The Role of Extracellular Proteases in Tumor Progression and the Development of Innovative Metal Ion Chelators That Inhibit Their Activity


Abstract
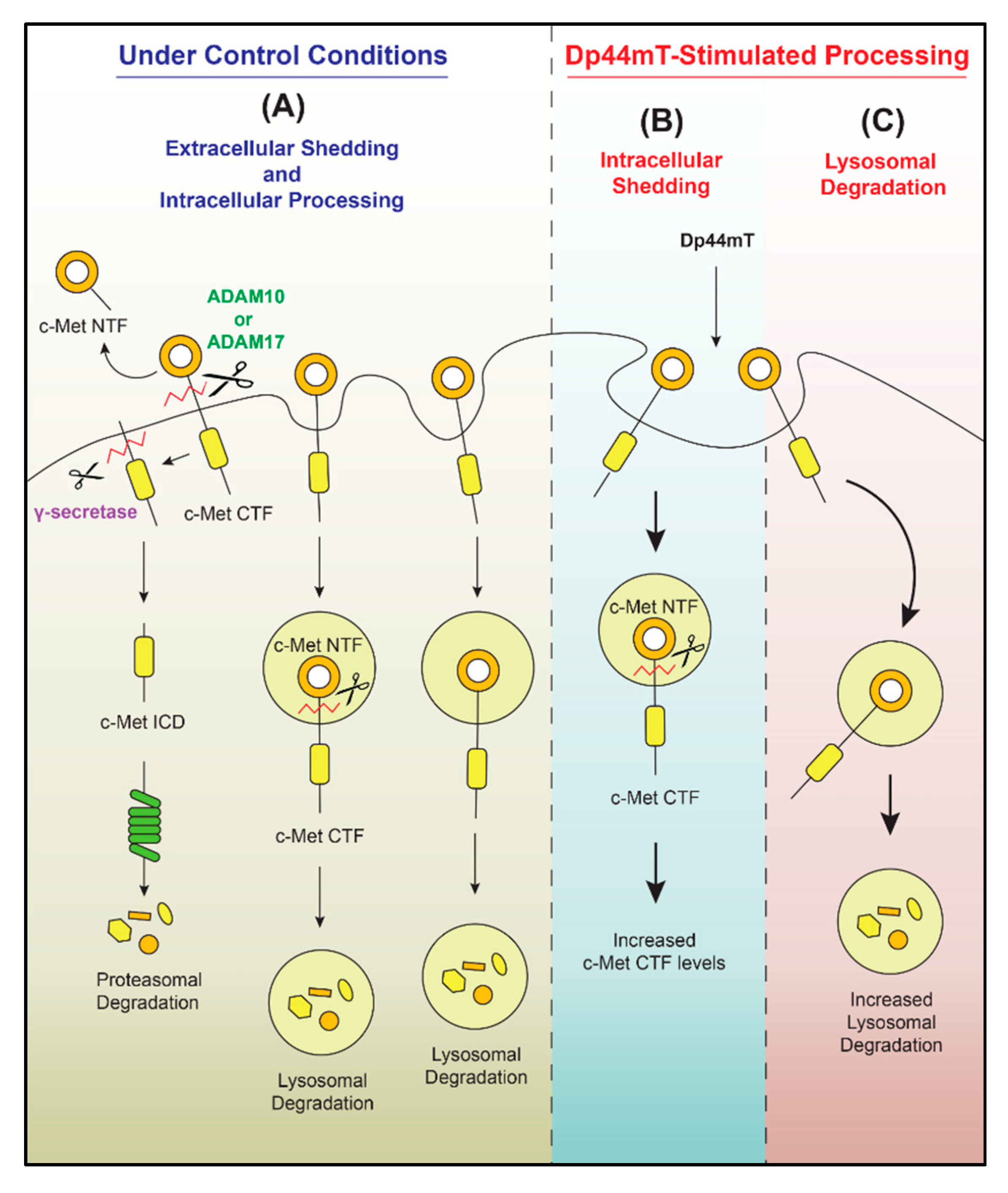
:1. Introduction
2. Functions of Extracellular Proteases in Cancer Progression
3. MMPs: Regulation and Function in Cancer
4. Tumor-Associated Trypsinogen (TAT) and Kallikrein-Related Peptidases (KLKs) in Cancer
5. Cross-Talk between Proteases and Kinases
6. The Proteases of Extracellular Vesicles (EVs) and Their Roles in Tumorigenesis
7. Therapeutics Targeting Proteases in Cancer
8. Thiosemicarbazones and Other Chelators Target Proteases by Indirect and Direct Mechanisms
8.1. Zinc(II) Chelators That Target MMPs
8.2. Thiosemicarbazones: Complex Inhibition of MMPs by Multiple Mechanisms
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lopez-Otin, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Matrisian, L.M. Emerging roles of proteases in tunour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Moali, C.; Hulmes, D.J. Extracellular and cell surface proteases in wound healing: New players are still emerging. Eur. J. Dermatol. 2009, 19, 552–564. [Google Scholar] [PubMed] [Green Version]
- Sanderson, R.D.; Bandari, S.K.; Vlodavsky, I. Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix Biol. 2019, 75, 160–169. [Google Scholar] [CrossRef]
- Lee, M.; Fridman, R.; Mobashery, S. Extracellular proteases as targets for treatment of cancer metastases. Chem. Soc. Rev. 2004, 33, 401–409. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Sullivan, R.J.; Lauffenburger, D.A. Molecular pathways: Receptor ectodomain shedding in treatment, resistance, and monitoring of cancer. Clin. Cancer Res. 2017, 23, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wong, J.; Lovejoy, D.B.; Kalinowski, D.S.; Richardson, D.R. Chelators at the cancer coalface: Desferrioxamine to Triapine and beyond. Clin. Cancer Res. 2006, 12, 6876–6883. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Kalinowski, D.S.; Kovacevic, Z.; Siafakas, A.R.; Jansson, P.J.; Stefani, C.; Lovejoy, D.B.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Thiosemicarbazones from the old to new: Iron chelators that are more than just ribonucleotide reductase inhibitors. J. Med. Chem. 2009, 52, 5271–5294. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Novel chelators for cancer treatment: Where are we now? Antioxid. Redox Signal 2013, 18, 973–1006. [Google Scholar] [CrossRef]
- Nyberg, P.; Ylipalosaari, M.; Sorsa, T.; Salo, T. Trypsins and their role in carcinoma growth. Exp. Cell Res. 2006, 312, 1219–1228. [Google Scholar] [CrossRef]
- Vilen, S.T.; Nyberg, P.; Hukkanen, M.; Sutinen, M.; Ylipalosaari, M.; Bjartell, A.; Paju, A.; Haaparanta, V.; Stenman, U.H.; Sorsa, T.; et al. Intracellular co-localization of trypsin-2 and matrix metalloprotease-9: Possible proteolytic cascade of trypsin-2, MMP-9 and enterokinase in carcinoma. Exp. Cell Res. 2008, 314, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, E.; Itkonen, O.; Halila, H.; Stenman, U.H. Cyst fluid of ovarian cancer patients contains high concentrations of trypsinogen-2. Cancer Res. 1990, 50, 2375–2378. [Google Scholar] [PubMed]
- Kopitz, C.; Gerg, M.; Bandapalli, O.R.; Ister, D.; Pennington, C.J.; Hauser, S.; Flechsig, C.; Krell, H.W.; Antolovic, D.; Brew, K.; et al. Tissue inhibitor of metalloproteinases-1 promotes liver metastasis by induction of hepatocyte growth factor signaling. Cancer Res. 2007, 67, 8615–8623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orme, J.J.; Du, Y.; Vanarsa, K.; Mayeux, J.; Li, L.; Mutwally, A.; Arriens, C.; Min, S.; Hutcheson, J.; Davis, L.S.; et al. Heightened cleavage of Axl receptor tyrosine kinase by ADAM metalloproteases may contribute to disease pathogenesis in SLE. Clin. Immunol. 2016, 169, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Khokha, R.; Hill, R.P. Molecular mechanisms of tumor invasion and metastasis: An integrated view. Curr. Mol. Med. 2003, 3, 659–671. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Liotta, L.A.; Tryggvason, K.; Garbisa, S.; Hart, I.; Foltz, C.M.; Shafie, S. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature 1980, 284, 67–68. [Google Scholar] [CrossRef]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of matrix metalloproteinases in angiogenesis and cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [PubMed] [Green Version]
- Littlepage, L.E.; Sternlicht, M.D.; Rougier, N.; Phillips, J.; Gallo, E.; Yu, Y.; Williams, K.; Brenot, A.; Gordon, J.I.; Werb, Z. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 2010, 70, 2224–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Woessner, J.F., Jr. Matrix metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, S.D. Matrix metalloproteinase degradation of extracellular matrix: Biological consequences. Curr. Opin. Cell Biol. 1998, 10, 602–608. [Google Scholar] [CrossRef]
- Nuti, E.; Tuccinardi, T.; Rossello, A. Matrix metalloproteinase inhibitors: New challenges in the era of post broad-spectrum inhibitors. Curr. Pharm. Des. 2007, 13, 2087–2100. [Google Scholar] [CrossRef]
- Whittaker, M.; Floyd, C.D.; Brown, P.; Gearing, A.J. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999, 99, 2735–2776. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.E.; Alonso, D.F.; Yoshiji, H.; Thorgeirsson, U.P. Tissue inhibitors of metalloproteinases: Structure, regulation and biological functions. Eur. J. Cell Biol. 1997, 74, 111–122. [Google Scholar]
