DNA Methylome Distinguishes Head and Neck Cancer from Potentially Malignant Oral Lesions and Healthy Oral Mucosa


 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. HPV Status
2.2. Overall DNA Methylation Findings
2.3. Differentially Methylated Gene Promoters in HNSCC Tissue Compared to Control Tissue
2.4. Differentially Methylated CPG Sites in HNSCC Tissue Compared to Control Tissue
2.5. Aberrant Methylation in Potentially Premalignant Oral Lesions
2.6. Validation Panel
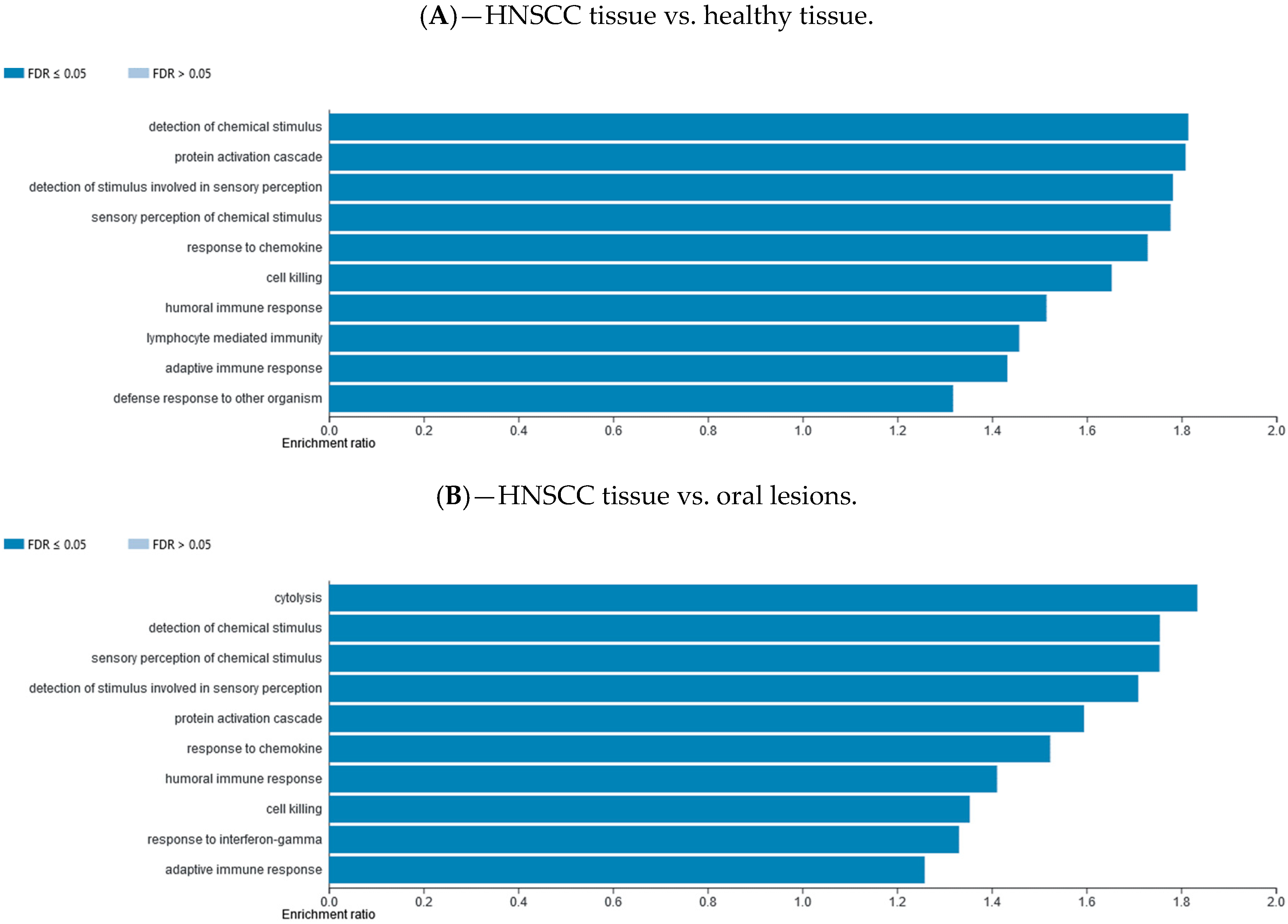
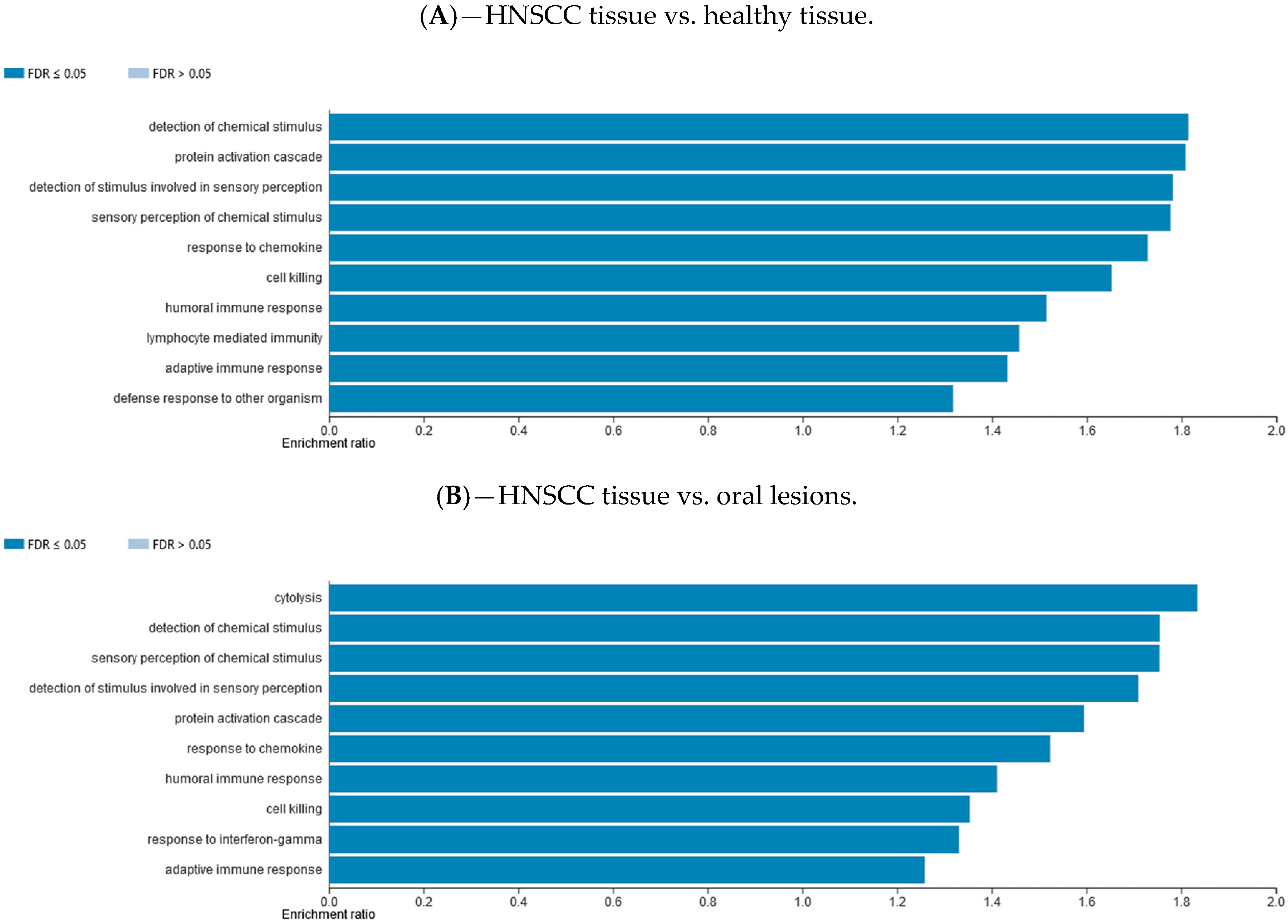
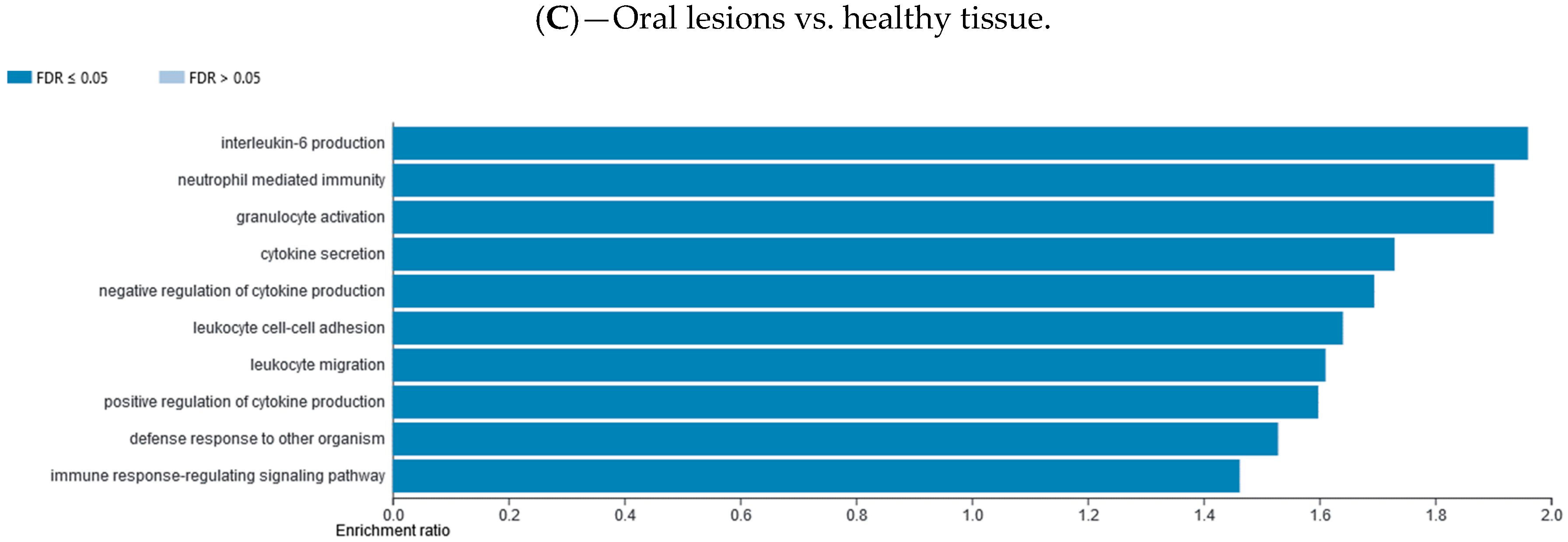
2.7. Gene Set Enrichment Data
2.8. External Database Validation
3. Discussion
4. Materials and Methods
4.1. Study Group
4.2. DNA Preparation
4.3. HPV Detection and Typing
4.4. Methylation Array Analysis
4.5. Data Processing and Statistical Analysis
4.6. Differentially Methylated Gene Promoters and Individual CPG Sites
4.7. Validation of Methylation by Pyrosequencing
4.8. Gene Set Enrichment Analysis
4.9. External Validation of Differentially Methylated Gene Promoters
4.10. Ethics Approval and Consent to Participate
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| comb.p.adj.fdr | FDR adjusted combined p-value; |
| CpG | 5′-cytosine-phosphate-guanine-3′ |
| FDR | false discovery rate |
| GSEA | gene set enrichment analysis |
| HNSCC | head and neck squamous cell carcinoma |
| HPV | human papillomavirus |
| KEGG | Kyoto encyclopedia of genes and genomes |
| limma | linear models for microarray data |
| mean.mean.diff | value of the mean difference across all sites in a region |
| miRNA | micro ribonucleic acids |
| NA | not annotated; NTA, network topology-based analysis |
| OLL | oral lichenoid lesions |
| OLP | oral lichen planus |
| OPSCC | oropharyngeal squamous cell carcinoma |
| ORA | over-representation enrichment analysis |
| OSCC | oral squamous cell carcinoma |
| PCA | principal component analysis |
| PCR | polymerase chain reaction |
| RnBeads | integrated software package for the analysis and interpretation of DNA methylation data |
| SNPs | single nucleotide polymorphisms |
| TCGA | The Cancer Genome Atlas |
| TS | type-specific |
References
- International Agency for Research on Cancer, Global Cancer Observatory. 2012. Available online: http://globocan.iarc.fr/Default.aspx (accessed on 13 June 2018).
