Cell-Penetrable Peptide-Conjugated FADD Induces Apoptosis and Regulates Inflammatory Signaling in Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
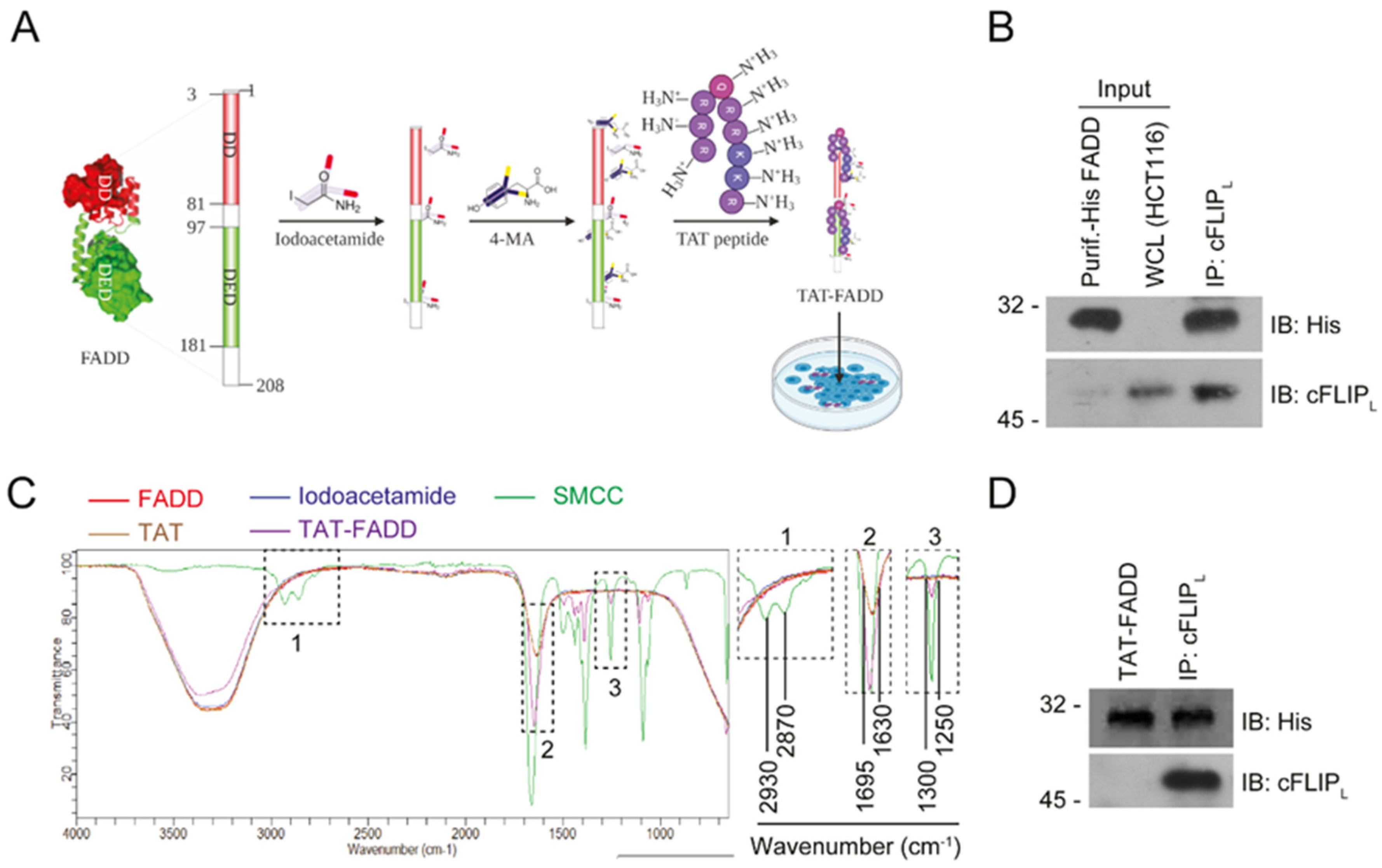
