Caffeine: An Overview of Its Beneficial Effects in Experimental Models and Clinical Trials of Parkinson’s Disease


Abstract
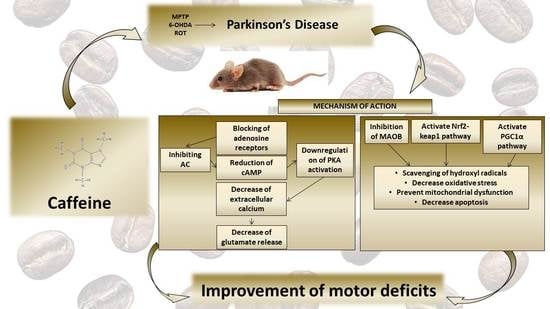
1. Introduction
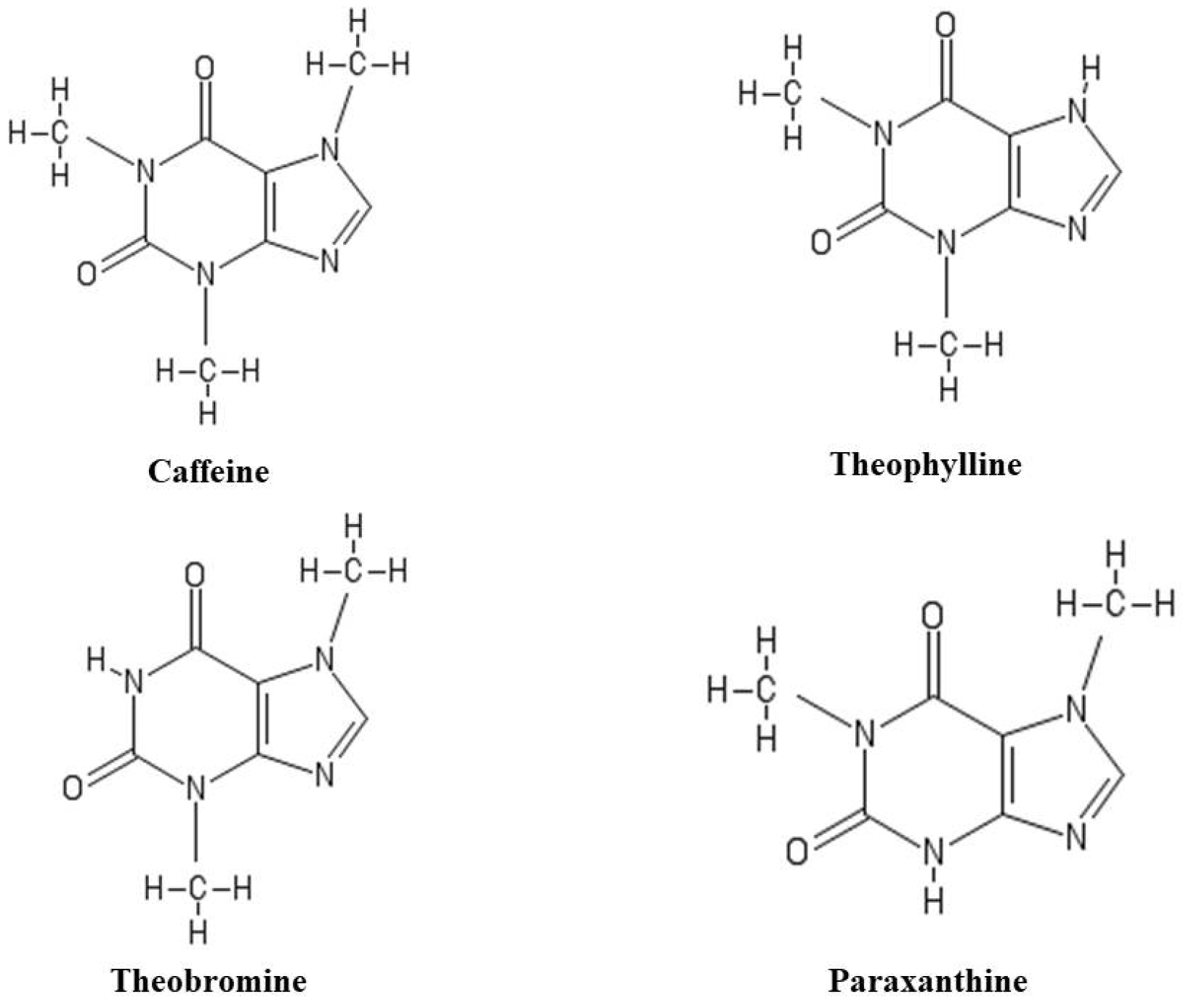
2. Caffeine
3. Neuroprotective Role of Caffeine in Experimental Studies of Parkinson’s Disease
4. Neuroprotective Role of Caffeine in Animal Models of Parkinson’s Disease
5. Evaluation of the Effects of Caffeine in Clinical Trials of Parkinson’s Disease
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s Disease |
| α-Syn | α-synuclein |
| ROT | rotenone |
| PQ | paraquat |
| MB | maneb |
| ROS | reactive oxygen species |
| H2O2 | hydrogen peroxide |
| GSH | Glutathione |
| BBB | blood brain barrier |
| CNS | central nervous system |
| ARs | adenosine receptors |
| MAO-B | monoamine oxidase-B |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PGC-1α | peroxisome proliferator-activated receptor-gamma coactivator 1-α |
| KO | knockout |
| PDE | phosphodiesterase |
| A2AR | adenosine 2A receptor |
| PKA | protein kinase A |
| TNF-α | tumor necrosis factor alpha |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| GABA | γ-aminobutyric acid |
| A1R | adenosine 1 receptor |
| MPTP | N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| 6-OHDA | 6-hydroxydopamine |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| DOPAC | 3,4 dihydroxyphenylacetic acid |
| CSC | 8-(-3-chlorostyryl)-caffeine |
| AIMs | abnormal involuntary movements |
| THP | trihexyphenidyl |
| ESS | Epworth Sleepiness Scale |
| UPDRS | unified Parkinson’s Disease rating scale |
| HC | healthy control |
| HPFS | Study of Health Professionals |
| NHS | Nurses’ Health Study |
References
- Beitz, J.M. Parkinson’s disease disease: A review. Front. Biosci. 2014, 6, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, H. Diagnosis and treatment of Parkinson’s disease. MMW Fortschr. Med. 2017, 159, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.B.; Iourinets, J.; Richard, I.H. Parkinson’s disease psychosis: Presentation, diagnosis and management. Neurodegener. Dis. Manag. 2017, 7, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Greenamyre, J.T. Gene-environment interactions in Parkinson’s disease: Specific evidence in humans and mammalian models. Neurobiol. Dis. 2013, 57, 38–46. [Google Scholar] [CrossRef]
- Shahpiri, Z.; Bahramsoltani, R.; Farzaei, M.H.; Farzaei, F.; Rahimi, R. Phytochemicals as future drugs for Parkinson’s disease: A comprehensive review. Rev. Neurosci. 2016, 27, 651–668. [Google Scholar] [CrossRef]
- Spencer, J.P.; Jenner, A.; Aruoma, O.I.; Evans, P.J.; Kaur, H.; Dexter, D.T.; Jenner, P.; Lees, A.J.; Marsden, D.C.; Halliwell, B. Intense oxidative DNA damage promoted by L-dopa and its metabolites. Implications for neurodegenerative disease. FEBS Lett. 1994, 353, 246–250. [Google Scholar] [CrossRef]
- Xu, F.; Liua, P.Y.; Pekar, J.J.; Lu, H.Z. Does acute caffeine ingestion alter brain metabolism in young adults? Neuroimage 2015, 110, 39–47. [Google Scholar] [CrossRef]
- Devasagayam, T.P.A.; Kamat, J.P.; Mohan, H.; Kesavan, P.C. Caffeine as an antioxidant: Inhibition of lipid peroxidation induced by reactive oxygen species. BBA Biomembr. 1996, 1282, 63–70. [Google Scholar] [CrossRef]
- Kolahdouzan, M.; Hamadeh, M.J. The neuroprotective effects of caffeine in neurodegenerative diseases. CNS Neurosci. Ther. 2017, 23, 272–290. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9. [Google Scholar] [CrossRef]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Lunet, N.; Santos, C.; Santos, J.; Vaz-Carneiro, A. Caffeine exposure and the risk of Parkinson’s disease: A systematic review and meta-analysis of observational studies. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S221–S238. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Saaksjarvi, K.; Knekt, P.; Rissanen, H.; Laaksonen, M.A.; Reunanen, A.; Mannisto, S. Prospective study of coffee consumption and risk of Parkinson’s disease. Eur. J. Clin. Nutr. 2008, 62, 908–915. [Google Scholar] [CrossRef]
- Ascherio, A.; Zhang, S.M.; Hernan, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef]
- Grosso, G.; Godos, J.; Galvano, F.; Giovannucci, E.L. Coffee, Caffeine, and Health Outcomes: An Umbrella Review. Annu. Rev. Nutr. 2017, 37, 131–156. [Google Scholar] [CrossRef]
- Gurley, B.J.; Steelman, S.C.; Thomas, S.L. Multi-ingredient, caffeine-containing dietary supplements: History, safety, and efficacy. Clin. Ther. 2015, 37, 275–301. [Google Scholar] [CrossRef]
- Talik, P.; Krzek, J.; Ekiert, R.J. Analytical Techniques Used for Determination of Methylxanthines and their Analogues-Recent Advances. Sep. Purif. Rev. 2012, 41, 1–61. [Google Scholar] [CrossRef]
- Monteiro, J.P.; Alves, M.G.; Oliveira, P.F.; Silva, B.M. Structure-Bioactivity Relationships of Methylxanthines: Trying to Make Sense of All the Promises and the Drawbacks. Molecules 2016, 21, 974. [Google Scholar] [CrossRef] [PubMed]