- Arantes, L.M.R.B.; De Carvalho, A.C.; Melendez, M.E.; Carvalho, A.L. Serum, plasma and saliva biomarkers for head and neck cancer. Expert Rev. Mol. Diagn. 2017, 18, 85–112. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Burris, J.L.; Studts, J.L.; DeRosa, A.; Ostroff, J.S. Systematic Review of Tobacco Use after Lung or Head/Neck Cancer Diagnosis: Results and Recommendations for Future Research. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1450–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakita, D.; Matsuo, K. Alcohol and head and neck cancer. Cancer Metastasis Rev. 2017, 36, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Hashibe, M.; Brennan, P.; Benhamou, S.; Castellsagué, X.; Chen, C.; Curado, M.P.; Maso, L.D.; Daudt, A.W.; Fabianova, E.; Wunsch-Filho, V.; et al. Alcohol Drinking in Never Users of Tobacco, Cigarette Smoking in Never Drinkers, and the Risk of Head and Neck Cancer: Pooled Analysis in the International Head and Neck Cancer Epidemiology Consortium. J. Natl. Cancer Inst. 2007, 99, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Hashibe, M.; Brennan, P.; Chuang, S.-C.; Boccia, S.; Castellsagué, X.; Chen, C.; Curado, M.P.; Maso, L.D.; Daudt, A.W.; Fabianova, E.; et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol. Biomark. Prev. 2009, 18, 541–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. HPV-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, G.; Dempsey, A. The role of HPV in head and neck cancer and review of the HPV vaccine. Prev. Med. 2011, 53, S5–S11. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Hisamatsu, K.; Suzui, N.; Hara, A.; Tomita, H.; Miyazaki, T. A Review of HPV-Related Head and Neck Cancer. J. Clin. Med. 2018, 7, 241. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Malik, A.; Garg, A.; Mair, M.; Nair, S.; Chaturvedi, P. Oral sex and human papilloma virus-related head and neck squamous cell cancer: A review of the literature. Postgrad. Med J. 2017, 93, 704–709. [Google Scholar] [CrossRef]
- Ghantous, Y.; Schussel, J.L.; Brait, M. Tobacco and alcohol-induced epigenetic changes in oral carcinoma. Curr. Opin. Oncol. 2018, 30, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.A.; Lechner, M.; Köferle, A.; Carén, H.; Butcher, L.M.; Feber, A.; Fenton, T.R.; Jay, A.; Boshoff, C.; Beck, S. Integrated virus-host methylome analysis in head and neck squamous cell carcinoma. Epigenetics 2013, 8, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, R.J.; Ironside, J.A.D.; Wei, W.I. Squamous cell carcinomas of the head and neck * Commentary: Head and neck carcinomas in the developing world. BMJ 2002, 325, 822–827. [Google Scholar] [CrossRef] [Green Version]
- Langevin, S.M.; Kuhnell, D.; Niu, L.; Biesiada, J.; Leung, Y.-K.; Deka, R.; Chen, A.; Medvedovic, M.; Kelsey, K.T.; Kasper, S.; et al. Comprehensive mapping of the methylation landscape of 16 CpG-dense regions in oral and pharyngeal squamous cell carcinoma. Epigenomics 2019, 11, 987–1002. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Herceg, Z.; Hainaut, P. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Mol. Oncol. 2007, 1, 26–41. [Google Scholar] [CrossRef] [Green Version]
- Van Der Meij, E.; Mast, H.; Van Der Waal, I. The possible premalignant character of oral lichen planus and oral lichenoid lesions: A prospective five-year follow-up study of 192 patients. Oral Oncol. 2007, 43, 742–748. [Google Scholar] [CrossRef]
- Casparis, S.; Borm, J.M.; Tektas, S.; Kamarashev, J.; Locher, M.; Damerau, G.; Grätz, K.W.; Stadlinger, B. Oral lichen planus (OLP), oral lichenoid lesions (OLL), oral dysplasia, and oral cancer: Retrospective analysis of clinicopathological data from 2002–2011. Oral Maxillofac. Surg. 2014, 19, 149–156. [Google Scholar] [CrossRef]
- Patel, P.S.; Dudhia, B.B.; Dudhia, S.B.; Jani, Y.V. Oral lichen planus to oral lichenoid lesions: Evolution or revolution. J. Oral Maxillofac. Pathol. 2015, 19, 364–370. [Google Scholar] [CrossRef]
- Stipetić, M.M.; Sabol, I.; Kranjčić, J.; Knežević, M.; Grce, M. Human Papillomavirus in the Lesions of the Oral Mucosa According to Topography. PLoS ONE 2013, 8, e69736. [Google Scholar] [CrossRef] [Green Version]
- Ambrosi, C.; Manzo, M.; Baubec, T. Dynamics and Context-Dependent Roles of DNA Methylation. J. Mol. Boil. 2017, 429, 1459–1475. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-S. Decreased expression of SPRR3 in Chinese human oesophageal cancer. Carcinogenesis 2000, 21, 2147–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-J.; Tian, J.; Qi, X.-K.; Xiang, T.; He, G.-P.; Zhang, H.; Yu, X.; Zhang, X.; Zhao, B.; Feng, Q.-S.; et al. Epstein-Barr virus activates F-box protein FBXO2 to limit viral infectivity by targeting glycoprotein B for degradation. PLoS Pathog. 2018, 14, e1007208. [Google Scholar] [CrossRef] [PubMed]
- Gene Cards—The Human Genes Gene Database. Available online: https://www.genecards.org/ (accessed on 22 August 2018).