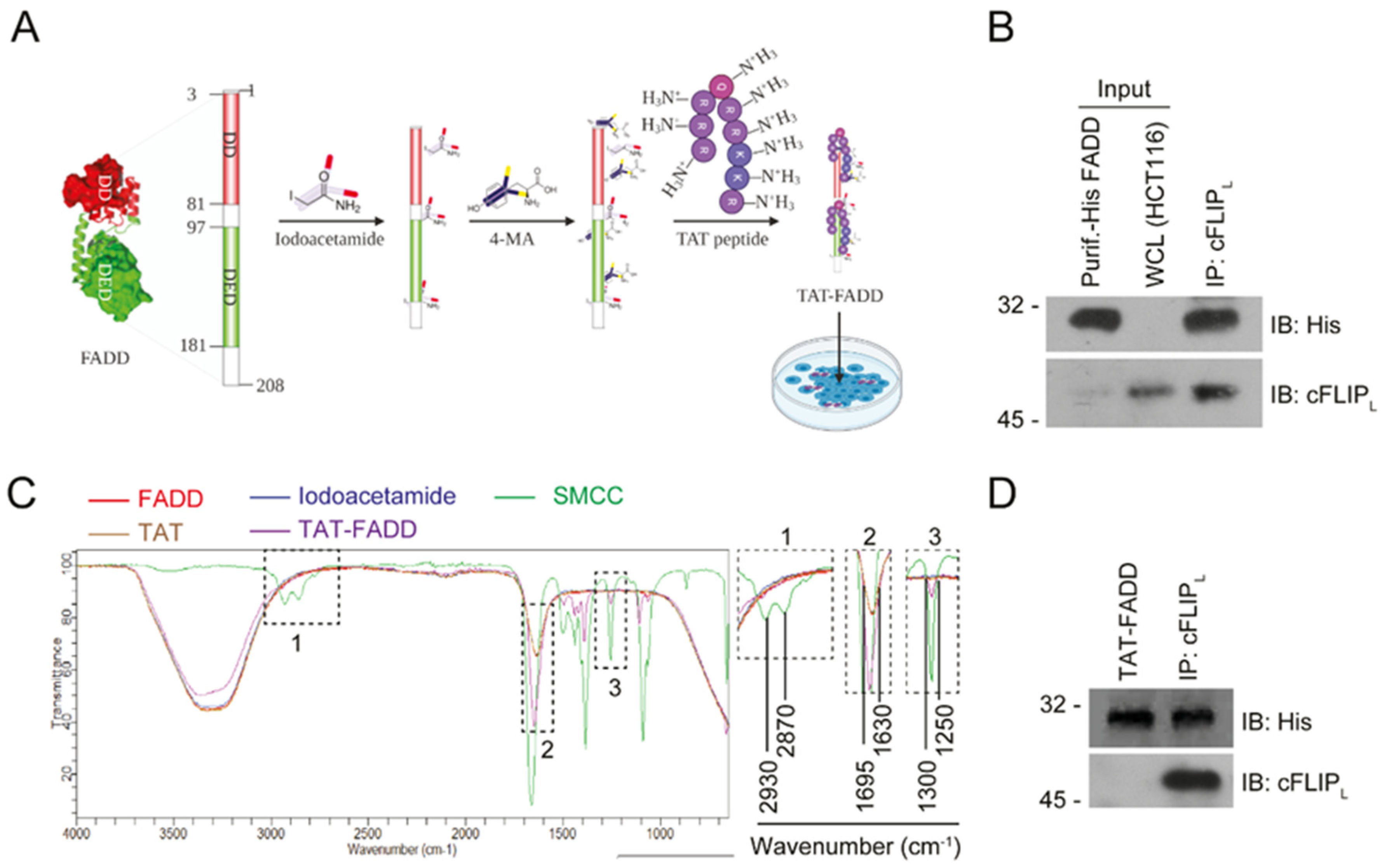
2.1. Conjugation and Characterization of TAT-FADD
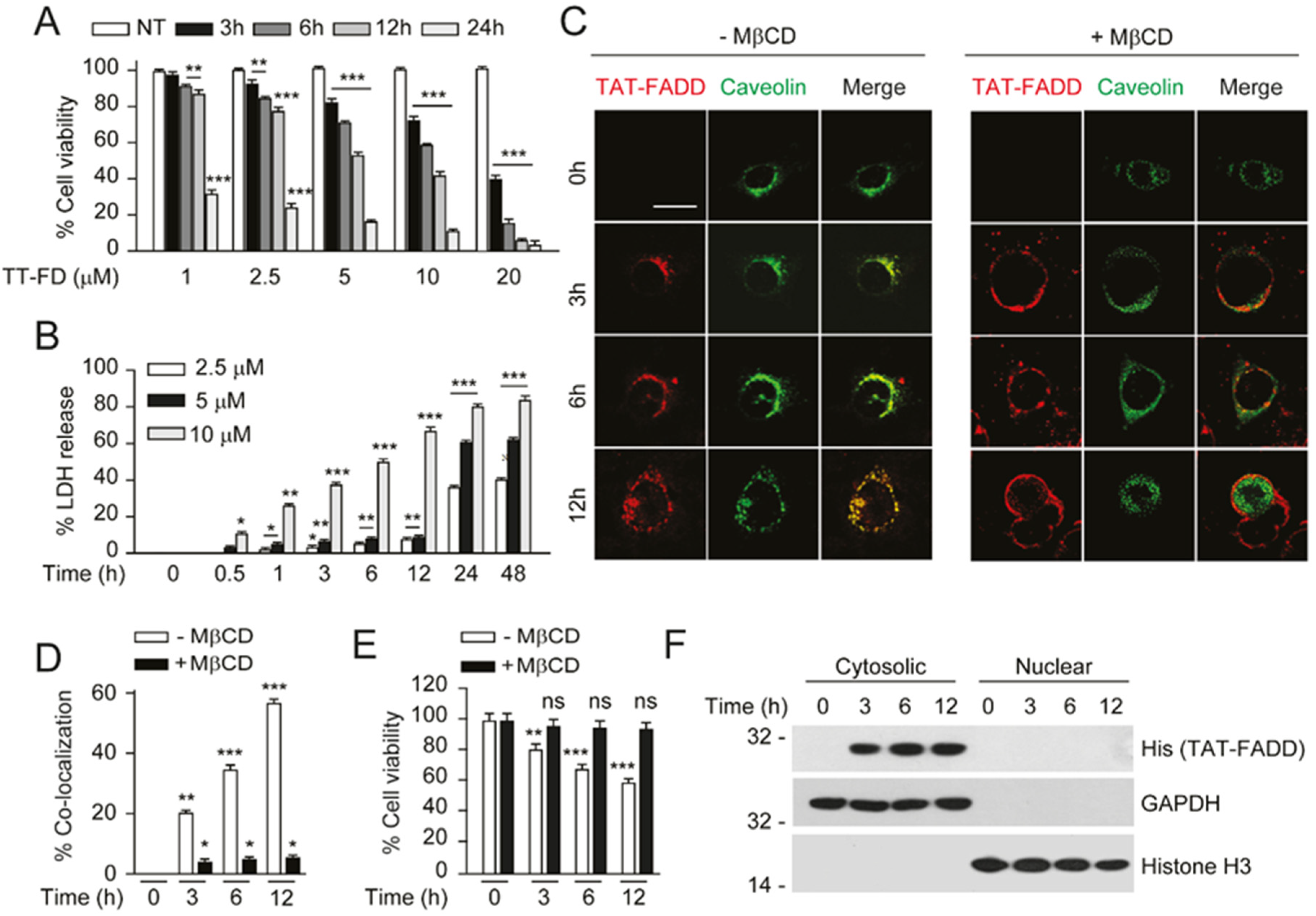