- Frary, C.D.; Johnson, R.K.; Wang, M.Q. Food sources and intakes of caffeine in the diets of persons in the United States. J. Am. Diet. Assoc. 2005, 105, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Heckman, M.A.; Weil, J.; Gonzalez de Mejia, E. Caffeine (1, 3, 7-trimethylxanthine) in foods: A comprehensive review on consumption, functionality, safety, and regulatory matters. J. Food Sci. 2010, 75, R77–R87. [Google Scholar] [CrossRef] [PubMed]
- Mandel, H.G. Update on caffeine consumption, disposition and action. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2002, 40, 1231–1234. [Google Scholar] [CrossRef]
- Orru, M.; Guitart, X.; Karcz-Kubicha, M.; Solinas, M.; Justinova, Z.; Barodia, S.K.; Zanoveli, J.; Cortes, A.; Lluis, C.; Casado, V.; et al. Psychostimulant pharmacological profile of paraxanthine, the main metabolite of caffeine in humans. Neuropharmacology 2013, 67, 476–484. [Google Scholar] [CrossRef]
- Lelo, A.; Birkett, D.J.; Robson, R.A.; Miners, J.O. Comparative pharmacokinetics of caffeine and its primary demethylated metabolites paraxanthine, theobromine and theophylline in man. Br. J. Clin. Pharmacol. 1986, 22, 177–182. [Google Scholar] [CrossRef]
- Che, B.; Wang, L.; Zhang, Z.; Zhang, Y.; Deng, Y. Distribution and accumulation of caffeine in rat tissues and its inhibition on semicarbazide-sensitive amine oxidase. Neurotoxicology 2012, 33, 1248–1253. [Google Scholar] [CrossRef]
- Kaplan, G.B.; Greenblatt, D.J.; Leduc, B.W.; Thompson, M.L.; Shader, R.I. Relationship of plasma and brain concentrations of caffeine and metabolites to benzodiazepine receptor binding and locomotor activity. J. Pharmacol. Exp. Ther. 1989, 248, 1078–1083. [Google Scholar]
- Arnaud, M.J. Pharmacokinetics and metabolism of natural methylxanthines in animal and man. Handb. Exp. Pharmacol. 2011, 33–91. [Google Scholar] [CrossRef]
- Stavric, B. Methylxanthines: Toxicity to humans. 3. Theobromine, paraxanthine and the combined effects of methylxanthines. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 1988, 26, 725–733. [Google Scholar] [CrossRef]
- Okuro, M.; Fujiki, N.; Kotorii, N.; Ishimaru, Y.; Sokoloff, P.; Nishino, S. Effects of paraxanthine and caffeine on sleep, locomotor activity, and body temperature in orexin/ataxin-3 transgenic narcoleptic mice. Sleep 2010, 33, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Hetzler, R.K.; Knowlton, R.G.; Somani, S.M.; Brown, D.D.; Perkins, R.M., 3rd. Effect of paraxanthine on FFA mobilization after intravenous caffeine administration in humans. J. Appl. Physiol. 1990, 68, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Sansone, R.; Ottaviani, J.I.; Rodriguez-Mateos, A.; Heinen, Y.; Noske, D.; Spencer, J.P.; Crozier, A.; Merx, M.W.; Kelm, M.; Schroeter, H.; et al. Methylxanthines enhance the effects of cocoa flavanols on cardiovascular function: Randomized, double-masked controlled studies. Am. J. Clin. Nutr. 2017, 105, 352–360. [Google Scholar] [CrossRef]
- Fredholm, B.B. On the mechanism of action of theophylline and caffeine. Acta Med. Scand. 1985, 217, 149–153. [Google Scholar] [CrossRef]
- Schnermann, J.; Osswald, H.; Hermle, M. Inhibitory effect of methylxanthines on feedback control of glomerular filtration rate in the rat kidney. Pflug. Arch. Eur. J. Physiol. 1977, 369, 39–48. [Google Scholar] [CrossRef]
- Ribeiro, J.A.; Sebastiao, A.M. Caffeine and adenosine. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S3–S15. [Google Scholar] [CrossRef]
- Riksen, N.P.; Smits, P.; Rongen, G.A. The cardiovascular effects of methylxanthines. Handb. Exp. Pharmacol. 2011, 413–437. [Google Scholar] [CrossRef]
- James, J.E. Critical review of dietary caffeine and blood pressure: A relationship that should be taken more seriously. Psychosom. Med. 2004, 66, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Teo, K.C.; Ho, S.L. Monoamine oxidase-B (MAO-B) inhibitors: Implications for disease-modification in Parkinson’s disease. Transl. Neurodegener. 2013, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, X.; Hu, X.; Fassett, J.; Zhu, G.; Tao, Y.; Li, J.; Huang, Y.; Zhang, P.; Zhao, B.; et al. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 2010, 13, 1011–1022. [Google Scholar] [CrossRef]
- Cohen, G. Monoamine oxidase and oxidative stress at dopaminergic synapses. J. Neural Transm. Suppl. 1990, 32, 229–238. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Xie, S.P.; Saw, W.T.; Ho, P.G.H.; Wang, H.; Lei, Z.; Yi, Z.; Tan, E.K. The Therapeutic Implications of Tea Polyphenols Against Dopamine (DA) Neuron Degeneration in Parkinson’s Disease (PD). Cells 2019, 8, 911. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A. Transcription Factor Nrf2: A novel target to modulate inflammatory and neuroprotective responses in Parkinson’s disease. SpringerPlus 2015, 4, L43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barone, M.C.; Sykiotis, G.P.; Bohmann, D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis. Models Mech. 2011, 4, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Simon, D.K. Transcribe to survive: Transcriptional control of antioxidant defense programs for neuroprotection in Parkinson’s disease. Antioxid. Redox Signal. 2009, 11, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Choi, O.H.; Shamim, M.T.; Padgett, W.L.; Daly, J.W. Caffeine and Theophylline Analogs-Correlation of Behavioral-Effects with Activity as Adenosine Receptor Antagonists and as Phosphodiesterase Inhibitors. Life Sci. 1988, 43, 387–398. [Google Scholar] [CrossRef]