- Taberna, M.; Mena, M.; Pavón, M.A.; Alemany, L.; Gillison, M.L.; Mesía, R. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. 2017, 28, 2386–2398. [Google Scholar] [CrossRef]
- Božinović, K.; Sabol, I.; Rakusic, Z.; Jakovčević, A.; Šekerija, M.; Lukinović, J.; Prgomet, D.; Grce, M. HPV-driven oropharyngeal squamous cell cancer in Croatia—Demography and survival. PLoS ONE 2019, 14, e0211577. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.; Fenton, T.R.; West, J.; Wilson, G.A.; Feber, A.; Henderson, S.; Thirlwell, C.; Dibra, H.K.; Jay, A.; Butcher, L.M.; et al. Identification and functional validation of HPV-mediated hypermethylation in head and neck squamous cell carcinoma. Genome Med. 2013, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, C.; Ma, G.; Zhang, Q. Expression of SPRR3 is associated with tumor cell proliferation and invasion in glioblastoma multiforme. Oncol. Lett. 2013, 7, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, Y.-B.; Shen, X.-M.; Chen, B.-S.; Du, X.-L.; Luo, M.-L.; Cai, Y.; Han, Y.-L.; Xu, X.; Zhan, Q.-M.; et al. Exogenous expression of Esophagin/SPRR3 attenuates the tumorigenicity of esophageal squamous cell carcinoma cellsviapromoting apoptosis. Int. J. Cancer 2007, 122, 260–266. [Google Scholar] [CrossRef]
- Solomon, B.; Young, R.J.; Rischin, D. Head and neck squamous cell carcinoma: Genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin. Cancer Boil. 2018, 52. [Google Scholar] [CrossRef]
- Strzelczyk, J.K.; Krakowczyk, Ł.; Owczarek, A.J. Aberrant DNA methylation of the p16, APC, MGMT, TIMP3 and CDH1 gene promoters in tumours and the surgical margins of patients with oral cavity cancer. J. Cancer 2018, 9, 1896–1904. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-S.; Kim, S.Y.; Lee, M.; Kim, S.H.; Kim, S.-M.; Kim, E.J. Radioresistance in a human laryngeal squamous cell carcinoma cell line is associated with DNA methylation changes and topoisomerase II α. Cancer Boil. Ther. 2015, 16, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentilini, D.; Scala, S.; Gaudenzi, G.; Garagnani, P.; Capri, M.; Cescon, M.; Grazi, G.L.; Bacalini, M.G.; Pisoni, S.; Dicitore, A.; et al. Epigenome-wide association study in hepatocellular carcinoma: Identification of stochastic epigenetic mutations through an innovative statistical approach. Oncotarget 2017, 8, 41890–41902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, N.; Levanon, E.Y.; Amariglio, N.; Heimberger, A.B.; Ram, Z.; Constantini, S.; Barbash, Z.S.; Adamsky, K.; Safran, M.; Hirschberg, A.; et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007, 17, 1586–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, G.; Wang, H.; Zhong, Z. Associations of RASSF1A, RARβ, and CDH1 promoter hypermethylation with oral cancer risk. Medicine (Baltimore) 2018, 97, e9971. [Google Scholar] [CrossRef]
- Liu, G. CDH1promoter methylation in patients with cervical carcinoma: A systematic meta-analysis with trial sequential analysis. Futur. Oncol. 2018, 14, 51–63. [Google Scholar] [CrossRef]
- Lee, J.-J.; Kao, K.-C.; Chiu, Y.-L.; Jung, C.-J.; Liu, C.-J.; Cheng, S.-J.; Chang, Y.-L.; Ko, J.-Y.; Chia, J.-S. Enrichment of Human CCR6+ Regulatory T Cells with Superior Suppressive Activity in Oral Cancer. J. Immunol. 2017, 199, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Xu, X.; Lee, E.-J.; Shull, A.Y.; Pei, L.; Awan, F.; Wang, X.; Choi, J.-H.; Deng, L.; Xin, H.-B.; et al. Phenotypic alteration of CD8+ T cells in chronic lymphocytic leukemia is associated with epigenetic reprogramming. Oncotarget 2016, 7, 40558–40570. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Tseng, R.-C.; Hsu, H.-S.; Wang, Y.-C.; Hsu, M.-T. Frequent down-regulation of hRAB37 in metastatic tumor by genetic and epigenetic mechanisms in lung cancer. Lung Cancer 2009, 63, 360–367. [Google Scholar] [CrossRef]