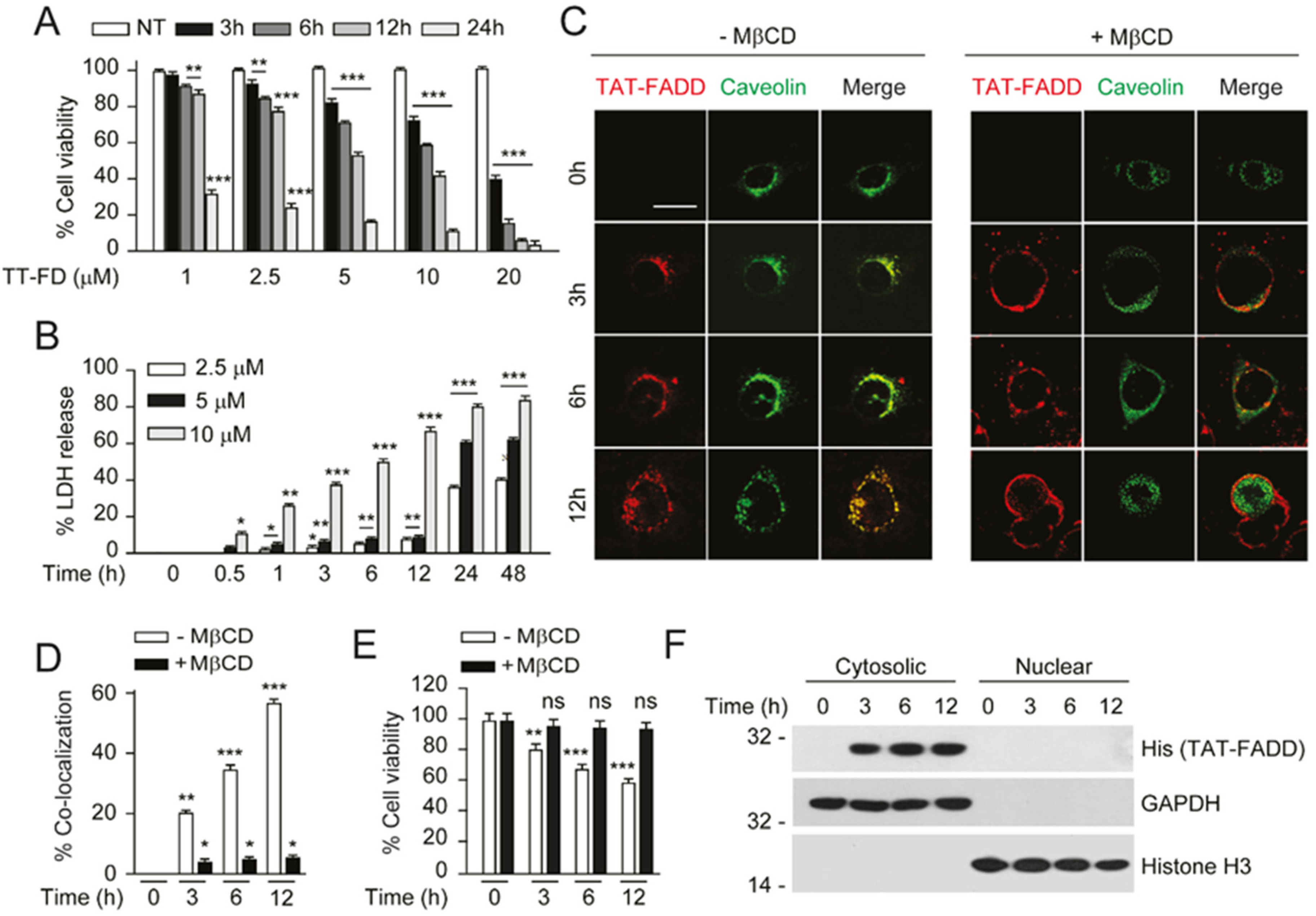
2.2. TAT-FADD Efficiently Internalized in the Cells and