Dissecting the Seed Maturation and Germination Processes in the Non-Orthodox Quercus ilex Species Based on Protein Signatures as Revealed by 2-DE Coupled to MALDI-TOF/TOF Proteomics Strategy

 ,
,
Abstract
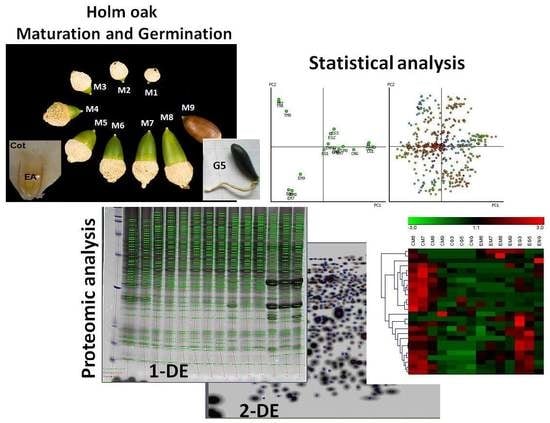
:
1. Introduction
2. Results
2.1. Acorn Maturation and Germination Stages
2.2. Protein Extraction, 1- and 2-D Gel Electrophoresis
2.3. Statistical Analysis of the Data
2.4. Protein Identification, Functional Classification of the Differentially Abundant Proteins, and Changes in Abundance Throughout the Maturation and Germination Stages
3. Discussion
4. Material and Methods
4.1. Plant Material
4.2. Protein Extraction and Gel Electrophoresis
4.3. Mass Spectrometry Analysis and Protein Identification
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1-DE | one dimensional gel electrophoresis |
| 2-DE | two-dimensional gel electrophoresis |
| WC | water content |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| IPG | immobilized pH gradient |
| IEF | isoelectric focusing |
| ROS | Reactive oxygen species |
| EA | embryo axis |
| Cot | cotyledon |
| NG | non-germinated acorns |
| MALDI | Matrix-assisted laser desorption ionization |
| TOF | Time of flight |
References
- Rey, M.D.; Castillejo, M.Á.; Sánchez-Lucas, R.; Guerrero-Sanchez, V.M.; López-Hidalgo, C.; Romero-Rodríguez, C.; Valero-Galván, J.; Sghaier-Hammami, B.; Simova-Stoilova, L.; Echevarría-Zomeño, S.; et al. Proteomics, holm oak (Quercus ilex L.) and other recalcitrant and orphan forest tree species: How do they see each other? Int. J. Mol. Sci. 2019, 20, 692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bareke, T. Biology of seed development and germination physiology. Adv. Plants Agric. Res. 2018, 8, 336–346. [Google Scholar] [CrossRef]
- Walters, C. Orthodoxy, recalcitrance and in-between: Describing variation in seed storage characteristics using threshold responses to water loss. Planta 2015, 242, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Berjak, P.; Pammenter, N.W. From avicennia to zizania: Seed recalcitrance in perspective. Ann. Bot. 2008, 101, 213–228. [Google Scholar] [CrossRef]
- Pammenter, N.W.; Berjak, P. A review of recalcitrant seed physiology in relation to desiccation-tolerance mechanisms. Seed Sci. Res. 1999, 9, 13–37. [Google Scholar] [CrossRef]
- Bai, F.; Settles, A.M. Imprinting in plants as a mechanism to generate seed phenotypic diversity. Front. Plant Sci. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Leprince, O.; Pellizzaro, A.; Berriri, S.; Buitink, J. Late seed maturation: Drying without dying. J. Exp. Bot. 2017, 68, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oracz, K.; Karpiński, S. Phytohormones signaling pathways and ROS involvement in seed germination. Front. Plant Sci. 2016, 7, 864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Bartels, A.; Cheng, X.; Meyer, A.; Charles An, Y.Q.; Hsieh, T.F.; Xiao, W. Epigenetics regulates reproductive development in plants. Plants 2019, 8, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajjou, L.; Duval, M.; Gallardo, K.; Catusse, J.; Bally, J.; Job, C.; Job, D. Seed Germination and Vigor. Annu. Rev. Plant Biol. 2012, 63, 507–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch-Savage, W.E.; Footitt, S. Seed dormancy cycling and the regulation of dormancy mechanisms to time germination in variable field environments. J. Exp. Bot. 2017, 68, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Angelovici, R.; Galili, G.; Fernie, A.R.; Fait, A. Seed desiccation: A bridge between maturation and germination. Trends Plant Sci. 2010, 15, 211–218. [Google Scholar] [CrossRef]