- Bond, H.M.; Scicchitano, S.; Chiarella, E.; Amodio, N.; Lucchino, V.; Aloisio, A.; Montalcini, Y.; Mesuraca, M.; Morrone, G. ZNF423: A New Player in Estrogen Receptor-Positive Breast Cancer. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Ortiz, C.I.; Lopez-Vidal, Y.; Arredondo-Hernandez, L.J.R.; Castillo-Rojas, G. Genetic Alterations in Gastric Cancer Associated with Helicobacter pylori Infection. Front. Med. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Peng, H.; Huang, X.; Zhao, M.; Li, Z.; Yin, N.; Wang, X.; Yu, F.; Yin, B.; Yuan, Y.; et al. Genome-wide profiling of DNA methylation and gene expression in esophageal squamous cell carcinoma. Oncotarget 2015, 7, 4507–4521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, Y.; Semi, K.; Hashimoto, K.; Lee, M.S.; Hirata, A.; Tomita, H.; Kuno, T.; Takamatsu, M.; Aoki, K.; Taketo, M.M.; et al. Reducing DNA methylation suppresses colon carcinogenesis by inducing tumor cell differentiation. Carcinogenesis 2015, 36, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Karnovsky, A.; Mahavisno, V.; Weymouth, T.; Pande, M.; Dolinoy, D.C.; Rozek, L.S.; Sartor, M.A. LRpath analysis reveals common pathways dysregulated via DNA methylation across cancer types. BMC Genom. 2012, 13, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielcarek-Kuchta, D.; Paluszczak, J.; Seget, M.; Kiwerska, K.; Biczysko, W.; Szyfter, K.; Szyfter, W. Prognostic factors in oral and oropharyngeal cancer based on ultrastructural analysis and DNA methylation of the tumor and surgical margin. Tumor Boil. 2014, 35, 7441–7449. [Google Scholar] [CrossRef] [PubMed]
- Juodzbalys, G.; Kasradze, D.; Cicciù, M.; Sudeikis, A.; Banys, L.; Galindo-Moreno, P.; Guobis, Z. Modern molecular biomarkers of head and neck cancer. Part I. Epigenetic diagnostics and prognostics: Systematic review. Cancer Biomark. 2017, 17, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.-S.; Gao, W.; Chan, J.Y.-W. Interactions between E-Cadherin and MicroRNA Deregulation in Head and Neck Cancers: The Potential Interplay. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [Green Version]
- Manos, M.M.; Ting, Y.; Wright, D.K.; Levwis, A.J.; Broker, T.R.; Wolinsky, S.M. The use of polymerase chain reaction amplification for the detection of genital human papillomaviruses. U Cancer Cells. 1989, 7, 209–214. [Google Scholar]
- Gašperov, N.M.; Sabol, I.; Halec, G.; Matovina, M.; Grce, M. Retrospective study of the prevalence of high-risk human papillomaviruses among Croatian women. Coll. Antropol. 2007, 31, doi. [Google Scholar]
- Gasperov, N.M.; Sabol, I.; Planinić, P.; Grubisic, G.; Fistonić, I.; Corusić, A.; Grce, M. Methylated Host Cell Gene Promoters and Human Papillomavirus Type 16 and 18 Predicting Cervical Lesions and Cancer. PLoS ONE 2015, 10, e0129452. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.L.; Van Dijk, S.J.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Boil. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2013, 30, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Müller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedeurwaerder‡, S.; Defrance, M.; Calonne, E.; Denis, H.; Sotiriou, C.; Fuks, F. Evaluation of the Infinium Methylation 450K technology. Epigenomics 2011, 3, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef]
- Smyth, G.K.; Michaud, J.; Scott, H. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef]
- Mikeska, T.; Felsberg, J.; Hewitt, C.A.; Dobrovic, A. Analysing DNA Methylation Using Bisulphite Pyrosequencing. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin, Germany, 2011; Volume 791, pp. 33–53. [Google Scholar]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Díez-Villanueva, A.; Mallona, I.; Peinado, M.A. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin 2015, 8, 22. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Function | MMD * | p-Value ** |
|---|---|---|---|
| HNSCC Tissue vs. Healthy Tissue | |||
| GPRC5D | G protein-coupled receptor | 0.78 | 6.49 × 10−7 |
| TMPRSS11B | Transmembrane protease | 0.76 | 2.04 × 10−8 |
| PIAS2 | Sumoylation | 0.66 | 1.22 × 10−6 |
| ARG1 | Arginase activity | 0.64 | 2.50 × 10−7 |
| SRPK2 | Protein kinase | 0.62 | 1.30 × 10−6 |
| AADACL2 | Hydrolase activity | 0.59 | 7.38 × 10−7 |
| RGPD4 | RNA transport | 0.58 | 4.71 × 10−6 |
| SPRR3 | Structural molecule activity | 0.53 | 2.44 × 10−7 |
| DEGS1 | Desaturase activity | 0.49 | 2.04 × 10−8 |
| TXNDC8 | Oxidoreductase activity | 0.48 | 1.15 × 10−5 |
| SH3TC1 | Myelination | 0.47 | 2.05 × 10−6 |
| ZPLD1 | Cerebral malformations | 0.47 | 6.17 × 10−6 |
| FBXO2 | Ubiquitination | 0.46 | 0.000236 |
| ATG16L1 | Autophagy | 0.46 | 0.000306 |
| GRHL1 | Transcription factor | 0.44 | 5.12 × 10−7 |
| HNSCC Tissue vs. Oral Lesions | |||
| RAD51B | RAD51 Paralog B | 0.85 | 2.53 × 10−8 |
| BARX2 | BARX Homeobox 2 | 0.81 | 3.94 × 10−8 |
| SLC5A10;FAM83G | Solute Carrier Family 5 Member 10 | 0.78 | 2.58 × 10−8 |
| NINL | Ninein Like | 0.77 | 3.96 × 10−8 |