- McPherson, P.S.; Kim, Y.K.; Valdivia, H.; Knudson, C.M.; Takekura, H.; Franzini-Armstrong, C.; Coronado, R.; Campbell, K.P. The brain ryanodine receptor: A caffeine-sensitive calcium release channel. Neuron 1991, 7, 17–25. [Google Scholar] [CrossRef]
- Liu, W.; Meissner, G. Structure-activity relationship of xanthines and skeletal muscle ryanodine receptor/Ca2+ release channel. Pharmacology 1997, 54, 135–143. [Google Scholar] [CrossRef]
- Lopez, F.; Miller, L.G.; Greenblatt, D.J.; Kaplan, G.B.; Shader, R.I. Interaction of caffeine with the GABAA receptor complex: Alterations in receptor function but not ligand binding. Eur. J. Pharmacol. 1989, 172, 453–459. [Google Scholar] [CrossRef]
- Roca, D.J.; Schiller, G.D.; Farb, D.H. Chronic caffeine or theophylline exposure reduces gamma-aminobutyric acid/benzodiazepine receptor site interactions. Mol. Pharmacol. 1988, 33, 481–485. [Google Scholar]
- Shi, D.; Padgett, W.L.; Daly, J.W. Caffeine analogs: Effects on ryanodine-sensitive calcium-release channels and GABAA receptors. Cell. Mol. Neurobiol. 2003, 23, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.E.; Daly, J.W. Stimulation of calcium release by caffeine analogs in pheochromocytoma cells. Biochem. Pharmacol. 1993, 46, 1825–1829. [Google Scholar] [CrossRef]
- Beavo, J.A.; Rogers, N.L.; Crofford, O.B.; Hardman, J.G.; Sutherland, E.W.; Newman, E.V. Effects of xanthine derivatives on lipolysis and on adenosine 3’,5’-monophosphate phosphodiesterase activity. Mol. Pharmacol. 1970, 6, 597–603. [Google Scholar] [PubMed]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Battig, K.; Holmen, J.; Nehlig, A.; Zvartau, E.E. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol. Rev. 1999, 51, 83–133. [Google Scholar]
- Stefanello, N.; Spanevello, R.M.; Passamonti, S.; Porciuncula, L.; Bonan, C.D.; Olabiyi, A.A.; Teixeira da Rocha, J.B.; Assmann, C.E.; Morsch, V.M.; Schetinger, M.R.C. Coffee, caffeine, chlorogenic acid, and the purinergic system. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2019, 123, 298–313. [Google Scholar] [CrossRef]
- Chee, H.K.; Oh, S.J. Molecular vibration-activity relationship in the agonism of adenosine receptors. Genom. Inform. 2013, 11, 282–288. [Google Scholar] [CrossRef]
- Dias, R.B.; Rombo, D.M.; Ribeiro, J.A.; Henley, J.M.; Sebastiao, A.M. Adenosine: Setting the stage for plasticity. Trends Neurosci. 2013, 36, 248–257. [Google Scholar] [CrossRef]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef]
- Halldner, L.; Aden, U.; Dahlberg, V.; Johansson, B.; Ledent, C.; Fredholm, B.B. The adenosine A1 receptor contributes to the stimulatory, but not the inhibitory effect of caffeine on locomotion: A study in mice lacking adenosine A1 and/or A2A receptors. Neuropharmacology 2004, 46, 1008–1017. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Porciuncula, L.O.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Neuroprotection by caffeine and adenosine A2A receptor blockade of beta-amyloid neurotoxicity. Br. J. Pharmacol. 2003, 138, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Nikodijevic, O.; Jacobson, K.A.; Daly, J.W. Chronic caffeine alters the density of adenosine, adrenergic, cholinergic, GABA, and serotonin receptors and calcium channels in mouse brain. Cell. Mol. Neurobiol. 1993, 13, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, S.; Schnermann, J.; Noorbakhsh, F.; Henry, S.; Yong, V.W.; Winston, B.W.; Warren, K.; Power, C. A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.A.; Agha, A.M.; Abdel-Rahman, A.A.; Nassar, N.N. Role of adenosine A2A receptor in cerebral ischemia reperfusion injury: Signaling to phosphorylated extracellular signal-regulated protein kinase (pERK1/2). Neuroscience 2016, 314, 145–159. [Google Scholar] [CrossRef]
- Mohamed, R.A.; Agha, A.M.; Nassar, N.N. SCH58261 the selective adenosine A(2A) receptor blocker modulates ischemia reperfusion injury following bilateral carotid occlusion: Role of inflammatory mediators. Neurochem. Res. 2012, 37, 538–547. [Google Scholar] [CrossRef]
- Pedata, F.; Pugliese, A.M.; Coppi, E.; Dettori, I.; Maraula, G.; Cellai, L.; Melani, A. Adenosine A2A receptors modulate acute injury and neuroinflammation in brain ischemia. Mediat. Inflamm. 2014, 2014, 805198. [Google Scholar] [CrossRef]
- Bonfoco, E.; Krainc, D.; Ankarcrona, M.; Nicotera, P.; Lipton, S.A. Apoptosis and Necrosis-2 Distinct Events Induced, Respectively, by Mild and Intense Insults with N-Methyl-D-Aspartate or Nitric-Oxide Superoxide in Cortical Cell-Cultures. Proc. Natl. Acad. Sci. USA 1995, 92, 7162–7166. [Google Scholar] [CrossRef]
- Matute, C.; Domercq, M.; Sanchez-Gomez, M.V. Glutamate-mediated glial injury: Mechanisms and clinical importance. Glia 2006, 53, 212–224. [Google Scholar] [CrossRef]