- Sghaier-Hammami, B.; Redondo-López, I.; Valero-Galvàn, J.; Jorrín-Novo, J.V. Protein profile of cotyledon, tegument, and embryonic axis of mature acorns from a non-orthodox plant species: Quercus ilex. Planta 2016, 243, 369–396. [Google Scholar] [CrossRef]
- Romero-Rodríguez, M.C.; Archidona-Yuste, A.; Abril, N.; Gil-Serrano, A.M.; Meijón, M.; Jorrín-Novo, J.V. Germination and early seedling development in quercus ilex recalcitrant and non-dormant seeds: Targeted transcriptional, hormonal, and sugar analysis. Front. Plant Sci. 2018, 9, 1508. [Google Scholar] [CrossRef]
- Galván, J.V.; Valledor, L.; Cerrillo, R.M.N.; Gil-Pelegrin, E.; Jorrín-Novo, J.V. Studies of variability in Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) through acorn protein profile analysis. J. Proteom. 2011, 74, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Miernyk, J.A.; Hajduch, M. Seed proteomics. J. Proteom. 2011, 74, 389–400. [Google Scholar] [CrossRef]
- Finch-Savage, W.E.; Blake, P.S. Indeterminate development in desiccation-sensitive seeds of Quercus robur L. Seed Sci. Res. 1994, 4, 127–133. [Google Scholar] [CrossRef]
- Xia, K.; Hill, L.M.; Li, D.Z.; Walters, C. Factors affecting stress tolerance in recalcitrant embryonic axes from seeds of four Quercus (Fagaceae) species native to the USA or China. Ann. Bot. 2014, 114, 1747–1759. [Google Scholar] [CrossRef]
- Sghaier-Hammami, B.; Drira, N.; Jorrín-Novo, J.V. Comparative 2-DE proteomic analysis of date palm (Phoenix dactylifera L.) somatic and zygotic embryos. J. Proteom. 2009, 73, 161–177. [Google Scholar] [CrossRef]
- Miguel, A.; de Vega-Bartol, J.; Marum, L.; Chaves, I.; Santo, T.; Leitão, J.; Varela, M.C.; Miguel, C.M. Characterization of the cork oak transcriptome dynamics during acorn development. BMC Plant Biol. 2015, 15, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobrino-Vesperinas, E.; Viviani, A.B. Pericarp micromorphology and dehydration characteristics of Quercus suber L. acorns. Seed Sci. Res. 2000, 10, 401–407. [Google Scholar] [CrossRef]
- Romero-Rodríguez, M.C.; Jorrín-novo, J.V.; Castillejo, M.A.; Angeles, M. Toward characterizing germination and early growth in the non-orthodox forest tree species Quercus ilex through complementary gel and gel-free proteomic analysis of embryo and seedlings. J. Proteom. 2019, 197, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Coulter, K.M.; Krochko, J.E.; Bewley, J.D. Morphological stages and storage protein accumulation in developing alfalfa (Medicago sativa L.) seeds. Seed Sci. Res. 1991, 1, 119–125. [Google Scholar] [CrossRef]
- Mouzo, D.; Bernal, J.; López-Pedrouso, M.; Franco, D.; Zapata, C. Advances in the biology of seed and vegetative storage proteins based on two-dimensional electrophoresis coupled to mass spectrometry. Molecules 2018, 23, 2462. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Q.; Ye, J.Q.; Rogowska-Wrzesinska, A.; Wojdyla, K.I.; Jensen, O.N.; Møller, I.M.; Song, S.Q. Proteomic comparison between maturation drying and prematurely imposed drying of zea mays seeds reveals a potential role of maturation drying in preparing proteins for seed germination, seedling vigor, and pathogen resistance. J. Proteome Res. 2014, 13, 606–626. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, K.; Le Signor, C.; Vandekerckhove, J.; Thompson, R.D.; Burstin, J. Proteomics of medicago truncatula seed development establishes the time frame of diverse metabolic processes related to reserve accumulation. Plant Physiol. 2003, 133, 664–682. [Google Scholar] [CrossRef] [Green Version]
