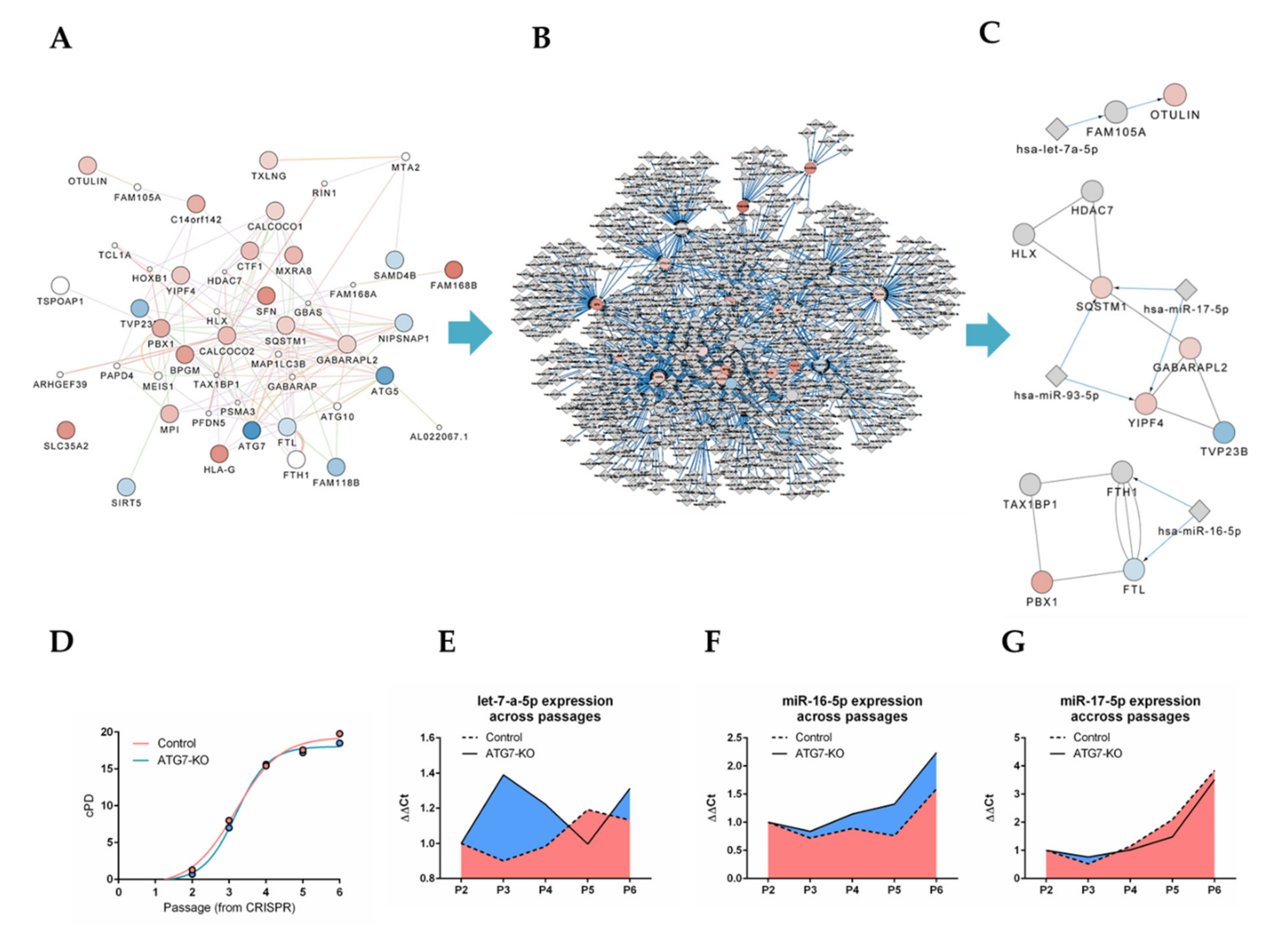
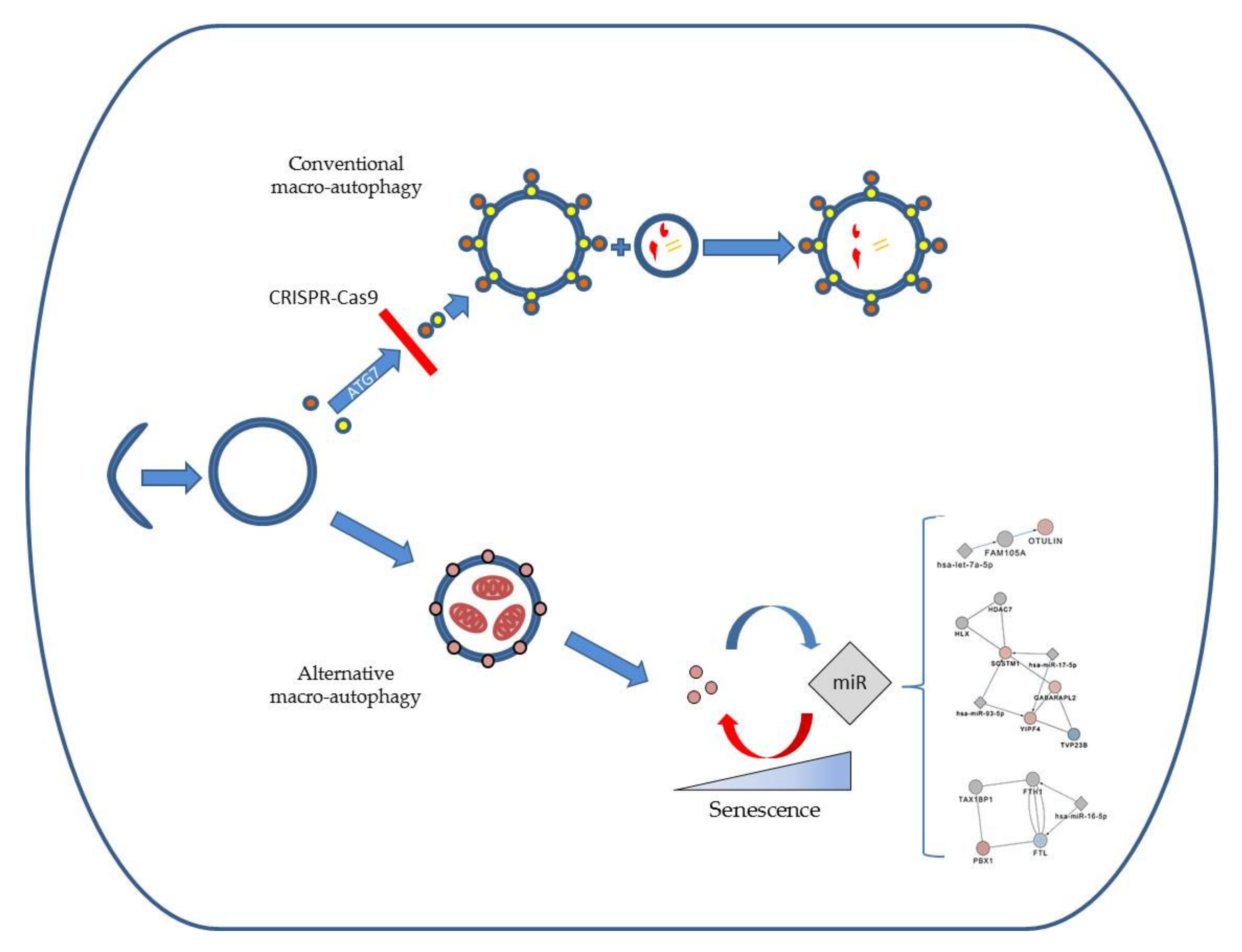
2.4. Bioinformatics Approach Predicts miR-16-5p, miR-17-5p, let-7a-5p and miR-93-5p as Key Regulators of ATG5/7alt with Biological Relevance in Senescence
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture and Genome Editing
4.3. Protein Isolation and Western Blot
4.4. RNA Isolation and RT-qPCR
4.5. FACS for Autophagosomal Fluorescence
4.6. Immunofluorescence
4.7. LC-MS
4.8. Bioinformatics Proteome Profiling Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.; Choi, M.E.; Hashimoto, S.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Autophagy: Friend or foe in lung disease? Ann. Am. Thorac. Soc. 2016, 13, S40–S47. [Google Scholar] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Green, D.R. Autophagy-independent functions of the autophagy machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Romero, Y.; Bueno, M.; Ramirez, R.; Alvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. Mtorc1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in ipf fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef]
- Hill, C.; Li, J.; Liu, D.; Conforti, F.; Brereton, C.J.; Yao, L.; Zhou, Y.; Alzetani, A.; Chee, S.J.; Marshall, B.G.; et al. Autophagy inhibition-mediated epithelial-mesenchymal transition augments local myofibroblast differentiation in pulmonary fibrosis. Cell Death Dis. 2019, 10, 591. [Google Scholar] [CrossRef]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The essential autophagy gene atg7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J. Biol. Chem. 2015, 290, 2547–2559. [Google Scholar] [CrossRef] [Green Version]
- Surolia, R.; Li, F.J.; Wang, Z.; Li, H.; Dsouza, K.; Thomas, V.; Mirov, S.; Pérez-Sala, D.; Athar, M.; Thannickal, V.J.; et al. Vimentin intermediate filament assembly regulates fibroblast invasion in fibrogenic lung injury. JCI Insight 2019, 4, e123253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of atg5/atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Invest. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, C.; Montano, M.; Garcia-Alvarez, J.; Ruiz, V.; Uhal, B.D.; Selman, M.; Pardo, A. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Ame. J. Respir. Cell Mol. Biol. 2001, 24, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Pardo, A.; Selman, M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann. Am. Thorac. Soc. 2016, 13, S417–S421. [Google Scholar] [CrossRef]