Retained in the Cytosol
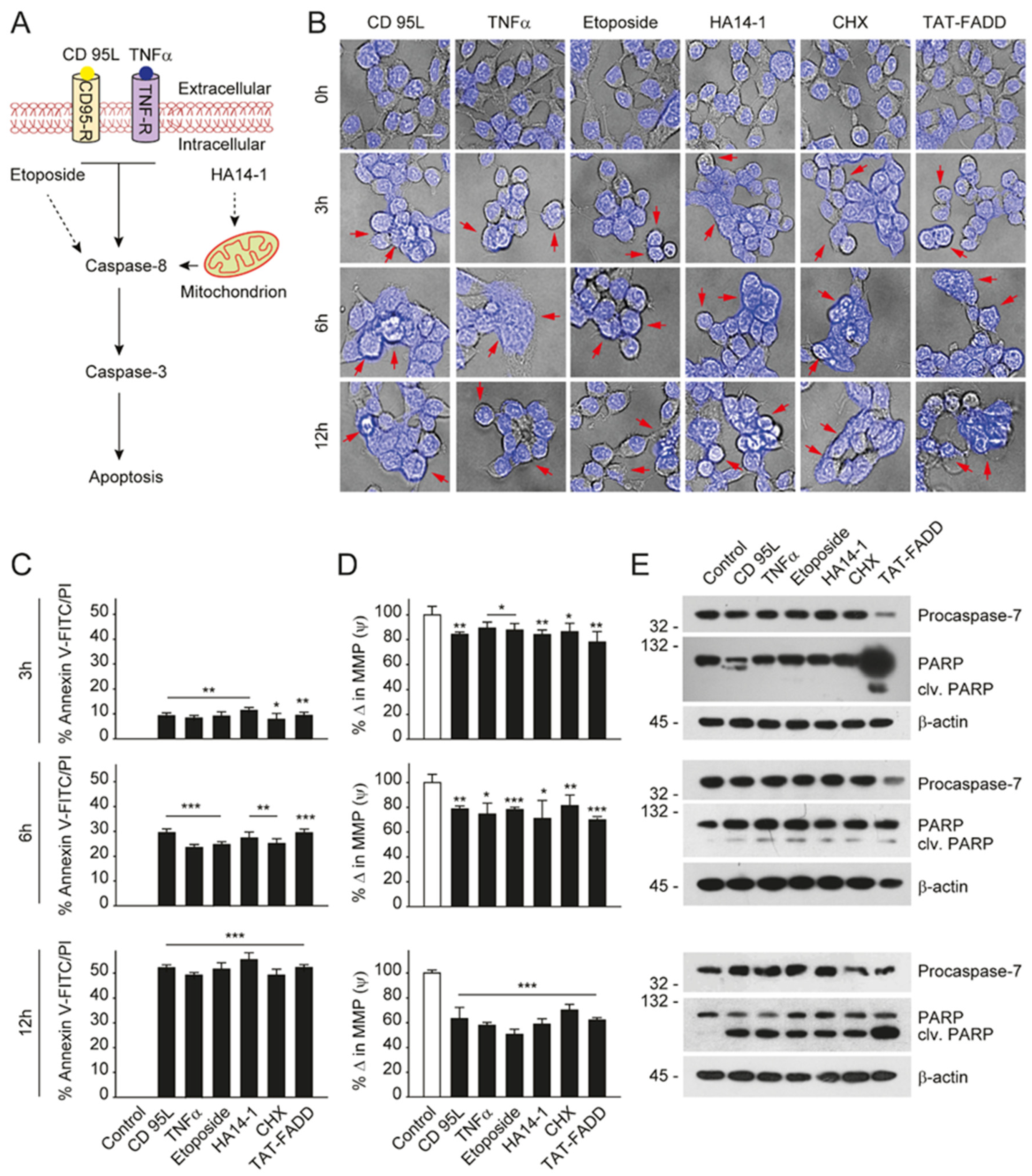
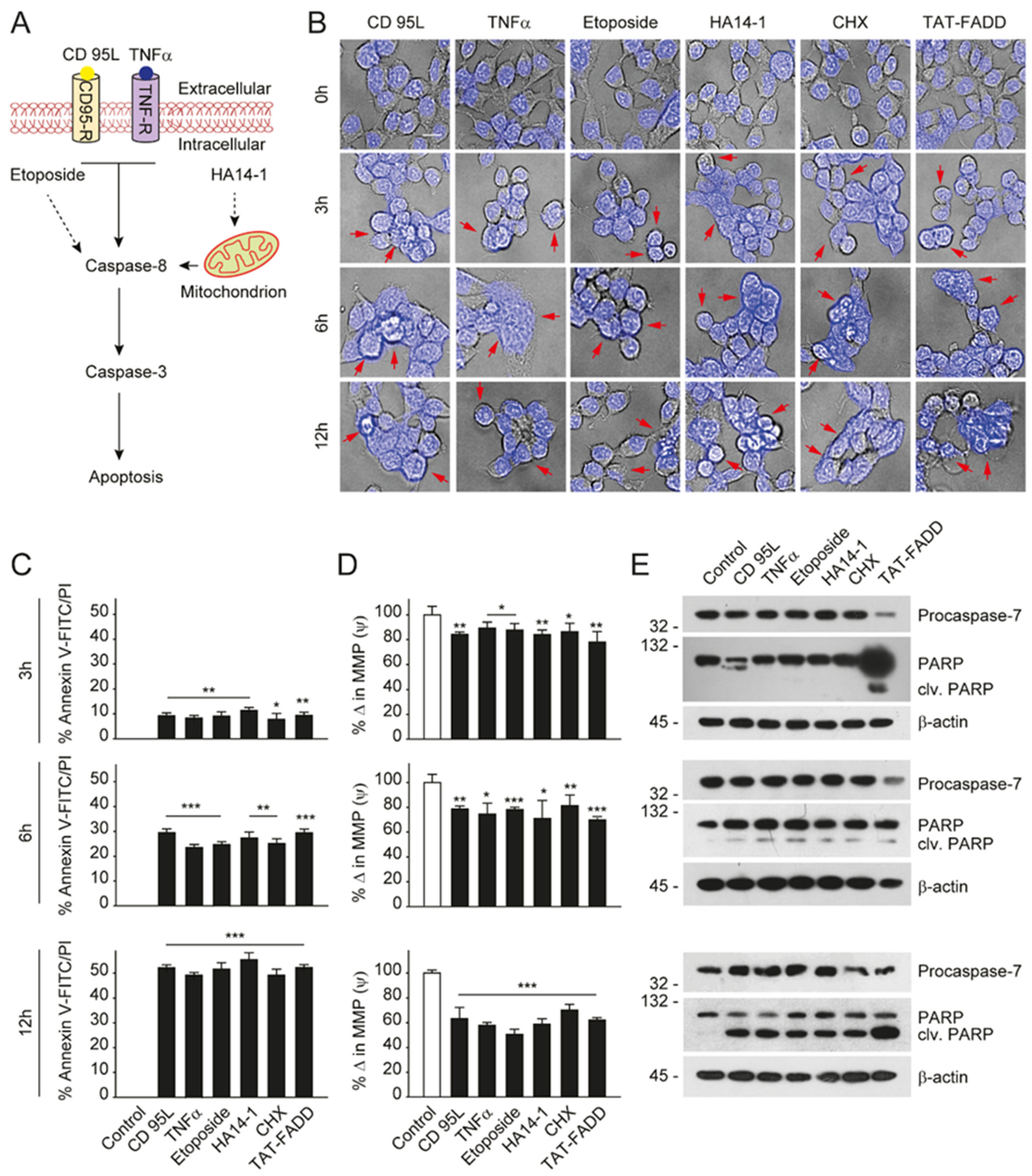