| NSMCE2 | NSE2/MMS21 Homolog, SMC5-SMC6 Complex SUMO Ligase | 0.76 | 6.36 × 10−7 |
| PGAP2 | Post-GPI Attachment to Proteins 2 | 0.75 | 9.48 × 10−8 |
| INO80C | INO80 Complex Subunit C | 0.74 | 1.96 × 10−9 |
| IL34 | Interleukin 34 | 0.74 | 2.20 × 10−9 |
| ZNF516 | Zinc Finger Protein 516 | 0.73 | 4.90 × 10−8 |
| GFOD2 | Glucose-Fructose Oxidoreductase Domain Containing 2 | 0.73 | 1.36 × 10−7 |
| PARD3 | Par-3 Family Cell Polarity Regulator | 0.73 | 1.36 × 10−7 |
| MCEE | Methylmalonyl-CoA Epimerase | 0.72 | 2.89 × 10−8 |
| POLM | DNA Polymerase Mu | 0.72 | 3.93 × 10−7 |
| ASPG | Asparaginase | 0.71 | 4.43 × 10−8 |
| TBC1D2 | TBC1 Domain Family Member 2 | 0.71 | 3.74 × 10−7 |
| Oral Lesions vs. Healthy Tissue | |||
| SLC5A10;FAM83G | Solute Carrier Family 5 Member 10 | 0.78 | 2.58 × 10−8 |
| TBC1D2 | TBC1 Domain Family Member 2 | 0.71 | 3.74 × 10−7 |
| SH3BP5L | SH3 Binding Domain Protein 5 Like | 0.70 | 2.68 × 10−7 |
| VANGL1 | VANGL Planar Cell Polarity Protein 1 | 0.69 | 6.49 × 10−7 |
| DLEC1 | Deleted in Lung And Esophageal Cancer 1 | 0.61 | 3.84 × 10−6 |
| TGOLN2 | Trans-Golgi Network Protein 2 | 0.61 | 3.79 × 10−7 |
| CTBP2 | C-Terminal Binding Protein 2 | 0.59 | 4.99 × 10−6 |
| PPP1CB | Protein Phosphatase 1 Catalytic Subunit Beta | 0.56 | 2.20 × 10−6 |
| VPS52 | VPS52, GARP Complex Subunit | 0.53 | 8.05 × 10−6 |
| MEPCE | Methylphosphate Capping Enzyme | 0.52 | 2.89 × 10−7 |
| HDAC4 | Histone Deacetylase 4 | 0.51 | 6.47 × 10−6 |
| ARAP1 | ArfGAP with RhoGAP Domain, Ankyrin Repeat, and PH Domain 1 | 0.50 | 4.77 × 10−7 |
| TCF20 | Transcription Factor 20 | 0.49 | 4.14 × 10−5 |
| NDUFS7 | NADH:Ubiquinone Oxidoreductase Core Subunit S7 | 0.49 | 0.00017 |
| GATAD2A | GATA Zinc Finger Domain Containing 2A | 0.47 | 3.58 × 10−8 |
| Gene Name | Function | MMD * | p-Value ** |
|---|---|---|---|
| HNSCC Tissue vs. Healthy Tissue | |||
| TRBC2 | T cell receptor | −0.80 | 2.48 × 10−7 |
| DGAT2 | Acyltransferase activity | −0.70 | 2.48 × 10−7 |
| ALG1L | Transferase activity | −0.70 | 3.29 × 10−5 |
| PDE4D | Enzyme binding | −0.68 | 1.06 × 10−5 |
| TRDC | T cell receptor | −0.67 | 1.46 × 10−5 |
| DNAJC6 | Phosphatase activity | −0.67 | 1.63 × 10−6 |
| IGKV3-20 | Immunoglobulin receptor binding | −0.66 | 1.71 × 10−5 |
| TMEM150B | Transmembrane protein | −0.66 | 8.13 × 10−5 |
| LAIR2 | Innate immune response | −0.65 | 1.81 × 10−5 |
| UBQLN3 | Protein degradation | −0.64 | 2.57 × 10−6 |
| ANKFN1 | Not known | −0.64 | 1.71 × 10−7 |
| MS4A1 | Differentiation of B-cells | −0.63 | 3.80 × 10−5 |
| CCT8L2 | Channel activity | −0.62 | 3.59 × 10−6 |
| SPOCK1 | Not known | −0.61 | 5.06 × 10−5 |
| IGHV4-39 | Antigen recognition | −0.61 | 9.44 × 10−7 |
| HNSCC Tissue vs. Oral Lesions | |||
| ART4 | ADP-Ribosyltransferase 4 (Dombrock Blood Group) | −0.88 | 7.92 × 10−11 |
| EPB41L3 | Erythrocyte Membrane Protein Band 4.1 Like 3 | −0.87 | 6.18 × 10−11 |
| ESRRG | Estrogen Related Receptor Gamma | −0.86 | 8.51 × 10−9 |
| ENPP1 | Ectonucleotide Pyrophosphatase/Phosphodiesterase 1 | −0.86 | 1.26 × 10−9 |
| GNG7 | G Protein Subunit Gamma 7 | −0.86 | 4.75 × 10−9 |
| PAPSS2 | 3′-Phosphoadenosine 5′-Phosphosulfate Synthase 2 | −0.85 | 4.10 × 10−9 |
| NGEF | Neuronal Guanine Nucleotide Exchange Factor | −0.84 | 1.87 × 10−9 |
| HIPK4 | Homeodomain Interacting Protein Kinase 4 | −0.84 | 6.69 × 10−9 |
| GPR158 | G Protein-Coupled Receptor 158 | −0.83 | 9.82 × 10−10 |
| GSG1L | GSG1 Like | −0.83 | 1.04 × 10−8 |
| SMPD3 | Sphingomyelin Phosphodiesterase 3 | −0.83 | 1.64 × 10−8 |
| GDF2 | Growth Differentiation Factor 2 | −0.83 | 5.15 × 10−10 |
| RERE | Arginine-Glutamic Acid Dipeptide Repeats | −0.82 | 2.19 × 10−8 |
| CDH13 | Cadherin 13 | −0.82 | 1.81 × 10−10 |
| HS3ST4 | Heparan Sulfate-Glucosamine 3-Sulfotransferase 4 | −0.82 | 1.02 × 10−8 |
| Oral Lesions vs. Healthy Tissue | |||
| ART4 | ADP-Ribosyltransferase 4 (Dombrock Blood Group) | −0.88 | 7.92 × 10−11 |
| ENPP1 | Ectonucleotide Pyrophosphatase/Phosphodiesterase 1 | −0.86 | 1.26 × 10−9 |
| GNG7 | G Protein Subunit Gamma 7 | −0.86 | 4.75 × 10−9 |
| PKD1L3 | Polycystin 1 Like 3, Transient Receptor Potential Channel Interacting | −0.81 | 2.20 × 10−9 |
| PLXNC1 | Plexin C1 | −0.81 | 1.41 × 10−9 |
| CAMK2B | Calcium/Calmodulin Dependent Protein Kinase II Beta | −0.79 | 1.05 × 10−8 |
| CACNA1S | Calcium Voltage–Gated Channel Subunit Alpha1 S | −0.78 | 8.18 × 10−9 |