- Maskos, K. Crystal structures of MMPs in complex with physiological and pharmacological inhibitors. Biochimie 2005, 87, 249–263. [Google Scholar] [CrossRef]
- Jablonska-Trypuc, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (extracellular matrix) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Loffek, S.; Schilling, O.; Franzke, C.W. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobin, E.; Bagwell, K.; Wagner, J.; Mysona, D.; Sandirasegarane, S.; Smith, N.; Bai, S.; Sharma, A.; Schleifer, R.; She, J.X. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer 2019, 19, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilles, C.; Polette, M.; Piette, J.; Birembaut, P.; Foidart, J.M. Epithelial-to-mesenchymal transition in HPV-33-transfected cervical keratinocytes is associated with increased invasiveness and expression of gelatinase A. Int. J. Cancer 1994, 59, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Gilles, C.; Polette, M.; Piette, J.; Munaut, C.; Thompson, E.W.; Birembaut, P.; Foidart, J.M. High level of MT-MMP expression is associated with invasiveness of cervical cancer cells. Int. J. Cancer 1996, 65, 209–213. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, H.; Itoh, F.; Hinoda, Y.; Okada, Y.; Imai, K. Contribution of matrilysin (MMP-7) to the metastatic pathway of human colorectal cancers. Gut 1999, 45, 252–258. [Google Scholar] [CrossRef]
- Itoh, T.; Tanioka, M.; Matsuda, H.; Nishimoto, H.; Yoshioka, T.; Suzuki, R.; Uehira, M. Experimental metastasis is suppressed in MMP-9-deficient mice. Clin. Exp. Metastasis 1999, 17, 177–181. [Google Scholar] [CrossRef]
- Monig, S.P.; Baldus, S.E.; Hennecken, J.K.; Spiecker, D.B.; Grass, G.; Schneider, P.M.; Thiele, J.; Dienes, H.P.; Holscher, A.H. Expression of MMP-2 is associated with progression and lymph node metastasis of gastric carcinoma. Histopathology 2001, 39, 597–602. [Google Scholar] [CrossRef]
- Moses, M.A.; Wiederschain, D.; Loughlin, K.R.; Zurakowski, D.; Lamb, C.C.; Freeman, M.R. Increased incidence of matrix metalloproteinases in urine of cancer patients. Cancer Res. 1998, 58, 1395–1399. [Google Scholar] [PubMed]
- Zucker, S.; Hymowitz, M.; Conner, C.; Zarrabi, H.M.; Hurewitz, A.N.; Matrisian, L.; Boyd, D.; Nicolson, G.; Montana, S. Measurement of matrix metalloproteinases and tissue inhibitors of metalloproteinases in blood and tissues. Clinical and experimental applications. Ann. N. Y. Acad. Sci. 1999, 878, 212–227. [Google Scholar] [CrossRef]
- Fang, J.; Shing, Y.; Wiederschain, D.; Yan, L.; Butterfield, C.; Jackson, G.; Harper, J.; Tamvakopoulos, G.; Moses, M.A. Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proc. Natl. Acad. Sci. USA 2000, 97, 3884–3889. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [PubMed]
- Lokeshwar, B.L.; Selzer, M.G.; Block, N.L.; Gunja-Smith, Z. Secretion of matrix metalloproteinases and their inhibitors (tissue inhibitor of metalloproteinases) by human prostate in explant cultures: Reduced tissue inhibitor of metalloproteinase secretion by malignant tissues. Cancer Res. 1993, 53, 4493–4498. [Google Scholar]
- Patterson, B.C.; Sang, Q.A. Angiostatin-converting enzyme activities of human matrilysin (MMP-7) and gelatinase B/type IV collagenase (MMP-9). J. Biol. Chem. 1997, 272, 28823–28825. [Google Scholar] [PubMed] [Green Version]
- Hamano, Y.; Zeisberg, M.; Sugimoto, H.; Lively, J.C.; Maeshima, Y.; Yang, C.; Hynes, R.O.; Werb, Z.; Sudhakar, A.; Kalluri, R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 2003, 3, 589–601. [Google Scholar] [PubMed] [Green Version]
- Young, D.; Das, N.; Anowai, A.; Dufour, A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. Int. J. Mol. Sci. 2019, 20, 3847. [Google Scholar]
- Jobin, P.G.; Butler, G.S.; Overall, C.M. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 Pt 11, 2043–2055. [Google Scholar]
- Basset, P.; Wolf, C.; Chambon, P. Expression of the stromelysin-3 gene in fibroblastic cells of invasive carcinomas of the breast and other human tissues: A review. Breast Cancer Res. Treat. 1993, 24, 185–193. [Google Scholar]
- Okada, A.; Bellocq, J.P.; Rouyer, N.; Chenard, M.P.; Rio, M.C.; Chambon, P.; Basset, P. Membrane-type matrix metalloproteinase (MT-MMP) gene is expressed in stromal cells of human colon, breast, and head and neck carcinomas. Proc. Natl. Acad. Sci. USA 1995, 92, 2730–2734. [Google Scholar]
- Majmudar, G.; Nelson, B.R.; Jensen, T.C.; Voorhees, J.J.; Johnson, T.M. Increased expression of stromelysin-3 in basal cell carcinomas. Mol. Carcinog. 1994, 9, 17–23. [Google Scholar]
- Polette, M.; Gilles, C.; Marchand, V.; Lorenzato, M.; Toole, B.; Tournier, J.M.; Zucker, S.; Birembaut, P. Tumor collagenase stimulatory factor (TCSF) expression and localization in human lung and breast cancers. J. Histochem. Cytochem. 1997, 45, 703–709. [Google Scholar]
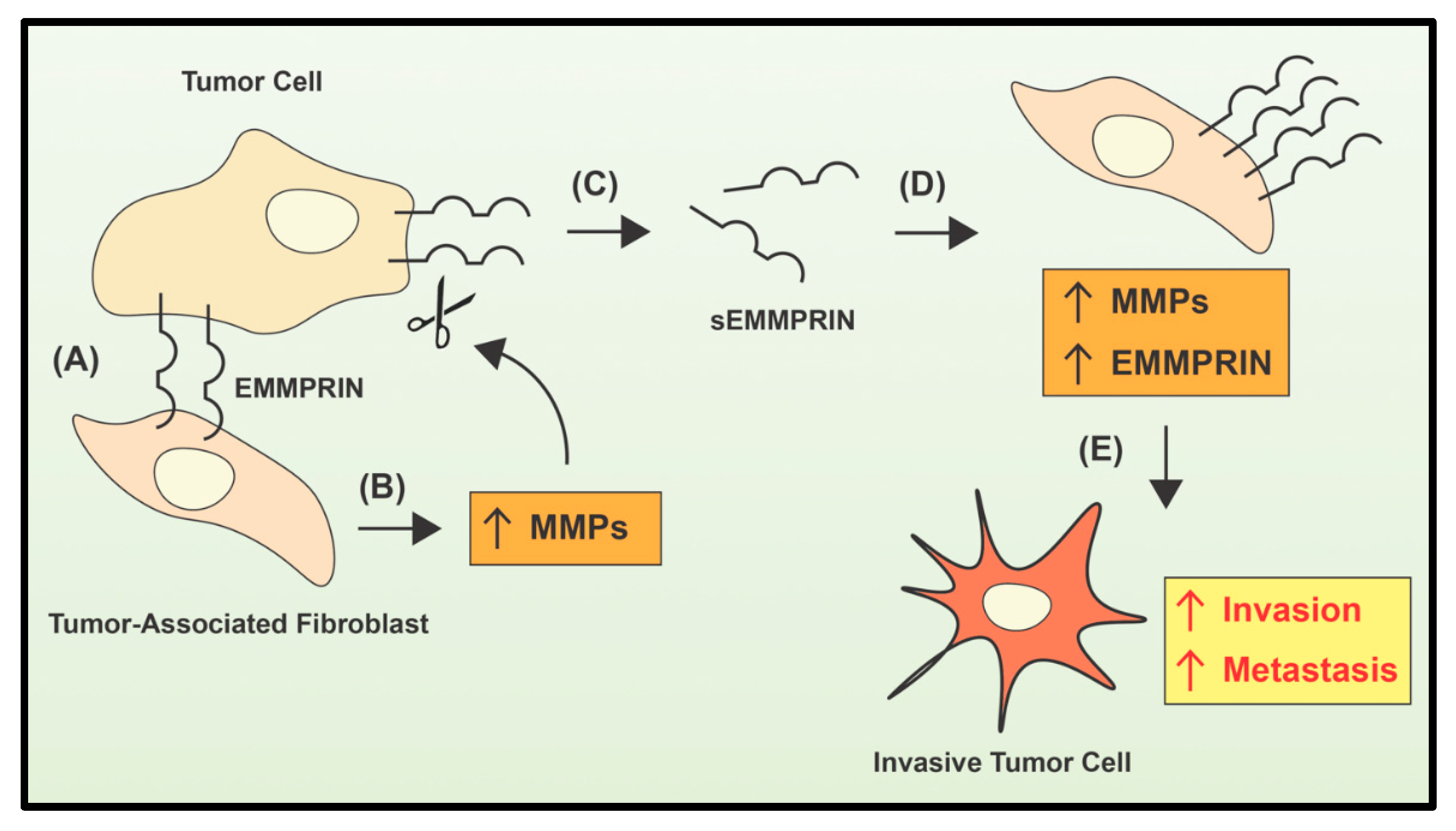
- Guo, H.; Zucker, S.; Gordon, M.K.; Toole, B.P.; Biswas, C. Stimulation of matrix metalloproteinase production by recombinant extracellular matrix metalloproteinase inducer from transfected Chinese hamster ovary cells. J. Biol. Chem. 1997, 272, 24–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.; Martinez, T.; Jablons, D.; Cameron, R.; Guo, H.; Toole, B.; Li, J.D.; Basbaum, C. Tumor-derived EMMPRIN (extracellular matrix metalloproteinase inducer) stimulates collagenase transcription through MAPK p38. FEBS Lett. 1998, 441, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Biswas, C.; Zhang, Y.; DeCastro, R.; Guo, H.; Nakamura, T.; Kataoka, H.; Nabeshima, K. The human tumor cell-derived collagenase stimulatory factor (renamed EMMPRIN) is a member of the immunoglobulin superfamily. Cancer Res. 1995, 55, 434–439. [Google Scholar] [PubMed]