- Popoli, P.; Pintor, A.; Domenici, M.R.; Frank, C.; Tebano, M.T.; Pezzola, A.; Scarchilli, L.; Quarta, D.; Reggio, R.; Malchiodi-Albedi, F.; et al. Blockade of striatal adenosine A2A receptor reduces, through a presynaptic mechanism, quinolinic acid-induced excitotoxicity: Possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 1967–1975. [Google Scholar] [CrossRef]
- O’Regan, M.H.; Simpson, R.E.; Perkins, L.M.; Phillis, J.W. The selective A2 adenosine receptor agonist CGS 21680 enhances excitatory transmitter amino acid release from the ischemic rat cerebral cortex. Neuroscience Lett. 1992, 138, 169–172. [Google Scholar] [CrossRef]
- Melani, A.; Pantoni, L.; Bordoni, F.; Gianfriddo, M.; Bianchi, L.; Vannucchi, M.G.; Bertorelli, R.; Monopoli, A.; Pedata, F. The selective A2A receptor antagonist SCH 58261 reduces striatal transmitter outflow, turning behavior and ischemic brain damage induced by permanent focal ischemia in the rat. Brain Res. 2003, 959, 243–250. [Google Scholar] [CrossRef]
- Li, X.X.; Nomura, T.; Aihara, H.; Nishizaki, T. Adenosine enhances glial glutamate efflux via A2a adenosine receptors. Life Sci. 2001, 68, 1343–1350. [Google Scholar] [CrossRef]
- Nishizaki, T.; Nagai, K.; Nomura, T.; Tada, H.; Kanno, T.; Tozaki, H.; Li, X.X.; Kondoh, T.; Kodama, N.; Takahashi, E.; et al. A new neuromodulatory pathway with a glial contribution mediated via A(2a) adenosine receptors. Glia 2002, 39, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Muller, C.E.; Rodrigues, A.L.S.; Porciuncula, L.O.; et al. Caffeine acts through neuronal adenosine A(2A) receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef] [PubMed]
- Sonsalla, P.K.; Wong, L.Y.; Harris, S.L.; Richardson, J.R.; Khobahy, I.; Li, W.; Gadad, B.S.; German, D.C. Delayed caffeine treatment prevents nigral dopamine neuron loss in a progressive rat model of Parkinson’s disease. Exp. Neurol. 2012, 234, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Prediger, R.D.S. Effects of Caffeine in Parkinson’s Disease: From Neuroprotection to the Management of Motor and Non-Motor Symptoms. J. Alzheimers Dis. 2010, 20, S205–S220. [Google Scholar] [CrossRef]
- Kalda, A.; Yu, L.; Oztas, E.; Chen, J.F. Novel neuroprotection by caffeine and adenosine A(2A) receptor antagonists in animal models of Parkinson’s disease. J. Neurol. Sci. 2006, 248, 9–15. [Google Scholar] [CrossRef]
- Hauser, R.A.; Hubble, J.P.; Truong, D.D.; Istradefylline, U.S.S.G. Randomized trial of the adenosine A(2A) receptor antagonist istradefylline in advanced PD. Neurology 2003, 61, 297–303. [Google Scholar] [CrossRef]
- Bara-Jimenez, W.; Sherzai, A.; Dimitrova, T.; Favit, A.; Bibbiani, F.; Gillespie, M.; Morris, M.J.; Mouradian, M.M.; Chase, T.N. Adenosine A(2A) receptor antagonist treatment of Parkinson’s disease. Neurology 2003, 61, 293–296. [Google Scholar] [CrossRef]
- Negida, A.; Elfil, M.; Attia; Farahat, E.; Gabr, M.; Essam, A.; Attia, D.; Ahmed, H. Caffeine; the Forgotten Potential for Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2017, 16, 652–657. [Google Scholar] [CrossRef]
- Delamarre, A.; Meissner, W.G. Epidemiology, environmental risk factors and genetics of Parkinson’s disease. Presse Med. 2017, 46, 175–181. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, J.; Park, H.J.; Park, E.S.; Oh, S.; Zheng, H.; Junn, E.; Voronkov, M.; Stock, J.B.; Mouradian, M.M. Synergistic neuroprotection by coffee components eicosanoyl-5-hydroxytryptamide and caffeine in models of Parkinson’s disease and DLB. Proc. Natl. Acad. Sci. USA 2018, 115, E12053–E12062. [Google Scholar] [CrossRef]
- Luan, Y.; Ren, X.; Zheng, W.; Zeng, Z.; Guo, Y.; Hou, Z.; Guo, W.; Chen, X.; Li, F.; Chen, J.F. Chronic Caffeine Treatment Protects Against alpha-Synucleinopathy by Reestablishing Autophagy Activity in the Mouse Striatum. Front. Neurosci. 2018, 12, 301. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.G.; Batalha, V.L.; Vicente Miranda, H.; Coelho, J.E.; Gomes, R.; Goncalves, F.Q.; Real, J.I.; Rino, J.; Albino-Teixeira, A.; Cunha, R.A.; et al. Adenosine A2A Receptors Modulate alpha-Synuclein Aggregation and Toxicity. Cereb. Cortex 2017, 27, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.G.; Temido-Ferreira, M.; Vicente Miranda, H.; Batalha, V.L.; Coelho, J.E.; Szego, E.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.; Schwarzschild, M.A. Adenosine A2A receptor gene disruption protects in an alpha-synuclein model of Parkinson’s disease. Ann. Neurol. 2012, 71, 278–282. [Google Scholar] [CrossRef]
- Ravina, B.M.; Fagan, S.C.; Hart, R.G.; Hovinga, C.A.; Murphy, D.D.; Dawson, T.M.; Marler, J.R. Neuroprotective agents for clinical trials in Parkinson’s disease—A systematic assessment. Neurology 2003, 60, 1234–1240. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Ebadi, M.; Srinivasan, S.K.; Baxi, M.D. Oxidative stress and antioxidant therapy in Parkinson’s disease. Prog. Neurobiol. 1996, 48, 1–19. [Google Scholar] [CrossRef]
- Vingill, S.; Connor-Robson, N.; Wade-Martins, R. Are rodent models of Parkinson’s disease behaving as they should? Behav. Brain. Res. 2018, 352, 133–141. [Google Scholar] [CrossRef]