- Erbaş, S.; Tonguç, M.; Karakurt, Y.; Şanli, A. Mobilization of seed reserves during germination and early seedling growth of two sunflower cultivars. J. Appl. Bot. Food Qual. 2016, 89. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, H.; Yan, H.; Qiu, L.; Baskin, C.C. Mobilization and role of starch, protein, and fat reserves during seed germination of six wild grassland species. Front. Plant Sci. 2018, 9, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, R.P.; Battistelli, A.; Moscatello, S.; Chen, Z.H.; Leegood, R.C.; Famiani, F. Metabolism of the seed and endocarp of cherry (Prunus avium L.) during development. Plant Physiol. Biochem. 2011, 49, 923–930. [Google Scholar] [CrossRef]
- Kabeya, D.; Sakai, S. The role of roots and cotyledons as storage organs in early stages of establishment in Quercus crispula: A quantitative analysis of the nonstructural carbohydrate in cotyledons and roots. Ann. Bot. 2003, 92, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, B.; Siddiqui, H.; David, L.C.; Powers, S.J.; Borrill, P.; Uauy, C.; Smith, A.M. Final grain weight is not limited by the activity of key starch-synthesising enzymes during grain filling in wheat. J. Exp. Bot. 2018, 69, 5461–5475. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, B.C.; Xu, Y.; Zhu, Y.X. Systematic studies of 12S seed storage protein accumulation and degradation patterns during Arabidopsis seed maturation and early seedling germination stages. J. Biochem. Mol. Biol. 2007, 40, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Gábrišová, D.; Klubicová, K.; Danchenko, M.; Gömöry, D.; Berezhna, V.V.; Skultety, L.; Miernyk, J.A.; Rashydov, N.; Hajduch, M. Do cupins have a function beyond being seed storage proteins? Front. Plant Sci. 2016, 6, 1215. [Google Scholar] [CrossRef] [Green Version]
- Uwer, U.; Willmitzer, L.; Altmann, T. Inactivation of a glycyl-tRNA synthetase leads to an arrest in plant embryo development. Plant Cell 1998, 10, 1277–1294. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Wu, J.; Liu, Y.; Huang, X.; Li, F.; Wang, Z.; Sun, M.X. Ribosomal protein NtRPL17 interacts with kinesin-12 family protein NtKRP and functions in the regulation of embryo/seed size and radicle growth. J. Exp. Bot. 2017, 68, 5553–5564. [Google Scholar] [CrossRef] [Green Version]
- Oracz, K.; Stawska, M. Cellular recycling of proteins in seed dormancy alleviation and germination. Front. Plant Sci. 2016, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Yao, X.; Ye, T.; Ma, S.; Liu, X.; Yin, X.; Wu, Y. Arabidopsis aspartic protease ASPG1 affects seed dormancy, seed longevity and seed germination. Plant Cell Physiol. 2018, 59, 1415–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuesta-Seijo, J.A.; Ruzanski, C.; Krucewicz, K.; Meier, S.; Hägglund, P.; Svensson, B.; Palcic, M.M. Functional and structural characterization of plastidic starch phosphorylase during barley endosperm development. PLoS ONE 2017, 12, e0175488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.E.; Tian, L. Phylogenomic analysis of UDP-dependent glycosyltransferases provides insights into the evolutionary landscape of glycosylation in plant metabolism. Plant J. 2019, 100, 1273–1288. [Google Scholar] [CrossRef]