2.3. TAT-FADD Constitute DISC Assembly and Instigates Cell Death via Apoptosis Signaling
2.4. TAT-FADD Efficiently Induces Apoptosis Compared with Conventional Apoptosis Inducers
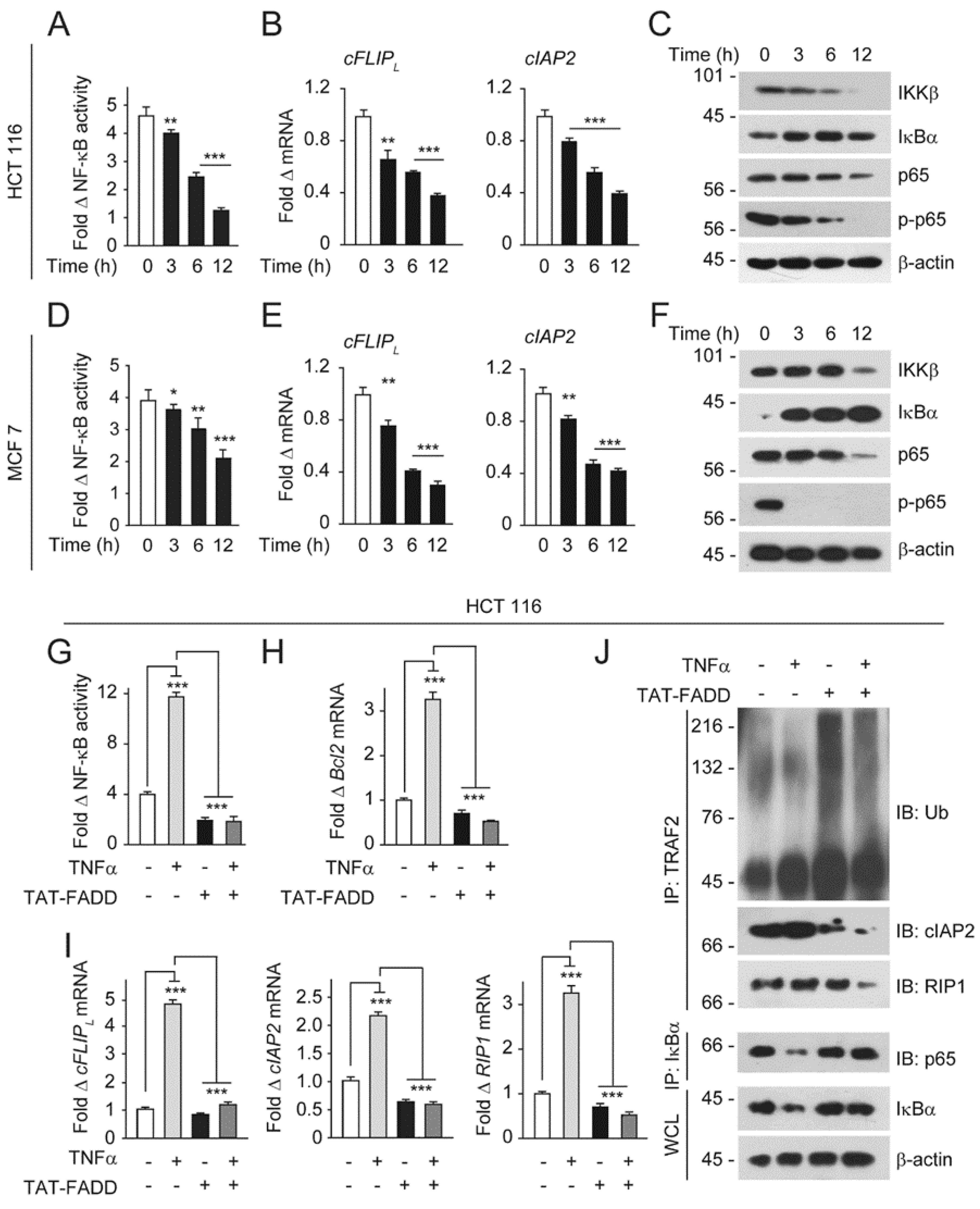