- Hayashi, A.; Matsunaga, N.; Okazaki, H.; Kakimoto, K.; Kimura, Y.; Azuma, H.; Ikeda, E.; Shiba, T.; Yamato, M.; Yamada, K.; et al. A Disruption Mechanism of the Molecular Clock in a MPTP Mouse Model of Parkinson’s Disease. Neuromol. Med. 2013, 15, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinson’s Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- Grunblatt, E.; Mandel, S.; Youdim, M.B.H. Neuroprotective strategies in Parkinson’s disease using the models of 6-hydroxydopamine and MPTP. Ann. N.Y. Acad. Sci. 2000, 899, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Zaitone, S.A.; Abo-Elmatty, D.M.; Shaalan, A.A. Acetyl-L-carnitine and alpha-lipoic acid affect rotenone-induced damage in nigral dopaminergic neurons of rat brain, implication for Parkinson’s disease therapy. Pharmacol. Biochem. Behav. 2012, 100, 347–360. [Google Scholar] [CrossRef]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N., Jr.; Schwarzschild, M.A. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, RC143. [Google Scholar] [CrossRef]
- Chen, X.; Lan, X.; Roche, I.; Liu, R.; Geiger, J.D. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J. Neurochem. 2008, 107, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Singh, S.; Singhal, N.K.; Sharma, A.; Parmar, D.; Singh, M.P. Nicotine- and caffeine-mediated changes in gene expression patterns of MPTP-lesioned mouse striatum: Implications in neuroprotection mechanism. Chem. Biol. Interact. 2010, 185, 81–93. [Google Scholar] [CrossRef]
- Singh, S.; Singh, K.; Patel, S.; Patel, D.K.; Singh, C.; Nath, C.; Singh, M.P. Nicotine and caffeine-mediated modulation in the expression of toxicant responsive genes and vesicular monoamine transporter-2 in 1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease phenotype in mouse. Brain Res. 2008, 1207, 193–206. [Google Scholar] [CrossRef]
- Aguiar, L.M.; Nobre, H.V., Jr.; Macedo, D.S.; Oliveira, A.A.; Freitas, R.M.; Vasconcelos, S.M.; Cunha, G.M.; Sousa, F.C.; Viana, G.S. Neuroprotective effects of caffeine in the model of 6-hydroxydopamine lesion in rats. Pharmacol. Biochem. Behav. 2006, 84, 415–419. [Google Scholar] [CrossRef]
- Aguiar, L.M.; Macedo, D.S.; Vasconcelos, S.M.; Oliveira, A.A.; de Sousa, F.C.; Viana, G.S. CSC, an adenosine A(2A) receptor antagonist and MAO B inhibitor, reverses behavior, monoamine neurotransmission, and amino acid alterations in the 6-OHDA-lesioned rats. Brain Res. 2008, 1191, 192–199. [Google Scholar] [CrossRef]
- Yu, L.Q.; Schwarzschild, M.A.; Chen, J.F. Cross-sensitization between caffeine- and L-dopa-induced behaviors in hemiparkinsonian mice. Neurosci. Lett. 2006, 393, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Reyhani-Rad, S.; Mahmoudi, J. Effect of adenosine A2A receptor antagonists on motor disorders induced by 6-hydroxydopamine in rat. Acta Cir. Bras. 2016, 31, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, J.E.; Langelier, N.A.; Oriel, B.S.; Reedy, C. The effects of systemic, intrastriatal, and intrapallidal injections of caffeine and systemic injections of A2A and A1 antagonists on forepaw stepping in the unilateral 6-OHDA-lesioned rat. Psychopharmacol. 2009, 201, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Bleickardt, C.; Mullins, D.; Parker, E.; Hodgson, R. A(2A) receptor antagonists do not induce dyskinesias in drug-naive or L-dopa sensitized rats. Brain Res. Bull. 2013, 98, 163–169. [Google Scholar] [CrossRef]
- Xiao, D.; Cassin, J.J.; Healy, B.; Burdett, T.C.; Chen, J.F.; Fredholm, B.B.; Schwarzschild, M.A. Deletion of adenosine A(1) or A((2)A) receptors reduces L-3,4-dihydroxyphenylalanine-induced dyskinesia in a model of Parkinson’s disease. Brain Res. 2011, 1367, 310–318. [Google Scholar] [CrossRef]
- Khadrawy, Y.A.; Salem, A.M.; El-Shamy, K.A.; Ahmed, E.K.; Fadl, N.N.; Hosny, E.N. Neuroprotective and Therapeutic Effect of Caffeine on the Rat Model of Parkinson’s Disease Induced by Rotenone. J. Diet. Suppl. 2017, 14, 553–572. [Google Scholar] [CrossRef]
- Kachroo, A.; Irizarry, M.C.; Schwarzschild, M.A. Caffeine protects against combined paraquat and maneb-induced dopaminergic neuron degeneration. Exp. Neurol. 2010, 223, 657–661. [Google Scholar] [CrossRef]
- Fernandes, V.S.; Santos, J.R.; Leao, A.H.F.F.; Medeiros, A.M.; Melo, T.G.; Izidio, G.S.; Cabral, A.; Ribeiro, R.A.; Abilio, V.C.; Ribeiro, A.M.; et al. Repeated treatment with a low dose of reserpine as a progressive model of Parkinson’s disease. Behav. Brain Res. 2012, 231, 154–163. [Google Scholar] [CrossRef]
- Colpaert, F.C. Pharmacological characteristics of tremor, rigidity and hypokinesia induced by reserpine in rat. Neuropharmacology 1987, 26, 1431–1440. [Google Scholar] [CrossRef]