- Woo, M.O.; Ham, T.H.; Ji, H.S.; Choi, M.S.; Jiang, W.; Chu, S.H.; Piao, R.; Chin, J.H.; Kim, J.A.; Park, B.S.; et al. Inactivation of the UGPase1 gene causes genic male sterility and endosperm chalkiness in rice (Oryza sativa L.). Plant J. 2008, 54, 190–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Verma, R.; Kim, M.; Lee, J.; Kim, Y.; Bang, J.; Reiter, W.; Pai, H. Depletion of UDP-D-apiose/UDP-D-xylose synthases results in rhamnogalacturonan-II deficiency, cell wall thickening, and cell death in higher plants. J. Biol. Chem. 2006, 281, 13708–13716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.R.; Hasenstein, K.H. Oxygen dependency of germinating Brassica seeds. Life Sci. Sp. Res. 2016, 8, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Cao, D.; Li, H.; Xue, H.; Chen, L.; Liu, B.; Cao, S. Quantitative proteomics of pomegranate varieties with contrasting seed hardness during seed development stages. Tree Genet. Genomes 2018, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Wang, J.; Zhang, Z.; Wu, Y. Transactivation of Sus1 and Sus2 by Opaque2 is an essential supplement to sucrose synthase-mediated endosperm filling in maize. Plant Biotechnol. J. 2020, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.; Alhagdow, M.; Nunes-Nesi, A.; Quemener, B.; Guillon, F.; Bouchet, B.; Faurobert, M.; Gouble, B.; Page, D.; Garcia, V.; et al. GDP-d-mannose 3,5-epimerase (GME) plays a key role at the intersection of ascorbate and non-cellulosic cell-wall biosynthesis in tomato. Plant J. 2009, 60, 499–508. [Google Scholar] [CrossRef]
- Amir, R.; Galili, G.; Cohen, H. The metabolic roles of free amino acids during seed development. Plant Sci. 2018, 275, 11–18. [Google Scholar] [CrossRef]
- Kamara, J.S.; Hoshino, M.; Satoh, Y.; Nayar, N.; Takaoka, M.; Sasanuma, T.; Abe, T. Japanese sake-brewing rice cultivars show high levels of globulin-like protein and a chloroplast stromal HSP70. Crop Sci. 2009, 49, 2198–2206. [Google Scholar] [CrossRef]
- De Ruiter, H.; Kollöffel, C. Activity of enzymes of arginine metabolism in the cotyledons of developing and germinating pea seeds. Plant Physiol. 1982, 70, 313–315. [Google Scholar] [CrossRef] [Green Version]
- Kollöffel, C.; Verkerk, B.C. Carbamoyl phosphate synthetase activity from the cotyledons of developing and germinating pea seeds. Plant Physiol. 1982, 69, 143–145. [Google Scholar] [CrossRef] [Green Version]
- Guan, M.; Møller, I.S.; Schjoerring, J.K. Two cytosolic glutamine synthetase isoforms play specific roles for seed germination and seed yield structure in Arabidopsis. J. Exp. Bot. 2015, 66, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Bertagnolli, B.L.; Wedding, R.T. Cystine content of legume seed proteins: Estimation by determination of cysteine with 2-vinylquinoline, and relation to protein content and activity of cysteine synthase. J. Nutr. 1977, 107, 2122–2127. [Google Scholar] [CrossRef]