- Ko, K.; Yazumi, S.; Yoshikawa, K.; Konda, Y.; Nakajima, M.; Chiba, T.; Takahashi, R. Activation of fibroblast-derived matrix metalloproteinase-2 by colon-cancer cells in non-contact Co-cultures. Int. J. Cancer 2000, 87, 165–171. [Google Scholar] [CrossRef]
- Bordador, L.C.; Li, X.; Toole, B.; Chen, B.; Regezi, J.; Zardi, L.; Hu, Y.; Ramos, D.M. Expression of emmprin by oral squamous cell carcinoma. Int. J. Cancer 2000, 85, 347–352. [Google Scholar] [CrossRef]
- Tang, Y.; Kesavan, P.; Nakada, M.T.; Yan, L. Tumor-stroma interaction: Positive feedback regulation of extracellular matrix metalloproteinase inducer (EMMPRIN) expression and matrix metalloproteinase-dependent generation of soluble EMMPRIN. Mol. Cancer Res. 2004, 2, 73–80. [Google Scholar]
- Stenman, U.H.; Koivunen, E.; Vuento, M. Characterization of a tumor-associated serine protease. Biol. Chem. Hoppe Seyler 1988, 369, 9–14. [Google Scholar]
- Koivunen, E.; Saksela, O.; Itkonen, O.; Osman, S.; Huhtala, M.L.; Stenman, U.H. Human colon carcinoma, fibrosarcoma and leukemia cell lines produce tumor-associated trypsinogen. Int. J. Cancer 1991, 47, 592–596. [Google Scholar] [CrossRef]
- Ohta, T.; Terada, T.; Nagakawa, T.; Tajima, H.; Itoh, H.; Fonseca, L.; Miyazaki, I. Pancreatic trypsinogen and cathepsin B in human pancreatic carcinomas and associated metastatic lesions. Br. J. Cancer 1994, 69, 152–156. [Google Scholar] [CrossRef]
- Terada, T.; Ohta, T.; Minato, H.; Nakanuma, Y. Expression of pancreatic trypsinogen/trypsin and cathepsin B in human cholangiocarcinomas and hepatocellular carcinomas. Hum. Pathol. 1995, 26, 746–752. [Google Scholar]
- Kawano, N.; Osawa, H.; Ito, T.; Nagashima, Y.; Hirahara, F.; Inayama, Y.; Nakatani, Y.; Kimura, S.; Kitajima, H.; Koshikawa, N.; et al. Expression of gelatinase A, tissue inhibitor of metalloproteinases-2, matrilysin, and trypsin(ogen) in lung neoplasms: An immunohistochemical study. Hum. Pathol. 1997, 28, 613–622. [Google Scholar] [CrossRef]
- Oyama, K.; Ohta, T.; Nishimura, G.I.; Elnemr, A.; Yasui, T.; Fujimura, T.; Fushida, S.; Kitagawa, H.; Kayahara, M.; Terada, T.; et al. Trypsinogen expression in colorectal cancers. Int. J. Mol. Med. 2000, 6, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Sorsa, T.; Salo, T.; Koivunen, E.; Tyynela, J.; Konttinen, Y.T.; Bergmann, U.; Tuuttila, A.; Niemi, E.; Teronen, O.; Heikkila, P.; et al. Activation of type IV procollagenases by human tumor-associated trypsin-2. J. Biol. Chem. 1997, 272, 21067–21074. [Google Scholar] [CrossRef] [Green Version]
- Miyata, S.; Koshikawa, N.; Yasumitsu, H.; Miyazaki, K. Trypsin stimulates integrin alpha(5)beta(1)-dependent adhesion to fibronectin and proliferation of human gastric carcinoma cells through activation of proteinase-activated receptor-2. J. Biol. Chem. 2000, 275, 4592–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjartell, A.; Paju, A.; Zhang, W.M.; Gadaleanu, V.; Hansson, J.; Landberg, G.; Stenman, U.H. Expression of tumor-associated trypsinogens (TAT-1 and TAT-2) in prostate cancer. Prostate 2005, 64, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, M.; Sorsa, T.; Stenman, M.; Nyberg, P.; Lindy, O.; Vesterinen, J.; Paju, A.; Konttinen, Y.T.; Stenman, U.H.; Salo, T. Tumor-associated trypsinogen-2 (trypsinogen-2) activates procollagenases (MMP-1, -8, -13) and stromelysin-1 (MMP-3) and degrades type I collagen. Biochemistry 2003, 42, 5414–5420. [Google Scholar] [CrossRef]
- Nyberg, P.; Moilanen, M.; Paju, A.; Sarin, A.; Stenman, U.H.; Sorsa, T.; Salo, T. MMP-9 activation by tumor trypsin-2 enhances in vivo invasion of human tongue carcinoma cells. J. Dent. Res. 2002, 81, 831–835. [Google Scholar] [CrossRef]
- Stefanini, A.C.; da Cunha, B.R.; Henrique, T.; Tajara, E.H. Involvement of kallikrein-related peptidases in normal and pathologic processes. Dis. Markers 2015, 2015, 946572. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Loessner, D.; Irving-Rodgers, H.; Obermair, A.; Nicklin, J.L.; Clements, J.A. Metastasis of ovarian cancer is mediated by kallikrein related peptidases. Clin. Exp. Metastasis 2014, 31, 135–147. [Google Scholar] [CrossRef]
- Kryza, T.; Bock, N.; Lovell, S.; Rockstroh, A.; Lehman, M.L.; Lesner, A.; Panchadsaram, J.; Silva, L.M.; Srinivasan, S.; Snell, C.E.; et al. The molecular function of kallikrein-related peptidase 14 demonstrates a key modulatory role in advanced prostate cancer. Mol. Oncol. 2020, 14, 105–128. [Google Scholar] [CrossRef]
- Iakovlev, V.; Siegel, E.R.; Tsao, M.S.; Haun, R.S. Expression of kallikrein-related peptidase 7 predicts poor prognosis in patients with unresectable pancreatic ductal adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1135–1142. [Google Scholar]