- Moo-Puc, R.E.; Villanueva-Toledo, J.; Arankowsky-Sandoval, G.; Alvarez-Cervera, F.; Gongora-Alfaro, J.L. Treatment with subthreshold doses of caffeine plus trihexyphenidyl fully restores locomotion and exploratory activity in reserpinized rats. Neurosci. Lett. 2004, 367, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Rios Romenets, S.; Altman, R.; et al. Caffeine for treatment of Parkinson disease: A randomized controlled trial. Neurology 2012, 79, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Anang, J.; Pelletier, A.; Joseph, L.; Moscovich, M.; Grimes, D.; Furtado, S.; Munhoz, R.P.; Appel-Cresswell, S.; Moro, A.; et al. Caffeine as symptomatic treatment for Parkinson disease (Cafe-PD): A randomized trial. Neurology 2017, 89, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Weisskopf, M.G.; O’Reilly, E.J.; McCullough, M.L.; Calle, E.E.; Rodriguez, C.; Thun, M.J. Coffee consumption, gender, and Parkinson’s disease mortality in the Cancer Prevention Study II cohort: The modifying effects of estrogen. Am. J. Epidemiol. 2004, 160, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Palacios, N.; Gao, X.; McCullough, M.L.; Schwarzschild, M.A.; Shah, R.; Gapstur, S.; Ascherio, A. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov. Disord. 2012, 27, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, R.; Macklin, E.A.; Hung, A.Y.; Hayes, M.T.; Hyman, B.T.; Wills, A.M.; Gomperts, S.N.; Growdon, J.H.; Ascherio, A.; Scherzer, C.R.; et al. Associations of Lower Caffeine Intake and Plasma Urate Levels with Idiopathic Parkinson?s Disease in the Harvard Biomarkers Study. J. Parkinson Dis. 2020, 10, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Paganini-Hill, A. Risk factors for Parkinson’s disease: The Leisure World cohort study. Neuroepidemiology 2001, 20, 118–124. [Google Scholar] [CrossRef]
- Hu, G.; Bidel, S.; Jousilahti, P.; Antikainen, R.; Tuomilehto, J. Coffee and tea consumption and the risk of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2007, 22, 2242–2248. [Google Scholar] [CrossRef]
- Tan, L.C.; Koh, W.P.; Yuan, J.M.; Wang, R.; Au, W.L.; Tan, J.H.; Tan, E.K.; Yu, M.C. Differential effects of black versus green tea on risk of Parkinson’s disease in the Singapore Chinese Health Study. Am. J. Epidemiol. 2008, 167, 553–560. [Google Scholar] [CrossRef]
- Hernan, M.A.; Takkouche, B.; Caamano-Isorna, F.; Gestal-Otero, J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002, 52, 276–284. [Google Scholar] [CrossRef]
- Kyrozis, A.; Ghika, A.; Stathopoulos, P.; Vassilopoulos, D.; Trichopoulos, D.; Trichopoulou, A. Dietary and lifestyle variables in relation to incidence of Parkinson’s disease in Greece. Eur. J. Epidemiol. 2013, 28, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Guo, X.; Park, Y.; Huang, X.; Sinha, R.; Freedman, N.D.; Hollenbeck, A.R.; Blair, A.; Chen, H. Caffeine intake, smoking, and risk of Parkinson disease in men and women. Am. J. Epidemiol. 2012, 175, 1200–1207. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Animal Models | Induced PD Models | Treatment | Dosage and Route of Administration | Therapeutic Effects | Ref. |
|---|---|---|---|---|---|
| C57BL/6 mice | MPTP (single dose 40 mg/kg) or (multiple-dose 20 mg/kg) via intraperitoneal | Caffeine and A2AR antagonists | 0–60 mg/kg via intraperitoneal 10 min before injury | Caffeine reduced MPTP-induced dopamine depletion, improving the locomotor activity. Caffeine exerts its action blocking the A2ARs, as demonstrated by using mice lacking the A2AR. The treatment with A2AR antagonists also confirmed this result. | [96] |
| male Sprague–Dawley rats | MPTP (75 μg/ day) unilateral intra-cerebroventricular infusion for 28 days | Caffeine | 60–80 mg/kg daily, after 1 to 3 weeks from the injury | After 7 and 21 days of caffeine administration, mice treated with MPTP showed a reduction in the loss of nigrostriatal dopamine neurons and the response of microglia in the substantia nigra, with a consequent decrease in neuroinflammation. | [75] |
| FVB mice | MPTP (20 mg/kg) daily via intraperitoneal for the second week | Caffeine | 10 mg/kg via intraperitoneal for the first 7 days and 10 min before MPTP treatment for the second week | The pretreatment with caffeine prevented the damage to the BBB induced by MPTP and decreased the activation of astrocytes and microglia. | [97] |
| Swiss albino mice | MPTP (20 mg/kg) daily for the subsequently 4 weeks | Caffeine and nicotine | Caffeine (20 mg/kg) and nicotine (1 mg/kg), via intraperitoneal, daily for the first 8 weeks | The consumption of caffeine restored the transcription of genes involved in several processes, including cell apoptosis, cell cycle regulation and oxidative stress. These transcripts were downregulated in MPTP mice. | [98] |
| Swiss albino mice | MPTP (20 mg/kg) from 1 day to 4 weeks | Caffeine and nicotine | Caffeine (10–30 mg/kg) and nicotine (0.5–1.5 mg/kg) pretreated daily via intraperitoneal for 8 weeks | Caffeine (20 mg/kg) or nicotine (1 mg/kg) treatments prevented the depletion of dopamine induced by MPTP. Both alkaloids reduced the expression of GSTA4-4, GST-ya, GST-yc and VMAT-2, reducing the MPTP-induced toxicity. | [99] |
| Wistar rats | 6-OHDA (12 μg/μL) single stereotaxic injection | Caffeine | 10 and 20 mg/kg via intraperitoneal for 13 days, one hour after injury | Caffeine prevented the MPTP-induced dopamine and DOPAC depletion. In this way, caffeine improved motor deficits. | [100] |
| Wistar rats | 6-OHDA (12 μg/μL) stereotaxic injection into the right corpus striatum | CSC and L-DOPA | Caffeine 1–5 mg/kg and L-DOPA (50 mg/kg + benserazide 12.5 mg/kg) administered alone or co-administered for 7 consecutive days | The CSC prevented dopamine depletion and DOPAC, 6-OHDA-induced. The CSC treatment promoted a decrease in monoamines, enhancing its neuroprotective effect. The effects of CSC were potentiated when administered together with L-DOPA. | [101] |