| SCGB1D1 | Secretoglobin Family 1D Member 1 | −0.78 | 2.34 × 10−7 |
| VPS13D | Vacuolar Protein Sorting 13 Homolog D | −0.76 | 6.12 × 10−8 |
| DLGAP4 | DLG Associated Protein 4 | −0.76 | 6.37 × 10−9 |
| LRP1B | LDL Receptor Related Protein 1B | −0.76 | 1.12 × 10−9 |
| COL2A1 | Collagen Type II Alpha 1 Chain | −0.75 | 1.30 × 10−8 |
| SLC24A3 | Solute Carrier Family 24 Member 3 | −0.74 | 3.07 × 10−9 |
| TBC1D8 | TBC1 Domain Family Member 8 | −0.74 | 1.84 × 10−8 |
| ABCC8 | ATP Binding Cassette Subfamily C Member 8 | −0.73 | 3.58 × 10−9 |
| Gene Name | Function | cg Position | MMD * | p-Value ** |
|---|---|---|---|---|
| HNSCC Tissue vs. Healthy Tissue | ||||
| LMBR1L | Probable receptor | cg12348519 | 0.93 | 4.59 × 10−8 |
| CDH1 | Adhesions, mobility, and proliferation | cg08285862 | 0.92 | 3.49 × 10−8 |
| EIF6 | Initiation of translation | cg09957666 | 0.92 | 1.96 × 10−8 |
| C16orf70 | Not known | cg03664901 | 0.92 | 3.48 × 10−8 |
| ETNK2 | Transferase and kinase activity | cg12142497 | 0.92 | 5.33 × 10−8 |
| C11orf73 | Cellular response to heat stress | cg23450586 | 0.91 | 5.01 × 10−9 |
| ADARB2 | RNA editing | cg26569590 | 0.91 | 2.10 × 10−8 |
| GAB1 | Cellular growth, transformation, and apoptosis | cg23020414 | 0.91 | 2.83 × 10−8 |
| ITPR3 | Metabolism and growth | cg05876496 | 0.91 | 6.35 × 10−8 |
| WDR61 | Transcriptional regulation | cg12339790 | 0.90 | 4.35 × 10−8 |
| PGAP2 | Protein transport | cg01156876 | 0.90 | 1.24 × 10−8 |
| DDX10 | RNA helicase | cg18585558 | 0.90 | 6.14 × 10−9 |
| DGKH | Kinase activity | cg22899750 | 0.90 | 1.09 × 10−7 |
| RAB40C | Oncogene | cg01770948 | 0.89 | 2.00 × 10−8 |
| BEAN1 | Not known | cg19471156 | 0.89 | 5.59 × 10−8 |
| HNSCC Tissue vs. Oral Lesions | ||||
| EIF6 | Eukaryotic Translation Initiation Factor 6 | cg09957666 | 0.91 | 5.68 × 10−10 |
| KANSL1 | KAT8 Regulatory NSL Complex Subunit 1 | cg07281649 | 0.91 | 1.43 × 10−9 |
| DDX10 | DEAD–Box Helicase 10 | cg18585558 | 0.89 | 3.58 × 10−10 |
| AP2A1 | Adaptor Related Protein Complex 2 Alpha 1 Subunit | cg08969148 | 0.89 | 8.97 × 10−10 |
| RAB40C | RAB40C, Member RAS Oncogene Family | cg01770948 | 0.89 | 1.84 × 10−9 |
| GAB1 | GRB2 Associated Binding Protein 1 | cg23020414 | 0.88 | 5.30 × 10−9 |
| ERGIC1 | Endoplasmic Reticulum-Golgi Intermediate Compartment 1 | cg07769006 | 0.88 | 1.25 × 10−9 |
| SNX14 | Sorting Nexin 14 | cg03776905 | 0.88 | 3.20 × 10−9 |
| PIGU | Phosphatidylinositol Glycan Anchor Biosynthesis Class U | cg09450087 | 0.88 | 1.22 × 10−10 |
| ARAP1 | ArfGAP with RhoGAP Domain, Ankyrin Repeat, and PH Domain 1 | cg09010791 | 0.87 | 1.56 × 10−9 |
| LMTK2 | Lemur Tyrosine Kinase 2 | cg05941925 | 0.87 | 2.59 × 10−9 |
| BEAN1 | Brain Expressed Associated with NEDD4 1 | cg19471156 | 0.87 | 6.96 × 10−9 |
| AP1S3 | Adaptor Related Protein Complex 1 Sigma 3 Subunit | cg25666945 | 0.87 | 1.66 × 10−9 |
| CDH1 | Cadherin 1 | cg08285862 | 0.87 | 2.28 × 10−8 |
| RYBP | RING1 and YY1 Binding Protein | cg08086385 | 0.86 | 3.11 × 10−10 |
| Oral Lesions vs. Healthy Tissue | ||||
| GRIP1 | Glutamate Receptor Interacting Protein 1 | cg09414535 | 0.68 | 0.000679 |
| MTMR10 | Myotubularin Related Protein 10 | cg25430175 | 0.66 | 0.000585 |
| RBM47 | RNA Binding Motif Protein 47 | cg11268702 | 0.66 | 0.000636 |
| MPHOSPH9 | M–Phase Phosphoprotein 9 | cg02132191 | 0.65 | 0.001055 |
| FOXK1 | Forkhead Box K1 | cg16026475 | 0.64 | 0.000765 |
| SNX3 | Sorting Nexin 3 | cg14452952 | 0.64 | 0.000825 |
| CIT | Citron Rho-Interacting Serine/Threonine Kinase | cg03601895 | 0.63 | 0.000866 |
| ZBTB38 | Zinc Finger and BTB Domain Containing 38 | cg13318410 | 0.63 | 0.001548 |
| DRD3 | Dopamine Receptor D3 | cg22253817 | 0.63 | 0.001115 |
| SPPL3 | Signal Peptide Peptidase Like 3 | cg11330512 | 0.63 | 0.001072 |
| ZNF407 | Zinc Finger Protein 407 | cg23863184 | 0.63 | 0.000942 |
| ADAMTSL1 | ADAMTS Like 1 | cg12699984 | 0.62 | 0.000767 |
| GNAT3 | G Protein Subunit Alpha Transducin 3 | cg10168361 | 0.62 | 0.000936 |
| L3MBTL3 | L3MBTL3, Histone Methyl-Lysine Binding Protein | cg22162357 | 0.62 | 0.001083 |
| EEPD1 | Endonuclease/Exonuclease/Phosphatase Family Domain Containing 1 | cg06387870 | 0.61 | 0.001083 |
| Gene Name | Function | cg Position | MMD * | p-Value ** |
|---|---|---|---|---|
| HNSCC Tissue vs. Healthy Tissue | ||||
| ATXN1 | Not known | cg07713291 | −0.96 | 2.97 × 10−9 |
| PPP2R2C | Cell growth | cg05805165 | −0.93 | 1.22 × 10−8 |