- Du, J.P.; Li, L.; Zheng, J.; Zhang, D.; Liu, W.; Zheng, W.H.; Li, X.S.; Yao, R.C.; Wang, F.; Liu, S.; et al. Kallikrein-related peptidase 7 is a potential target for the treatment of pancreatic cancer. Oncotarget 2018, 9, 12894–12906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamandis, E.P.; Scorilas, A.; Fracchioli, S.; Van Gramberen, M.; De Bruijn, H.; Henrik, A.; Soosaipillai, A.; Grass, L.; Yousef, G.M.; Stenman, U.H.; et al. Human kallikrein 6 (hK6): A new potential serum biomarker for diagnosis and prognosis of ovarian carcinoma. J. Clin. Oncol. 2003, 21, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.Y.; Katsaros, D.; Scorilas, A.; Fracchioli, S.; Bellino, R.; van Gramberen, M.; de Bruijn, H.; Henrik, A.; Stenman, U.H.; Massobrio, M.; et al. The serum concentration of human kallikrein 10 represents a novel biomarker for ovarian cancer diagnosis and prognosis. Cancer Res. 2003, 63, 807–811. [Google Scholar] [PubMed]
- Lopez-Otin, C.; Hunter, T. The regulatory crosstalk between kinases and proteases in cancer. Nat. Rev. Cancer 2010, 10, 278–292. [Google Scholar]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 929–941. [Google Scholar] [PubMed]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Ullrich, A. EGFR signal transactivation in cancer cells. Biochem. Soc. Trans. 2003, 31 Pt 6, 1203–1208. [Google Scholar] [CrossRef] [Green Version]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar]
- Borrell-Pages, M.; Rojo, F.; Albanell, J.; Baselga, J.; Arribas, J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 2003, 22, 1114–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiretti, F.; Deprez-Beauclair, P.; Bonardo, B.; Aubert, H.; Juhan-Vague, I.; Nalbone, G. Identification of SAP97 as an intracellular binding partner of TACE. J. Cell Sci. 2003, 116 Pt 10, 1949–1957. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.K.; Schlondorff, J.; Blobel, C.P. Evidence for an interaction of the metalloprotease-disintegrin tumour necrosis factor alpha convertase (TACE) with mitotic arrest deficient 2 (MAD2), and of the metalloprotease-disintegrin MDC9 with a novel MAD2-related protein, MAD2beta. Biochem. J. 1999, 343 Pt 3, 673–680. [Google Scholar]
- Zheng, Y.; Schlondorff, J.; Blobel, C.P. Evidence for regulation of the tumor necrosis factor alpha-convertase (TACE) by protein-tyrosine phosphatase PTPH1. J. Biol. Chem. 2002, 277, 42463–42470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudoin, A.R.; Grondin, G. Shedding of vesicular material from the cell surface of eukaryotic cells: Different cellular phenomena. Biochim. Biophys. Acta 1991, 1071, 203–219. [Google Scholar] [CrossRef]
- Denzer, K.; Kleijmeer, M.J.; Heijnen, H.F.; Stoorvogel, W.; Geuze, H.J. Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000, 113 Pt 9, 3365–3374. [Google Scholar]
- Shimoda, M.; Khokha, R. Metalloproteinases in extracellular vesicles. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 Pt 11, 1989–2000. [Google Scholar] [CrossRef]
- Nawaz, M.; Shah, N.; Zanetti, B.R.; Maugeri, M.; Silvestre, R.N.; Fatima, F.; Neder, L.; Valadi, H. Extracellular vesicles and matrix remodeling enzymes: The emerging roles in extracellular matrix remodeling, progression of diseases and tissue repair. Cells 2018, 7, 167. [Google Scholar]
- Remacle, A.; Murphy, G.; Roghi, C. Membrane type I-matrix metalloproteinase (MT1-MMP) is internalised by two different pathways and is recycled to the cell surface. J. Cell Sci. 2003, 116 Pt 19, 3905–3916. [Google Scholar] [CrossRef] [Green Version]
- Hakulinen, J.; Sankkila, L.; Sugiyama, N.; Lehti, K.; Keski-Oja, J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell Biochem. 2008, 105, 1211–1218. [Google Scholar]
- Han, K.Y.; Dugas-Ford, J.; Seiki, M.; Chang, J.H.; Azar, D.T. Evidence for the involvement of MMP14 in MMP2 processing and recruitment in exosomes of corneal fibroblasts. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5323–5329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2005, 113, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Redzic, J.S.; Kendrick, A.A.; Bahmed, K.; Dahl, K.D.; Pearson, C.G.; Robinson, W.A.; Robinson, S.E.; Graner, M.W.; Eisenmesser, E.Z. Extracellular vesicles secreted from cancer cell lines stimulate secretion of MMP-9, IL-6, TGF-beta1 and EMMPRIN. PLoS ONE 2013, 8, e71225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, N.; Ahonen, M.; Kahari, V.M. Matrix metalloproteinases in tumor invasion. Cell Mol. Life Sci. 2000, 57, 5–15. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Stetler-Stevenson, W.G. Matrix metalloproteinases and metastasis. Cancer Chemother. Pharmacol. 1999, 43, S42–S51. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.D.; Han, Z.D.; He, H.C.; Bi, X.C.; Dai, Q.S.; Zhu, G.; Ye, Y.K.; Liang, Y.X.; Qin, W.J.; Zhang, Z.; et al. CD147, MMP-1, MMP-2 and MMP-9 protein expression as significant prognostic factors in human prostate cancer. Oncology 2008, 75, 230–236. [Google Scholar] [CrossRef]
- Nikkola, J.; Vihinen, P.; Vuoristo, M.S.; Kellokumpu-Lehtinen, P.; Kahari, V.M.; Pyrhonen, S. High serum levels of matrix metalloproteinase-9 and matrix metalloproteinase-1 are associated with rapid progression in patients with metastatic melanoma. Clin. Cancer Res. 2005, 11, 5158–5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, A.; Adams, S.; Mignatti, P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Mullooly, M.; McGowan, P.M.; Crown, J.; Duffy, M.J. The ADAMs family of proteases as targets for the treatment of cancer. Cancer Biol. Ther. 2016, 17, 870–880. [Google Scholar]