| C57BL/6 mice | 6-OHDA (2.5 μg/μL) unilaterally into the left dorsal corpus striatum | Caffeine and L-DOPA | Caffeine (2.5 or 10 mg/kg) and L-DOPA (2.0 mg /kg) administered alone via intraperitoneal after the injury for 14–21 days, or co-administered at higher dosage for 26 days | The treatment of caffeine associated with L-DOPA led to an improvement in sensitized rotational behavior in mice induced with 6-OHDA compared to control mice. | [102] |
| Wistar rats | 6-OHDA (0.2 μL/min) unilaterally into the substantia nigra | Caffeine and SCH 5826 | Caffeine (30 mg/kg) and SCH 58261 (2 mg/kg) via intraperitoneal | The treatment with caffeine led to an improvement in balance disorders in mice treated with 6-OHDA. These results could be obtained through the inhibition of presynaptic A2AR, as demonstrated by the use of its antagonist, SCH 58261. | [103] |
| Long Evans rats | 6-OHDA (12 μg) unilaterally via intraperitoneal | Caffeine, SCH 5826, L-DOPA, N6-Cyclopentyladenosine and 8-Cyclopentyltheophylline | Caffeine (15 mg/kg), SCH 58261 (2 mg/kg), L- DOPA (8 mg/kg) N6-Cyclopentyladenosine (0.03–0.2 mg/kg) and 8-Cyclopentyltheophylline (3–7 mg/kg) via systemic intraperitoneal | The caffeine treatment associated with L-DOPA in rats induced with 6-OHDA improved the motor activity. These improvements could be obtained through the inhibition of A2AR, as demonstrated by the use of its antagonist SCH 5826. | [104] |
| Sprague Dawley rats | 6-OHDA (2 μg/μL) unilateral infusion into the nigrostriatal | L-DOPA, Caffeine, SCH 412348, Istradefillin and Vipadenant | L-DOPA (6 mg/kg), Caffeine (30 mg/kg), SCH 412348 (3 mg/kg), Istradefillin (3 mg/kg) and Vipadenant (10 mg/kg) after 14 days from the injury with L-DOPA and A2AR antagonists for 19–22 days | Chronic treatment with caffeine or A2AR antagonists (SCH 412348, vipadenant, or istradefillin) does not induce dyskinetic activity in rats treated with 6-OHDA, unlike L-DOPA. | [105] |
| C57BL/6 (A1−/−, A2A+/+) KO, (A1+/+, A2A−/−) KO and (A1−/−, A2A−/−) KO mice | 6-OHDA (10 μg) stereotactic unilateral injection | Caffeine and L-DOPA | Caffeine (3–15 mg/kg) and L-DOPA (2 mg/kg) via intraperitoneal with L-DOPA for 2–3 weeks after 14 days from the injury and 10 min before the administration of L-DOPA with two doses intraperitoneal of caffeine | Caffeine did not alleviate L-DOPA-induced dyskinesia, probably due to its general motor stimulation actions. The simultaneous blocking of A1R and A2AR, like caffeine, did not improve dyskinesia compared to the use of a specific A1R or A2AR antagonist alone. | [106] |
| Wistar rats | ROT (1.5 mg/kg) via intraperitoneal for 45 days | Caffeine | 30 mg/kg before or after the induction of ROT | Caffeine treatment restored dopamine levels in the corpus striatum and prevented motor and muscle deficits induced by ROT. Caffeine reduced the level of MDA and oxidative stress. | [107] |
| C57BL/6NCrl mice | PQ (10 mg/kg) and MB (30 mg/kg) via intraperitoneal 10 min after caffeine treatment | Caffeine | 5 or 20 mg/kg via intraperitoneal | Caffeine treatment prevented the neurodegeneration of dopaminergic neurons induced by chronic pesticide exposure. | [108] |
| Wistar rats | Reserpine (5 mg/kg) via intraperitoneal | Caffeine and THP | Caffeine (1 mg/kg) and THP (0.1 mg/kg), 24 h after the injury, the animals were treated alone with caffeine or THP, or caffeine and THP combined | In reserpine-induced rats, the co-treatment of caffeine and THP led to the recovery of motor and exploratory activities. The treatments with caffeine or THP did not invert hypokinesia induced by reserpine. | [111] |
| Title Study | Type Study | Patients | Age | Outcome | Results | Ref. |
|---|---|---|---|---|---|---|
| Caffeine for Motor Manifestations of Parkinson’s Disease (NCT01190735) | Complete phase 2 clinical study | 28 | aged 18 years and older | The aim of the study was to evaluate the tolerability and efficacy of the treatment at increasing doses of caffeine (100–200–300–500 mg). | NA | - |
| Caffeine for Excessive Daytime Somnolence in Parkinson’s Disease (NCT00459420) | Double-blind controlled phase 2/3 complete study | 58 | aged 18 years and older | The study evaluated the effects of caffeine in idiopathic PD patients treated daily with caffeine (100 mg) and for the following 3 weeks with caffeine (200 mg). | Caffeine treatment led to a reduction in the ESS score and the improvement of the Clinical Global Impression of Change outcomes and UPDRS. | [112] |
| Caffeine as a Therapy for Parkinson’s Disease (NCT01738178) | Complete phase 3 clinical study | 121 | aged 45–70 years | The aim of the study was to evaluate the effects of caffeine (200 mg) in idiopathic PD patients. | Caffeine did not improve motor parkinsonism, while it led to a slight improvement in sleepiness. Patients taking for long-time caffeine had a slight increase in dyskinesia and a worsening in cognitive tests. | [113] |
| Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women | Prospective cohort study | 10 men and 16 women | aged 40–75 years and aged 30–55 years | The study assessed the relationship between caffeine consumption with PD risk. | Moderate coffee consumption reduced the PD risk more in men than women. | [17] |
| Coffee Consumption, Gender, and Parkinson’s Disease Mortality in the Cancer Prevention Study II Cohort: The Modifying Effects of Estrogen | Cohort study | 909 men and 340 women | average aged 57 years men and aged 56 years women | The aim of the study was to evaluate the relationship between caffeine consumption and mortality rate for PD. | Caffeine consumption associated with smoking and alcohol led to a decrease in PD risk and a reduction in the rate of mortality. | [114] |
| Caffeine and Risk of Parkinson’s Disease in a Large Cohort of Men and Women | Prospective study | 197 men and 120 women | mean age 75 years men and 74 women | The study evaluated the relationship between caffeine consumption and PD risk. | Caffeine consumption reduced the risk of PD onset in both sexes. | [115] |
| Associations of Lower Caffeine Intake and Plasma Urate Levels with Idiopathic Parkinson’s Disease in the Harvard Biomarkers Study | Case-control study | 566 | average age 67 years | The aim of the study was to assess the effects of caffeine consumption and PD risk. | Caffeine consumption was associated with a lower risk of onset of idiopathic PD in the sex-independent way. | [116] |
| Association of Coffee and Caffeine Intake with the Risk of Parkinson Disease | Cohort study | 8004 | aged 45–68 years | The study assessed the relationship between caffeine consumption and PD risk. | The habitual caffeine consumers showed PD risk to be 5 times lower compared to non-coffee consumers. | [15] |
| Risk Factors for Parkinson’s Disease: The Leisure World Cohort Study | Case-control study | 395 | mean ages 75 years | The aim of the study was to evaluate the effects on caffeine consumption, as well as smoking and other risk factors related to PD risk. | Coffee consumption and smoking led to a reduction in the PD risk. | [117] |
| Coffee and tea consumption and the risk of Parkinson’s disease | Cohort study | 200 | aged 25–64 years | The study assessed the relationship between caffeine consumption and PD risk. | The higher caffeine consumption reduces the PD risk, especially in the 25–49 age range. | [118] |
| Prospective Study of Coffee Consumption and Risk of Parkinson’s Disease | Prospective Study | 101 | aged 50–79 years | The aim of the study was to analyze coffee consumption on the incidence of PD. | The daily consumers of coffee showed a lower risk of PD onset compared to non-coffee drinkers. | [16] |
| Differential Effects of Black Versus Green Tea on Risk of Parkinson’s Disease in the Singapore Chinese Health Study | Cohort study | 157 | aged 45–74 years | The study evaluated the relationship between caffeine consumption and PD risk. | The habitual coffee consumers or black tea, but not of green tea showed a low risk of developing PD. | [119] |
| A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease | Meta-analysis study | 18,605 | NA | The aim of the study was to assess coffee consumption and PD risk. | The daily coffee consumers showed a low PD risk compared to non-consumers. | [120] |
| Dietary and Lifestyle Variables in Relation to Incidence of Parkinson’s Disease in Greece | Cohort study | 118 | aged 20–86 years | The aim of the study was to examine the relationship between caffeine and eating habits as well as lifestyle and PD risk. | Caffeine consumption was associated with a lower risk of PD. | [121] |
| Caffeine Intake, Smoking, and Risk of Parkinson Disease in Men and Women | Cohort study | 318,260 | aged 50–71 years | The study was to evaluate the relationship between caffeine intake and PD risk. | Caffeine intake reduced the risk of PD onset independently by gender. | [122] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schepici, G.; Silvestro, S.; Bramanti, P.; Mazzon, E. Caffeine: An Overview of Its Beneficial Effects in Experimental Models and Clinical Trials of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 4766. https://doi.org/10.3390/ijms21134766
Schepici G, Silvestro S, Bramanti P, Mazzon E. Caffeine: An Overview of Its Beneficial Effects in Experimental Models and Clinical Trials of Parkinson’s Disease. International Journal of Molecular Sciences. 2020; 21(13):4766. https://doi.org/10.3390/ijms21134766
Chicago/Turabian StyleSchepici, Giovanni, Serena Silvestro, Placido Bramanti, and Emanuela Mazzon. 2020. "Caffeine: An Overview of Its Beneficial Effects in Experimental Models and Clinical Trials of Parkinson’s Disease" International Journal of Molecular Sciences 21, no. 13: 4766. https://doi.org/10.3390/ijms21134766
APA StyleSchepici, G., Silvestro, S., Bramanti, P., & Mazzon, E. (2020). Caffeine: An Overview of Its Beneficial Effects in Experimental Models and Clinical Trials of Parkinson’s Disease. International Journal of Molecular Sciences, 21(13), 4766. https://doi.org/10.3390/ijms21134766