| CCR6 | Immune regulation | cg05094429 | −0.92 | 1.08 × 10−7 |
| RAB37 | Oncogene | cg25267982 | −0.92 | 1.29 × 10−8 |
| DUSP27 | Phosphatase activity | cg23713934 | −0.91 | 1.94 × 10−7 |
| ZNF521 | Transcription factor | cg21830945 | −0.91 | 7.58 × 10−8 |
| SLC6A17 | Transporter | cg12072789 | −0.90 | 7.38 × 10−8 |
| SPIN1 | Methylated histone binding | cg13554018 | −0.90 | 1.63 × 10−8 |
| CXCR1 | Receptor | cg13519373 | −0.90 | 1.22 × 10−8 |
| SPTBN1 | Cell shape | cg06149826 | −0.89 | 6.91 × 10−9 |
| NBAS | Golgi to ER transport | cg27424261 | −0.89 | 5.31 × 10−7 |
| NRG3 | Ligand | cg10656958 | −0.89 | 9.45 × 10−8 |
| COL5A1 | Forming collagen | cg26087052 | −0.89 | 3.96 × 10−8 |
| CDX1 | Transcriptional regulation | cg12473781 | −0.89 | 5.77 × 10−8 |
| BATF3 | Transcriptional regulation | cg03219362 | −0.89 | 2.10 × 10−8 |
| HNSCC Tissue vs. Oral Lesions | ||||
| FAM69A | Family with Sequence Similarity 69 Member A | cg22727960 | −0.93 | 7.05 × 10−11 |
| ATP6V0A1 | ATPase H+ Transporting V0 Subunit A1 | cg19022525 | −0.92 | 9.01 × 10−11 |
| LBP | Lipopolysaccharide Binding Protein | cg18979491 | −0.92 | 3.02 × 10−10 |
| WDR25 | WD Repeat Domain 25 | cg24211276 | −0.91 | 6.22 × 10−11 |
| SH3RF3 | SH3 Domain Containing Ring Finger 3 | cg27294813 | −0.91 | 1.01 × 10−9 |
| NINJ2 | Ninjurin 2 | cg05534515 | −0.91 | 2.74 × 10−12 |
| RAB37 | RAB37, Member RAS Oncogene Family | cg25267982 | −0.90 | 1.17 × 10−9 |
| CXCR1 | C-X-C Motif Chemokine Receptor 1 | cg13519373 | −0.90 | 1.76 × 10−10 |
| SPTBN | Spectrin Beta, Non–Erythrocytic 1 | cg06149826 | −0.90 | 1.54 × 10−10 |
| RHOH | Ras Homolog Family Member H | cg15729055 | −0.90 | 1.90 × 10−9 |
| GRIK5 | Glutamate Ionotropic Receptor Kainate Type Subunit 5 | cg03100024 | −0.90 | 2.47 × 10−9 |
| KLRD1 | Killer Cell Lectin Like Receptor D1 | cg05377120 | −0.90 | 6.88 × 10−9 |
| TENM2 | Teneurin Transmembrane Protein 2 | cg26758826 | −0.89 | 3.56 × 10−11 |
| FAM69A | Family with Sequence Similarity 69 Member A | cg05172999 | −0.89 | 3.14 × 10−10 |
| ITK | IL2 Inducible T Cell Kinase | cg12250498 | −0.89 | 3.46 × 10−10 |
| Oral Lesions vs. Healthy Tissue | ||||
| PHACTR1 | Phosphatase and Actin Regulator 1 | cg02381687 | −0.80 | 0.000673 |
| MARCH8 | Membrane Associated Ring-CH-Type Finger 8 | cg26841425 | −0.80 | 0.000585 |
| PPP1R1B | Protein Phosphatase 1 Regulatory Inhibitor Subunit 1B | cg03104421 | −0.79 | 0.000585 |
| HDAC4 | Histone Deacetylase 4 | cg21190228 | −0.79 | 0.00057 |
| IL22RA2 | Interleukin 22 Receptor Subunit Alpha 2 | cg23507945 | −0.79 | 0.001772 |
| CAMKK2 | Calcium/Calmodulin Dependent Protein Kinase Kinase 2 | cg03391567 | −0.78 | 0.000679 |
| INPP5D | Inositol Polyphosphate-5-Phosphatase D | cg22666015 | −0.78 | 0.000709 |
| CSGALNACT1 | Chondroitin Sulfate N–Acetylgalactosaminyltransferase 1 | cg24423468 | −0.77 | 0.001266 |
| GTDC1 | Glycosyltransferase Like Domain Containing 1 | cg19251811 | −0.77 | 0.000676 |
| IGSF3 | Immunoglobulin Superfamily Member 3 | cg13004173 | −0.77 | 0.000585 |
| HELZ | Helicase with Zinc Finger | cg15015109 | −0.76 | 0.000772 |
| DEFA4 | Defensin Alpha 4 | cg06617936 | −0.76 | 0.000678 |
| AK5 | Adenylate Kinase 5 | cg21487631 | −0.76 | 0.000681 |
| LHFPL2 | LHFPL Tetraspan Subfamily Member 2 | cg20879720 | −0.76 | 0.000981 |
| STK10 | Serine/Threonine Kinase 10 | cg22406187 | −0.76 | 0.000765 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milutin Gašperov, N.; Sabol, I.; Božinović, K.; Dediol, E.; Mravak-Stipetić, M.; Licastro, D.; Dal Monego, S.; Grce, M. DNA Methylome Distinguishes Head and Neck Cancer from Potentially Malignant Oral Lesions and Healthy Oral Mucosa. Int. J. Mol. Sci. 2020, 21, 6853. https://doi.org/10.3390/ijms21186853
Milutin Gašperov N, Sabol I, Božinović K, Dediol E, Mravak-Stipetić M, Licastro D, Dal Monego S, Grce M. DNA Methylome Distinguishes Head and Neck Cancer from Potentially Malignant Oral Lesions and Healthy Oral Mucosa. International Journal of Molecular Sciences. 2020; 21(18):6853. https://doi.org/10.3390/ijms21186853
Chicago/Turabian StyleMilutin Gašperov, Nina, Ivan Sabol, Ksenija Božinović, Emil Dediol, Marinka Mravak-Stipetić, Danilo Licastro, Simeone Dal Monego, and Magdalena Grce. 2020. "DNA Methylome Distinguishes Head and Neck Cancer from Potentially Malignant Oral Lesions and Healthy Oral Mucosa" International Journal of Molecular Sciences 21, no. 18: 6853. https://doi.org/10.3390/ijms21186853