- Weber, S.; Saftig, P. Ectodomain shedding and ADAMs in development. Development 2012, 139, 3693–3709. [Google Scholar] [CrossRef] [Green Version]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 research: A promising target for cancer and inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witters, L.; Scherle, P.; Friedman, S.; Fridman, J.; Caulder, E.; Newton, R.; Lipton, A. Synergistic inhibition with a dual epidermal growth factor receptor/HER-2/neu tyrosine kinase inhibitor and a disintegrin and metalloprotease inhibitor. Cancer Res. 2008, 68, 7083–7089. [Google Scholar]
- Tape, C.J.; Willems, S.H.; Dombernowsky, S.L.; Stanley, P.L.; Fogarasi, M.; Ouwehand, W.; McCafferty, J.; Murphy, G. Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. USA 2011, 108, 5578–5583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, R.C.; Bradley, E.C.; Levy, R.S.; Doval, D.; Bondarde, S.; Sahoo, T.P.; Lokanatha, D.; Julka, P.K.; Nagarkar, R.; Friedman, S.M. Clinical benefit of INCB7839, a potent and selective ADAM inhibitor, in combination with trastuzumab in patients with metastatic HER2+ breast cancer. J. Clin. Oncol. 2010, 28, 3025. [Google Scholar]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtowicz-Praga, S.M.; Dickson, R.B.; Hawkins, M.J. Matrix metalloproteinase inhibitors. Investig. New Drugs 1997, 15, 61–75. [Google Scholar]
- Iwata, H.; Yamamoto, M.; Nemori, R.; Mizutani, M.; Iwase, T.; Miura, S.; Obata, Y.; Hara, Y.; Omoto, Y.; Toyama, T.; et al. Localization of gelatinolytic activity can be detected in breast cancer tissues by film in situ zymography. Breast Cancer 2001, 8, 111–115. [Google Scholar]
- Murnane, M.J.; Cai, J.; Shuja, S.; McAneny, D.; Klepeis, V.; Willett, J.B. Active MMP-2 effectively identifies the presence of colorectal cancer. Int. J. Cancer 2009, 125, 2893–2902. [Google Scholar] [CrossRef] [Green Version]
- Oh, L.Y.; Larsen, P.H.; Krekoski, C.A.; Edwards, D.R.; Donovan, F.; Werb, Z.; Yong, V.W. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J. Neurosci. 1999, 19, 8464–8475. [Google Scholar]
- Lam, C.; Jamerson, M.; Cabral, G.; Carlesso, A.M.; Marciano-Cabral, F. Expression of matrix metalloproteinases in Naegleria fowleri and their role in invasion of the central nervous system. Microbiology 2017, 163, 1436–1444. [Google Scholar]
- Hu, J.; Zhang, X.; Nothnick, W.B.; Spencer, T.E. Matrix metalloproteinases and their tissue inhibitors in the developing neonatal mouse uterus. Biol. Reprod. 2004, 71, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannikov, G.A.; Karelina, T.V.; Collier, I.E.; Marmer, B.L.; Goldberg, G.I. Substrate binding of gelatinase B induces its enzymatic activity in the presence of intact propeptide. J. Biol. Chem. 2002, 277, 16022–16027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
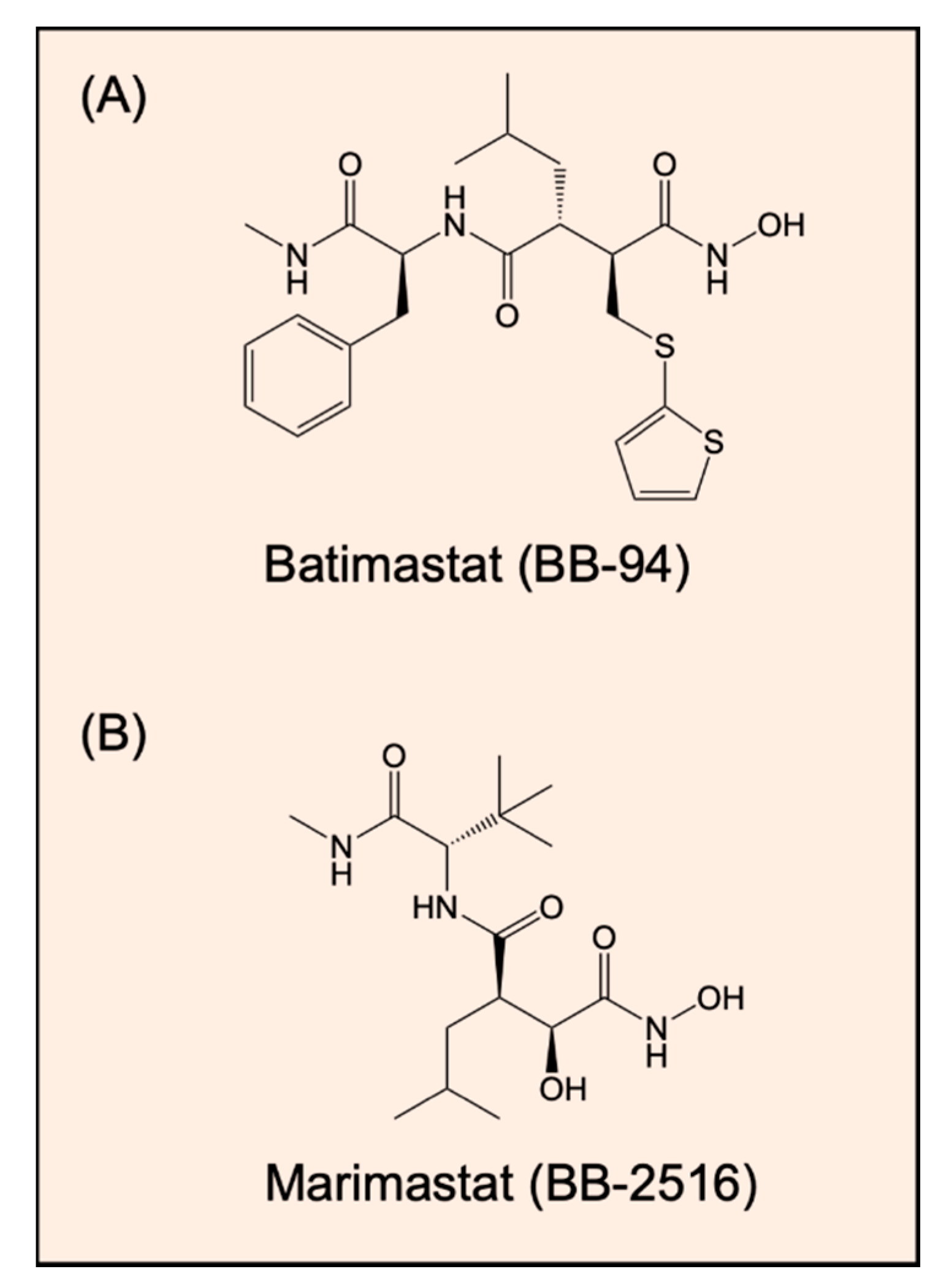
- Watson, S.A.; Morris, T.M.; Robinson, G.; Crimmin, M.J.; Brown, P.D.; Hardcastle, J.D. Inhibition of organ invasion by the matrix metalloproteinase inhibitor batimastat (BB-94) in two human colon carcinoma metastasis models. Cancer Res. 1995, 55, 3629–3633. [Google Scholar]
- Wojtowicz-Praga, S.; Low, J.; Marshall, J.; Ness, E.; Dickson, R.; Barter, J.; Sale, M.; McCann, P.; Moore, J.; Cole, A.; et al. Phase I trial of a novel matrix metalloproteinase inhibitor batimastat (BB-94) in patients with advanced cancer. Investig. New Drugs 1996, 14, 193–202. [Google Scholar] [CrossRef]
- Sparano, J.A.; Bernardo, P.; Stephenson, P.; Gradishar, W.J.; Ingle, J.N.; Zucker, S.; Davidson, N.E. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J. Clin. Oncol. 2004, 22, 4683–4690. [Google Scholar]
- Macaulay, V.M.; O’Byrne, K.J.; Saunders, M.P.; Braybrooke, J.P.; Long, L.; Gleeson, F.; Mason, C.S.; Harris, A.L.; Brown, P.; Talbot, D.C. Phase I study of intrapleural batimastat (BB-94), a matrix metalloproteinase inhibitor, in the treatment of malignant pleural effusions. Clin. Cancer Res. 1999, 5, 513–520. [Google Scholar]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [Green Version]
- Kohn, K.W. Mediation of divalent metal ions in the binding of tetracycline to macromolecules. Nature 1961, 191, 1156–1158. [Google Scholar] [CrossRef]
- Palm, G.J.; Lederer, T.; Orth, P.; Saenger, W.; Takahashi, M.; Hillen, W.; Hinrichs, W. Specific binding of divalent metal ions to tetracycline and to the Tet repressor/tetracycline complex. J. Biol. Inorg. Chem. 2008, 13, 1097–1110. [Google Scholar] [PubMed]
- Burns, F.R.; Stack, M.S.; Gray, R.D.; Paterson, C.A. Inhibition of purified collagenase from alkali-burned rabbit corneas. Investig. Ophthalmol. Vis. Sci. 1989, 30, 1569–1575. [Google Scholar]
- Xu, X.; Sutak, R.; Richardson, D.R. Iron chelation by clinically relevant anthracyclines: Alteration in expression of iron-regulated genes and atypical changes in intracellular iron distribution and trafficking. Mol. Pharmacol. 2008, 73, 833–844. [Google Scholar]
- Karakiulakis, G.; Missirlis, E.; Maragoudakis, M.E. Basement membrane collagen-degrading activity from a malignant tumor is inhibited by anthracycline antibiotics. Biochim. Biophys. Acta 1990, 1035, 218–222. [Google Scholar] [PubMed]
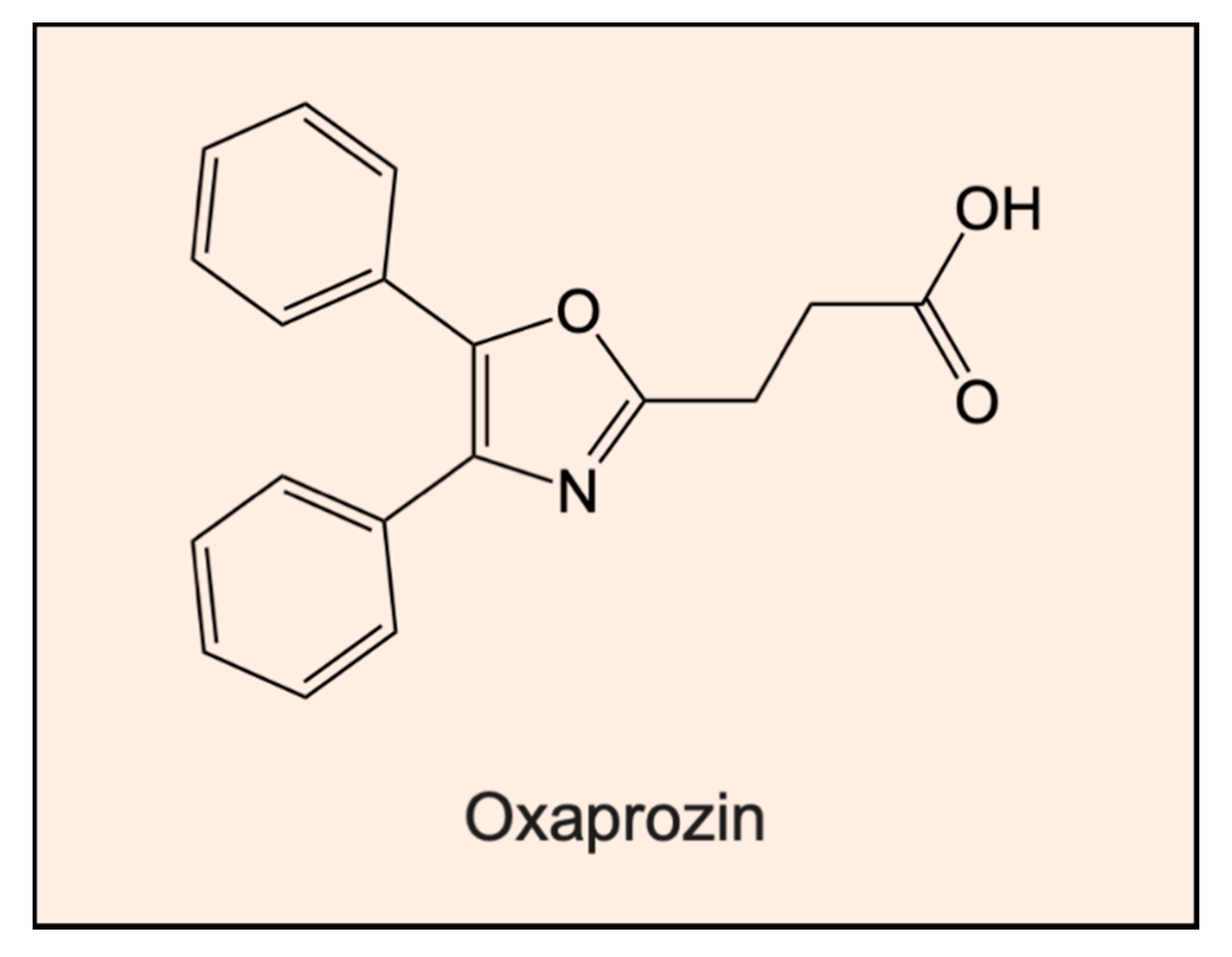
- Ianni, A.; Celenza, G.; Franceschini, N. Oxaprozin: A new hope in the modulation of matrix metalloproteinase 9 activity. Chem. Biol. Drug Des. 2019, 93, 811–817. [Google Scholar]
- Rao, B.G. Recent developments in the design of specific matrix metalloproteinase inhibitors aided by structural and computational studies. Curr. Pharm. Des. 2005, 11, 295–322. [Google Scholar]