- Cheung, G.P.; Rosenblum, I.Y.; Sallach, H.J. Comparative studies of enzymes related to serine metabolism in higher plants. Plant Physiol. 1968, 43, 1813–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Mawhinney, T.P.; Preuss, M.L.; Schroeder, A.C.; Chen, B.; Abraham, L.; Jez, J.M.; Chen, S. A redox-active isopropylmalate dehydrogenase functions in the biosynthesis of glucosinolates and leucine in Arabidopsis. Plant J. 2009, 60, 679–690. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Li, X.X.; Zhu, B.Q.; Wen, Y.Q.; Duan, C.Q.; Pan, Q.H. Molecular characterization and expression analysis on two isogenes encoding 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase in grapes. Mol. Biol. Rep. 2011, 38, 4739–4747. [Google Scholar] [CrossRef] [PubMed]
- Su, P.H.; Li, H.M. Arabidopsis stromal 70-kD heat shock proteins are essential for plant development and important for thermotolerance of germinating seeds. Plant Physiol. 2008, 146, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Ricardo, C.P.P.; Martins, I.; Francisco, R.; Sergeant, K.; Pinheiro, C.; Campos, A.; Renaut, J.; Fevereiro, P. Proteins associated with cork formation in Quercus suber L. stem tissues. J. Proteom. 2011, 74, 1266–1278. [Google Scholar] [CrossRef] [PubMed]
- Czolpinska, M.; Rurek, M. Plant glycine-rich proteins in stress response: An emerging, still prospective story. Front. Plant Sci. 2018, 9, 302. [Google Scholar] [CrossRef]
- Šunderlíková, V.; Salaj, J.; Kopecky, D.; Salaj, T.; Wilhem, E.; Matušíková, I. Dehydrin genes and their expression in recalcitrant oak (Quercus robur) embryos. Plant Cell Rep. 2009, 28, 1011. [Google Scholar] [CrossRef]
- Clark, G.B.; Morgan, R.O.; Fernandez, M.P.; Roux, S.J. Evolutionary adaptation of plant annexins has diversified their molecular structures, interactions and functional roles. New Phytol. 2012, 196, 695–712. [Google Scholar] [CrossRef]
- Anil Kumar, S.; Hima Kumari, P.; Shravan Kumar, G.; Mohanalatha, C.; Kavi Kishor, P.B. Osmotin: A plant sentinel and a possible agonist of mammalian adiponectin. Front. Plant Sci. 2015, 6, 163. [Google Scholar] [CrossRef] [Green Version]
- Sinha, M.; Singh, R.P.; Kushwaha, G.S.; Iqbal, N.; Singh, A.; Kaushik, S.; Kaur, P.; Sharma, S.; Singh, T.P. Current overview of allergens of plant pathogenesis related protein families. Sci. World J. 2014, 2014, 543195. [Google Scholar] [CrossRef] [Green Version]
- Catusse, J.; Job, C.; Job, D. Transcriptome- and proteome-wide analyses of seed germination. C. R.-Biol. 2008, 331, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Xin, X.; Fu, S.; An, M.; Wu, S.; Chen, X.; Zhang, J.; He, J.; Whelan, J.; Lu, X. Proteomic and carbonylation profile analysis at the critical node of seed ageing in Oryza sativa. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Ma, X.; Feng, Y.; Tian, Q.; Wang, Y.; Xu, N.; Tang, J.; Wang, G. Updating and interaction of polycomb repressive complex 2 components in maize (Zea mays). Planta 2019, 250, 573–588. [Google Scholar] [CrossRef]
- Sarkar Das, S.; Yadav, S.; Singh, A.; Gautam, V.; Sarkar, A.K.; Nandi, A.K.; Karmakar, P.; Majee, M.; Sanan-Mishra, N. Expression dynamics of miRNAs and their targets in seed germination conditions reveals miRNA-ta-siRNA crosstalk as regulator of seed germination. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Zhao, X.; Zhou, X.R.; Green, A.G.; Fan, Y.; Wang, L.; Singh, S.P.; Liu, Q. Comparative Transcriptomic Analysis of Developing Cotton Cotyledons and Embryo Axis. PLoS ONE 2013, 8, e71756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlereth, A.; Becker, C.; Horstmann, C.; Tiedemann, J.; Müntz, K. Comparison of globulin mobilization and cysteine proteinases in embryonic axes and cotyledons during germination and seedling growth of vetch (Vicia sativa L.). J. Exp. Bot. 2000, 51, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.M.; Wilson, A.T. Metabolism of the axis and cotyledons of phaseolus vulgaris seeds during early germination. Phytochemistry 1972, 11, 1931–1935. [Google Scholar] [CrossRef]