- Geng, Y.; Liu, X.; Liang, J.; Habiel, D.M.; Kulur, V.; Coelho, A.L.; Deng, N.; Xie, T.; Wang, Y.; Liu, N.; et al. Pd-l1 on invasive fibroblasts drives fibrosis in a humanized model of idiopathic pulmonary fibrosis. JCI Insight 2019, 4, e125326. [Google Scholar] [CrossRef] [Green Version]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The mirna-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding rnas in development and disease: Background, mechanisms, and therapeutic approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [Green Version]
- Sood, P.; Krek, A.; Zavolan, M.; Macino, G.; Rajewsky, N. Cell-type-specific signatures of micrornas on target mrna expression. Proc. Natl. Acad. Sci. USA 2006, 103, 2746–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. Microrna-21 contributes to myocardial disease by stimulating map kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Velikoff, M.; Canalis, E.; Horowitz, J.C.; Kim, K.K. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L786–L796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. Ctgf is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012, 5, S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Ichikawa, A.; Saito, N.; Kadota, T.; Tsubouchi, K.; Sato, N.; Yoshida, M.; et al. Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir. Res. 2017, 18, 114. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhou, X.J.; Zhang, H. Exploring the role of autophagy-related gene 5 (atg5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef]
- Sukseree, S.; Schwarze, U.Y.; Gruber, R.; Gruber, F.; Quiles Del Rey, M.; Mancias, J.D.; Bartlett, J.D.; Tschachler, E.; Eckhart, L. Atg7 is essential for secretion of iron from ameloblasts and normal growth of murine incisors during aging. Autophagy 2020. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Heart. J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Mailleux, A.A.; Crestani, B. Licence to kill senescent cells in idiopathic pulmonary fibrosis? Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef]
- Melk, A.; Baisantry, A.; Schmitt, R. The yin and yang of autophagy in acute kidney injury. Autophagy 2016, 12, 596–597. [Google Scholar] [CrossRef] [Green Version]
- Baisantry, A.; Bhayana, S.; Rong, S.; Ermeling, E.; Wrede, C.; Hegermann, J.; Pennekamp, P.; Sorensen-Zender, I.; Haller, H.; Melk, A.; et al. Autophagy induces prosenescent changes in proximal tubular s3 segments. J. Am. Soc. Nephrol. 2016, 27, 1609–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of gata4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Dalle Pezze, P.; Nelson, G.; Otten, E.G.; Korolchuk, V.I.; Kirkwood, T.B.; von Zglinicki, T.; Shanley, D.P. Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PLoS Comput. Biol. 2014, 10, e1003728. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in cell senescence: Is mitophagy the weakest link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Tsang, J.; Zhu, J.; van Oudenaarden, A. Microrna-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell 2007, 26, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, M.; Yoshida, M.; Hirata, K.; Furuyama, K.; Masui, T.; Uemoto, S.; Kawaguchi, Y. Senescence caused by inactivation of the homeodomain transcription factor pdx1 in adult pancreatic acinar cells in mice. FEBS Lett. 2019, 593, 2226–2234. [Google Scholar] [CrossRef]
- Soleimanpour, S.A.; Ferrari, A.M.; Raum, J.C.; Groff, D.N.; Yang, J.; Kaufman, B.A.; Stoffers, D.A. Diabetes susceptibility genes pdx1 and clec16a function in a pathway regulating mitophagy in β-cells. Diabetes 2015, 64, 3475–3484. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the crispr-cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic SD, F.M.; Kunz, M.; Xiao, K.; Just, A.; Pich, A.; Bauersachs, J.; Fiedler, J.; Sedding, D.; Thum, T. Inflammatory drivers of cardiovascular disease: Molecular characterization of senescent coronary vascular smooth muscle cells. Front. Physiol. Sect. Clin. Transl. Physiol. 2020. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2 Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2016. [Google Scholar]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The genemania prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Kutmon, M.; Ehrhart, F.; Willighagen, E.; Evelo, C.; Coort, S. Cytargetlinker app update: A flexible solution for network extension in cytoscape. F1000 Res. 2019, 7. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. Mirtarbase update 2018: A resource for experimentally validated microrna-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stojanović, S.D.; Fuchs, M.; Fiedler, J.; Xiao, K.; Meinecke, A.; Just, A.; Pich, A.; Thum, T.; Kunz, M. Comprehensive Bioinformatics Identifies Key microRNA Players in ATG7-Deficient Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 4126. https://doi.org/10.3390/ijms21114126
Stojanović SD, Fuchs M, Fiedler J, Xiao K, Meinecke A, Just A, Pich A, Thum T, Kunz M. Comprehensive Bioinformatics Identifies Key microRNA Players in ATG7-Deficient Lung Fibroblasts. International Journal of Molecular Sciences. 2020; 21(11):4126. https://doi.org/10.3390/ijms21114126
Chicago/Turabian StyleStojanović, Stevan D., Maximilian Fuchs, Jan Fiedler, Ke Xiao, Anna Meinecke, Annette Just, Andreas Pich, Thomas Thum, and Meik Kunz. 2020. "Comprehensive Bioinformatics Identifies Key microRNA Players in ATG7-Deficient Lung Fibroblasts" International Journal of Molecular Sciences 21, no. 11: 4126. https://doi.org/10.3390/ijms21114126