- Georgiadis, D.; Yiotakis, A. Specific targeting of metzincin family members with small-molecule inhibitors: Progress toward a multifarious challenge. Bioorg. Med. Chem. 2008, 16, 8781–8794. [Google Scholar]
- Overall, C.M.; Kleifeld, O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 2006, 94, 941–946. [Google Scholar]
- Fingleton, B. MMPs as therapeutic targets—Still a viable option? Semin. Cell Dev. Biol. 2008, 19, 61–68. [Google Scholar]
- Shah, M.A.; Starodub, A.; Sharma, S.; Berlin, J.; Patel, M.; Wainberg, Z.A.; Chaves, J.; Gordon, M.; Windsor, K.; Brachmann, C.B.; et al. Andecaliximab/GS-5745 Alone and Combined with mFOLFOX6 in Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: Results from a Phase I Study. Clin. Cancer Res. 2018, 24, 3829–3837. [Google Scholar]
- Shah, M.A.; Yanez Ruiz, E.P.; Bodoky, G.; Starodub, A.; Cunningham, D.; Yip, D.; Wainberg, Z.A.; Bendell, J.C.; Thai, D.; Bhargava, P.; et al. A phase III, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of andecaliximab combined with mFOLFOX6 as first-line treatment in patients with advanced gastric or gastroesophageal junction adenocarcinoma (GAMMA-1). J. Clin. Oncol. 2019, 37, 4. [Google Scholar] [CrossRef]
- Rossello, A.; Nuti, E.; Carelli, P.; Orlandini, E.; Macchia, M.; Nencetti, S.; Zandomeneghi, M.; Balzano, F.; Uccello Barretta, G.; Albini, A.; et al. N-i-Propoxy-N-biphenylsulfonylaminobutylhydroxamic acids as potent and selective inhibitors of MMP-2 and MT1-MMP. Bioorg. Med. Chem. Lett. 2005, 15, 1321–1326. [Google Scholar] [PubMed]
- Cherney, R.J.; Mo, R.; Meyer, D.T.; Hardman, K.D.; Liu, R.Q.; Covington, M.B.; Qian, M.; Wasserman, Z.R.; Christ, D.D.; Trzaskos, J.M.; et al. Sultam hydroxamates as novel matrix metalloproteinase inhibitors. J. Med. Chem. 2004, 47, 2981–2983. [Google Scholar] [PubMed]
- Nakatani, S.; Ikura, M.; Yamamoto, S.; Nishita, Y.; Itadani, S.; Habashita, H.; Sugiura, T.; Ogawa, K.; Ohno, H.; Takahashi, K.; et al. Design and synthesis of novel metalloproteinase inhibitors. Bioorg. Med. Chem. 2006, 14, 5402–5422. [Google Scholar] [PubMed]
- Subramaniam, R.; Haldar, M.K.; Tobwala, S.; Ganguly, B.; Srivastava, D.K.; Mallik, S. Novel bis-(arylsulfonamide) hydroxamate-based selective MMP inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 3333–3337. [Google Scholar] [PubMed] [Green Version]
- Whitlock, G.A.; Dack, K.N.; Dickinson, R.P.; Lewis, M.L. A novel series of highly selective inhibitors of MMP-3. Bioorg. Med. Chem. Lett. 2007, 17, 6750–6753. [Google Scholar]
- Jacobsen, F.E.; Buczynski, M.W.; Dennis, E.A.; Cohen, S.M. A macrophage cell model for selective metalloproteinase inhibitor design. Chembiochem 2008, 9, 2087–2095. [Google Scholar] [CrossRef] [Green Version]
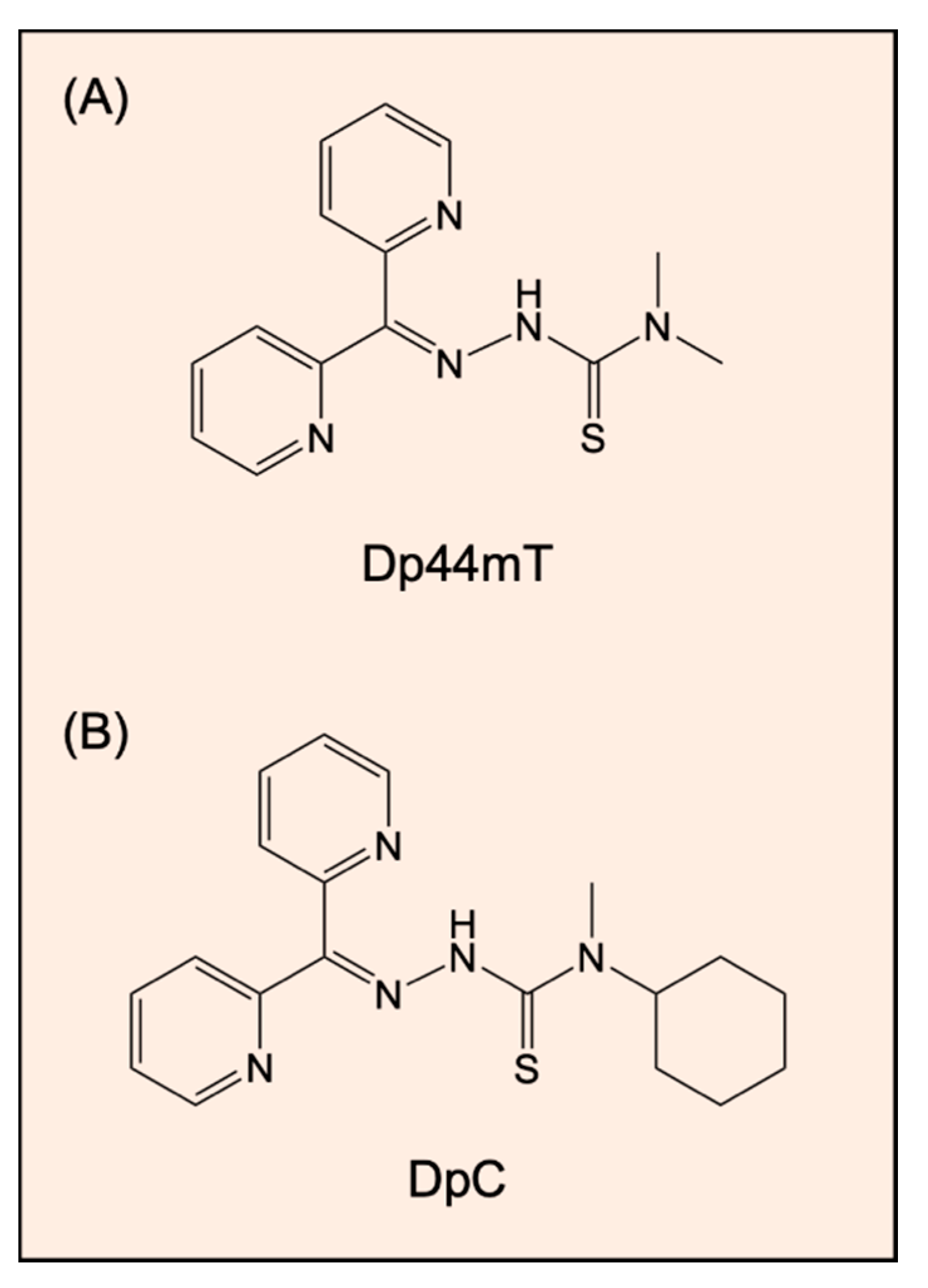
- Yuan, J.; Lovejoy, D.B.; Richardson, D.R. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: In vitro and in vivo assessment. Blood 2004, 104, 1450–1458. [Google Scholar] [CrossRef]
- Whitnall, M.; Howard, J.; Ponka, P.; Richardson, D.R. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. USA 2006, 103, 14901–14906. [Google Scholar] [CrossRef] [Green Version]
- Kovacevic, Z.; Chikhani, S.; Lovejoy, D.B.; Richardson, D.R. Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor N-myc down-stream regulated gene 1: A new strategy for the treatment of pancreatic cancer. Mol. Pharmacol. 2011, 80, 598–609. [Google Scholar]
- Lovejoy, D.B.; Sharp, D.M.; Seebacher, N.; Obeidy, P.; Prichard, T.; Stefani, C.; Basha, M.T.; Sharpe, P.C.; Jansson, P.J.; Kalinowski, D.S.; et al. Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J. Med. Chem. 2012, 55, 7230–7244. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.L.; Richardson, D.R.; Kalinowski, D.S.; Kovacevic, Z.; Tan-Un, K.C.; Chan, G.C. The novel thiosemicarbazone, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC), inhibits neuroblastoma growth in vitro and in vivo via multiple mechanisms. J. Hematol. Oncol. 2016, 9, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quach, P.; Gutierrez, E.; Basha, M.T.; Kalinowski, D.S.; Sharpe, P.C.; Lovejoy, D.B.; Bernhardt, P.V.; Jansson, P.J.; Richardson, D.R. Methemoglobin formation by triapine, di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT), and other anticancer thiosemicarbazones: Identification of novel thiosemicarbazones and therapeutics that prevent this effect. Mol. Pharmacol. 2012, 82, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Jansson, P.J.; Kalinowski, D.S.; Lane, D.J.; Kovacevic, Z.; Seebacher, N.A.; Fouani, L.; Sahni, S.; Merlot, A.M.; Richardson, D.R. The renaissance of polypharmacology in the development of anti-cancer therapeutics: Inhibition of the “Triad of Death” in cancer by Di-2-pyridylketone thiosemicarbazones. Pharmacol. Res. 2015, 100, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.J.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Novel thiosemicarbazones of the ApT and DpT series and their copper complexes: Identification of pronounced redox activity and characterization of their antitumor activity. J. Med. Chem. 2010, 53, 5759–5769. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.J.; Hawkins, C.L.; Lovejoy, D.B.; Richardson, D.R. The iron complex of Dp44mT is redox-active and induces hydroxyl radical formation: An EPR study. J. Inorg. Biochem. 2010, 104, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, D.B.; Jansson, P.J.; Brunk, U.T.; Wong, J.; Ponka, P.; Richardson, D.R. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 2011, 71, 5871–5880. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhang, D.; Yue, F.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J. Biol. Chem. 2012, 287, 17016–17028. [Google Scholar] [CrossRef] [Green Version]