- Roberts, E.H. Predicting the storage life of seeds. Seed Sci. Technol. 1973, 1, 499–514. [Google Scholar]
- Obroucheva, N.; Sinkevich, I.; Lityagina, S. Physiological aspects of seed recalcitrance: A case study on the tree Aesculus hippocastanum. Tree Physiol. 2016, 36, 1127–1150. [Google Scholar] [CrossRef] [Green Version]
- Angelovici, R.; Lipka, A.E.; Deason, N.; Gonzalez-Jorge, S.; Lin, H.; Cepela, J.; Buell, R.; Gore, M.A.; DellaPenna, D. Genome-wide analysis of branched-chain amino acid levels in Arabidopsis Seeds. Plant Cell 2013, 25, 4827–4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrant, J.M.; Berjak, P.; Pammenter, N.W. Proteins in development and germination of a desiccation sensitive (recalcitrant) seed species. Plant Growth Regul. 1992, 11, 257–265. [Google Scholar] [CrossRef]
- Moothoo-Padayachie, A.; Macdonald, A.; Varghese, B.; Pammenter, N.W.; Govender, P. Uncovering the basis of viability loss in desiccation sensitive Trichilia dregeana seeds using differential quantitative protein expression profiling by iTRAQ. J. Plant Physiol. 2018, 221, 119–131. [Google Scholar] [CrossRef]
- Pukacka, S.; Malec, M.; Ratajczak, E. ROS production and antioxidative system activity in embryonic axes of Quercus robur seeds under different desiccation rate conditions. Acta Physiol. Plant. 2011, 33, 2219. [Google Scholar] [CrossRef]
- Vishal, B.; Kumar, P.P. Regulation of seed germination and abiotic stresses by gibberellins and abscisic acid. Front. Plant Sci. 2018, 9, 838. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.; Makarevich, G. Epigenetic mechanisms governing seed development in plants. EMBO Rep. 2006, 7, 1223–1227. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Plitta-Michalak, B.P.; Naskręt-Barciszewska, M.; Barciszewski, J.; Bujarska-Borkowska, B.; Chmielarz, P. Global 5-methylcytosine alterations in DNA during ageing of Quercus robur seeds. Ann. Bot. 2015, 116, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Plitta-Michalak, B.P.; Naskret-Barciszewska, M.Z.; Kotlarski, S.; Tomaszewski, D.; Tylkowski, T.; Barciszewski, J.; Chmielarz, P.; Michalak, M. Changes in genomic 5-methylcytosine level mirror the response of orthodox (Acer platanoides L.) and recalcitrant (Acer pseudoplatanus L.) seeds to severe desiccation. Tree Physiol. 2018, 38, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.F.; Chen, P.Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L.; et al. Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. USA 2017, 114, E9730–E9739. [Google Scholar] [CrossRef] [Green Version]
- Maldonado, A.M.; Echevarría-Zomeño, S.; Jean-Baptiste, S.; Hernández, M.; Jorrín-Novo, J.V. Evaluation of three different protocols of protein extraction for Arabidopsis thaliana leaf proteome analysis by two-dimensional electrophoresis. J. Proteom. 2008, 71, 461–472. [Google Scholar] [CrossRef]