- Menezes, S.V.; Fouani, L.; Huang, M.L.H.; Geleta, B.; Maleki, S.; Richardson, A.; Richardson, D.R.; Kovacevic, Z. The metastasis suppressor, NDRG1, attenuates oncogenic TGF-beta and NF-kappaB signaling to enhance membrane E-cadherin expression in pancreatic cancer cells. Carcinogenesis 2019, 40, 805–818. [Google Scholar] [CrossRef]
- Wangpu, X.; Lu, J.; Xi, R.; Yue, F.; Sahni, S.; Park, K.C.; Menezes, S.; Huang, M.L.; Zheng, M.; Kovacevic, Z.; et al. Targeting the metastasis suppressor, N-Myc downstream regulated gene-1, with novel di-2-pyridylketone thiosemicarbazones: Suppression of tumor cell migration and cell-collagen adhesion by inhibiting focal adhesion kinase/paxillin signaling. Mol. Pharmacol. 2016, 89, 521–540. [Google Scholar] [CrossRef] [Green Version]
- Kovacevic, Z.; Chikhani, S.; Lui, G.Y.; Sivagurunathan, S.; Richardson, D.R. The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and Ras signaling pathways. Antioxid. Redox Signal 2013, 18, 874–887. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Geleta, B.; Leck, L.Y.W.; Paluncic, J.; Chiang, S.; Jansson, P.J.; Kovacevic, Z.; Richardson, D.R. Thiosemicarbazones suppress expression of the c-Met oncogene by mechanisms involving lysosomal degradation and intracellular shedding. J. Biol. Chem. 2020, 295, 481–503. [Google Scholar] [PubMed]
- Lim, S.C.; Jansson, P.J.; Assinder, S.J.; Maleki, S.; Richardson, D.R.; Kovacevic, Z. Unique targeting of androgen-dependent and -independent AR signaling in prostate cancer to overcome androgen resistance. FASEB J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xing, F.; Iiizumi-Gairani, M.; Okuda, H.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Hirota, S.; Kobayashi, A.; Mo, Y.Y.; et al. N-myc downstream regulated gene 1 modulates Wnt-beta-catenin signalling and pleiotropically suppresses metastasis. EMBO Mol. Med. 2012, 4, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T.; Richardson, D.R. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: A link between iron metabolism and proliferation. Blood 2004, 104, 2967–2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.; Saletta, F.; Suryo Rahmanto, Y.; Kovacevic, Z.; Richardson, D.R. N-myc downstream regulated 1 (NDRG1) is regulated by eukaryotic initiation factor 3a (eIF3a) during cellular stress caused by iron depletion. PLoS ONE 2013, 8, e57273. [Google Scholar]
- Sharma, A.; Mendonca, J.; Ying, J.; Kim, H.S.; Verdone, J.E.; Zarif, J.C.; Carducci, M.; Hammers, H.; Pienta, K.J.; Kachhap, S. The prostate metastasis suppressor gene NDRG1 differentially regulates cell motility and invasion. Mol. Oncol. 2017, 11, 655–669. [Google Scholar] [CrossRef]
- Liu, Y.L.; Bai, W.T.; Luo, W.; Zhang, D.X.; Yan, Y.; Xu, Z.K.; Zhang, F.L. Downregulation of NDRG1 promotes invasion of human gastric cancer AGS cells through MMP-2. Tumour Biol. 2011, 32, 99–105. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370, 61–65. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar]
- Sun, J.; Zhang, D.; Bae, D.H.; Sahni, S.; Jansson, P.; Zheng, Y.; Zhao, Q.; Yue, F.; Zheng, M.; Kovacevic, Z.; et al. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis 2013, 34, 1943–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, B.A.; Kovacevic, Z.; Park, K.C.; Kalinowski, D.S.; Jansson, P.J.; Lane, D.J.; Sahni, S.; Richardson, D.R. Molecular functions of the iron-regulated metastasis suppressor, NDRG1, and its potential as a molecular target for cancer therapy. Biochim. Biophys. Acta 2014, 1845, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, D.; Xie, C.; Zheng, T.; Liang, Y.; Hong, X.; Lu, Z.; Song, X.; Song, R.; Yang, H.; et al. The iron chelator Dp44mT inhibits hepatocellular carcinoma metastasis via N-Myc downstream-regulated gene 2 (NDRG2)/gp130/STAT3 pathway. Oncotarget 2014, 5, 8478–8491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.C.; Kang, Y.K.; Kim, W.H.; Jang, Y.J.; Kim, D.J.; Park, I.Y.; Sohn, B.H.; Sohn, H.A.; Lee, H.G.; Lim, J.S.; et al. Functional and clinical evidence for NDRG2 as a candidate suppressor of liver cancer metastasis. Cancer Res. 2008, 68, 4210–4220. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wu, G.J.; Liu, X.W.; Zhang, R.; Yu, L.; Zhang, G.; Liu, F.; Yu, C.G.; Yuan, J.L.; Wang, H.; et al. Suppression of invasion and metastasis of prostate cancer cells by overexpression of NDRG2 gene. Cancer Lett. 2011, 310, 94–100. [Google Scholar] [CrossRef]
- Shon, S.K.; Kim, A.; Kim, J.Y.; Kim, K.I.; Yang, Y.; Lim, J.S. Bone morphogenetic protein-4 induced by NDRG2 expression inhibits MMP-9 activity in breast cancer cells. Biochem. Biophys. Res. Commun. 2009, 385, 198–203. [Google Scholar] [CrossRef]
- Jin, R.; Liu, W.; Menezes, S.; Yue, F.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. The metastasis suppressor NDRG1 modulates the phosphorylation and nuclear translocation of beta-catenin through mechanisms involving FRAT1 and PAK4. J. Cell Sci. 2014, 127 Pt 14, 3116–3130. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Zhang, D.; Zheng, Y.; Zhao, Q.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. Targeting the metastasis suppressor, NDRG1, using novel iron chelators: Regulation of stress fiber-mediated tumor cell migration via modulation of the ROCK1/pMLC2 signaling pathway. Mol. Pharmacol. 2013, 83, 454–469. [Google Scholar] [CrossRef] [Green Version]
- Geleta, B.; Park, K.C.; Jansson, P.; Sahni, S.; Maleki, S.; Xu, Z.; Murakami, T.; Pajic, M.; Apte, M.V.; Richardson, D.R.; et al. Breaking the cycle: Bespoke targeting of NDRG1 to inhibit bi-directional oncogenic cross-1 talk between pancreatic cancer and stroma. 2020; submitted. [Google Scholar]
- Saxena, P.; Trerotola, M.; Wang, T.; Li, J.; Sayeed, A.; Vanoudenhove, J.; Adams, D.S.; Fitzgerald, T.J.; Altieri, D.C.; Languino, L.R. PSA regulates androgen receptor expression in prostate cancer cells. Prostate 2012, 72, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Veveris-Lowe, T.L.; Lawrence, M.G.; Collard, R.L.; Bui, L.; Herington, A.C.; Nicol, D.L.; Clements, J.A. Kallikrein 4 (hK4) and prostate-specific antigen (PSA) are associated with the loss of E-cadherin and an epithelial-mesenchymal transition (EMT)-like effect in prostate cancer cells. Endocr. Relat. Cancer 2005, 12, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Sanford, M. Enzalutamide: A review of its use in metastatic, castration-resistant prostate cancer. Drugs 2013, 73, 1723–1732. [Google Scholar] [CrossRef]
- Stacy, A.E.; Palanimuthu, D.; Bernhardt, P.V.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R. Zinc(II)-thiosemicarbazone complexes are localized to the lysosomal compartment where they transmetallate with copper ions to induce cytotoxicity. J. Med. Chem. 2016, 59, 4965–4984. [Google Scholar] [CrossRef] [PubMed]
- Kovala-Demertzi, D.; Yadav, P.N.; Wiecek, J.; Skoulika, S.; Varadinova, T.; Demertzis, M.A. zinc(II) complexes derived from pyridine-2-carbaldehyde thiosemicarbazone and (1E)-1-pyridin-2-ylethan-1-one thiosemicarbazone. Synthesis, crystal structures and antiproliferative activity of zinc(II) complexes. J. Inorg. Biochem. 2006, 100, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.B.; Pereira, T.M.; Souza, D.L.N.; Lopes, D.S.; Freitas, V.; Avila, V.M.R.; Kummerle, A.E.; Sant’Anna, C.M.R. Structure-based discovery of thiosemicarbazone metalloproteinase inhibitors for hemorrhage treatment in snakebites. ACS Med. Chem. Lett. 2017, 8, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Le, N.T.; Richardson, D.R. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim. Biophys. Acta 2002, 1603, 31–46. [Google Scholar] [CrossRef]
- Richardson, D.R.; Tran, E.H.; Ponka, P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood 1995, 86, 4295–4306. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.R.; Milnes, K. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents II: The mechanism of action of ligands derived from salicylaldehyde benzoyl hydrazone and 2-hydroxy-1-naphthylaldehyde benzoyl hydrazone. Blood 1997, 89, 3025–3038. [Google Scholar] [CrossRef]
- Dixon, K.M.; Lui, G.Y.; Kovacevic, Z.; Zhang, D.; Yao, M.; Chen, Z.; Dong, Q.; Assinder, S.J.; Richardson, D.R. Dp44mT targets the AKT, TGF-beta and ERK pathways via the metastasis suppressor NDRG1 in normal prostate epithelial cells and prostate cancer cells. Br. J. Cancer 2013, 108, 409–419. [Google Scholar] [CrossRef]
- Lui, G.Y.; Obeidy, P.; Ford, S.J.; Tselepis, C.; Sharp, D.M.; Jansson, P.J.; Kalinowski, D.S.; Kovacevic, Z.; Lovejoy, D.B.; Richardson, D.R. The iron chelator, deferasirox, as a novel strategy for cancer treatment: Oral activity against human lung tumor xenografts and molecular mechanism of action. Mol. Pharmacol. 2013, 83, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Suryo Rahmanto, Y.; Richardson, D.R. Bp44mT: An orally active iron chelator of the thiosemicarbazone class with potent anti-tumour efficacy. Br. J. Pharmacol. 2012, 165, 148–166. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MMPs | Function | Cancer Type |
|---|---|---|
| MMP-1 |
|
|
| MMP-2 |
|
|
| MMP-3 |
|
|
| MMP-7 |
|
|
| MMP-8 |
|
|
| MMP-9 |
|
|
| MMP-10 |
|
|
| MMP-11 |
|
|
| MMP-12 | Promotes tumor formation | Bronchioalveolar adenocarcinoma |
| MMP-13 | Promotes angiogenesis via stimulation of ERK-FAK signaling pathway and VEGF-A secretion | Head and neck squamous cell carcinoma |
| MMP-14 | Induces chromatin instability by cleaving pericentrin | Glioma, breast cancer and colon adenocarcinoma |
| MMP-17 |
|
|
| MMP-19 |
|
|
| MMP-26 |
|
|
| MMP-28 |
|
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.C.; Dharmasivam, M.; Richardson, D.R. The Role of Extracellular Proteases in Tumor Progression and the Development of Innovative Metal Ion Chelators That Inhibit Their Activity. Int. J. Mol. Sci. 2020, 21, 6805. https://doi.org/10.3390/ijms21186805
Park KC, Dharmasivam M, Richardson DR. The Role of Extracellular Proteases in Tumor Progression and the Development of Innovative Metal Ion Chelators That Inhibit Their Activity. International Journal of Molecular Sciences. 2020; 21(18):6805. https://doi.org/10.3390/ijms21186805
Chicago/Turabian StylePark, Kyung Chan, Mahendiran Dharmasivam, and Des R. Richardson. 2020. "The Role of Extracellular Proteases in Tumor Progression and the Development of Innovative Metal Ion Chelators That Inhibit Their Activity" International Journal of Molecular Sciences 21, no. 18: 6805. https://doi.org/10.3390/ijms21186805