- Wang, W.; Vignani, R.; Scali, M.; Cresti, M. A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis 2006, 27, 2782–2786. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Neuhoff, V.; Arold, N.; Taube, D.; Ehrhardt, W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 1988, 9, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, T.H.; Lauber, W.M.; Berggren, K.; Kemper, C.; Yue, S.; Patton, W.F. Fluorescence detection of proteins in sodium dodecyl sulfate-polyacrylamide gels using environmentally benign, nonfixative, saline solution. Electrophoresis 2000, 21, 497–508. [Google Scholar] [CrossRef]
- Chich, J.F.; David, O.; Villers, F.; Schaeffer, B.; Lutomski, D.; Huet, S. Statistics for proteomics: Experimental design and 2-DE differential analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 849, 261–272. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Valledor, L.; Romero-Rodríguez, M.C.; Jorrin-Novo, J.V. Standardization of data processing and statistical analysis in comparative plant proteomics experiment. Methods Mol. Biol. 2014, 1072, 51–60. [Google Scholar] [CrossRef]
- Sharov, A.A.; Dudekula, D.B.; Ko, M.S.H. A web-based tool for principal component and significance analysis of microarray data. Bioinformatics 2005, 21, 2548–2549. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998. [Google Scholar] [CrossRef] [Green Version]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stages | Length (mm) | Maximum Diameter (mm) | Weight (g) |
|---|---|---|---|
| M1 | 5.00 ± 0.28f | 3.07 ± 0.16g | 0.03 ± 0.0003e |
| M2 | 5.03 ± 0.24f | 3.55 ± 0.32g | 0.05 ± 0.0003de |
| M3 | 8.61 ± 0.36ef | 7.16 ± 0.34f | 0.19 ± 0.04de |
| M4 | 10.88 ± 0.67de | 8.95 ± 0.31e | 0.40 ± 0.06de |
| M5 | 14.28 ± 0.78cd | 10.65 ± 0.39d | 0.83 ± 0.068cd |
| M6 | 18.04 ± 1.04cd | 11.86 ± 0.42cd | 1.33 ± 0.11cd |
| M7 | 25.73 ± 1.56b | 13.42 ± 0.53bc | 2.72 ± 0.35b |
| M8 | 36.16 ± 1.48a | 15.77 ± 0.41a | 5.24 ± 0.17a |
| M9 | 33.12 ± 0.88a | 14.95 ± 1.20ab | 3.50 ± 0.30b |
| Matched Spots | M6 | M7 | M8 | M9 | G3 | G5 | NG | DP |
|---|---|---|---|---|---|---|---|---|
| Cotyledon | 504 ± 4.3a | 506.6 ± 1.5a | 489 ± 1ab | 456.6 ± 23b | 248.6 ± 46c | 230.6 ± 17c | 234.6 ± 23c | 558 |
| Embryo axis | 325.3 ± 0.53c | 382 ± 2bc | 422.6 ± 2.8b | 470 ± 1a | 385.3 ± 25b | 243 ± 53d | 388.3 ± 62b | 591 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sghaier-Hammami, B.; B.M. Hammami, S.; Baazaoui, N.; Gómez-Díaz, C.; Jorrín-Novo, J.V. Dissecting the Seed Maturation and Germination Processes in the Non-Orthodox Quercus ilex Species Based on Protein Signatures as Revealed by 2-DE Coupled to MALDI-TOF/TOF Proteomics Strategy. Int. J. Mol. Sci. 2020, 21, 4870. https://doi.org/10.3390/ijms21144870
Sghaier-Hammami B, B.M. Hammami S, Baazaoui N, Gómez-Díaz C, Jorrín-Novo JV. Dissecting the Seed Maturation and Germination Processes in the Non-Orthodox Quercus ilex Species Based on Protein Signatures as Revealed by 2-DE Coupled to MALDI-TOF/TOF Proteomics Strategy. International Journal of Molecular Sciences. 2020; 21(14):4870. https://doi.org/10.3390/ijms21144870
Chicago/Turabian StyleSghaier-Hammami, Besma, Sofiene B.M. Hammami, Narjes Baazaoui, Consuelo Gómez-Díaz, and Jesús V. Jorrín-Novo. 2020. "Dissecting the Seed Maturation and Germination Processes in the Non-Orthodox Quercus ilex Species Based on Protein Signatures as Revealed by 2-DE Coupled to MALDI-TOF/TOF Proteomics Strategy" International Journal of Molecular Sciences 21, no. 14: 4870. https://doi.org/10.3390/ijms21144870