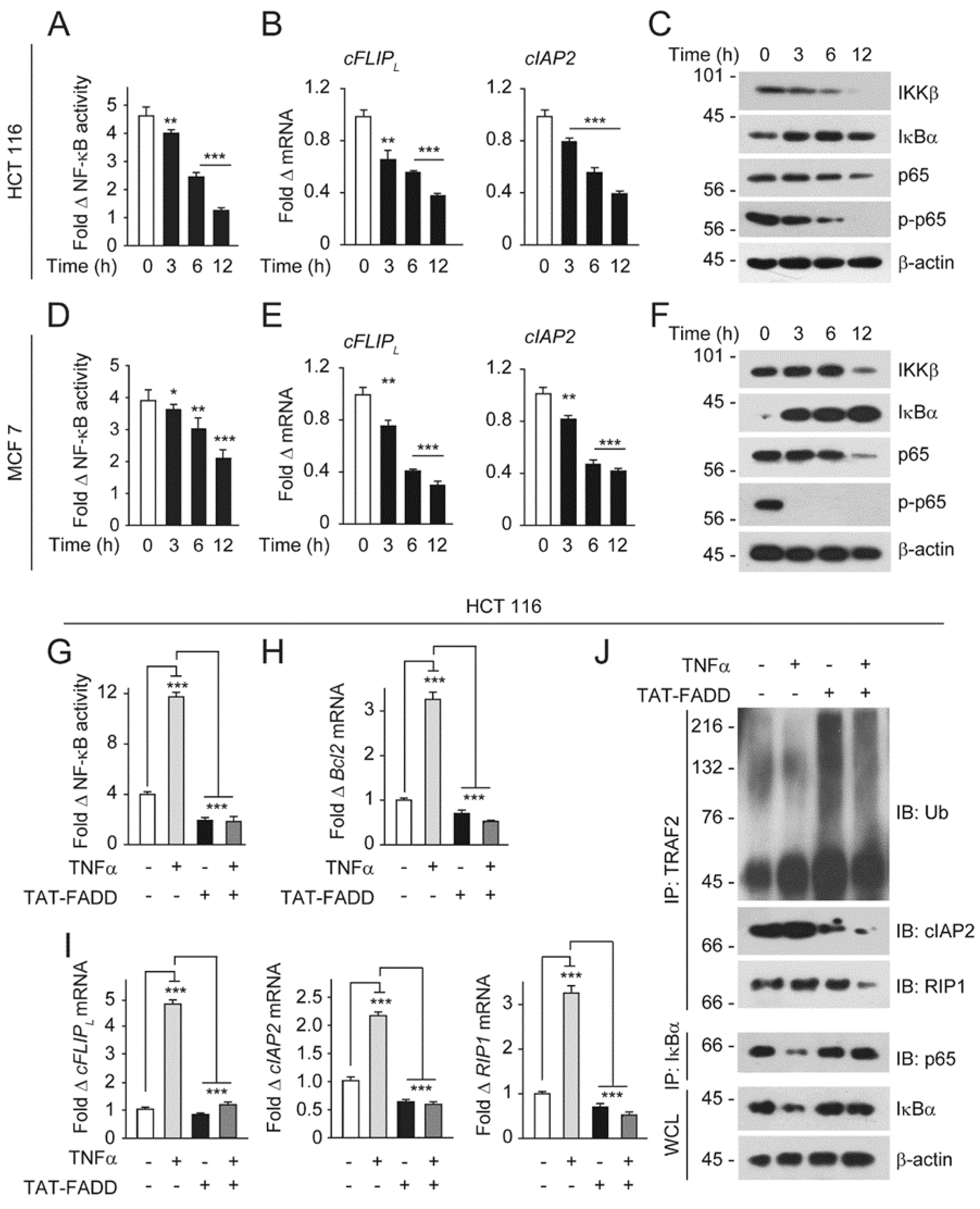
2.5. TAT-FADD Mitigates NF-κB Activation in Cancer Cells
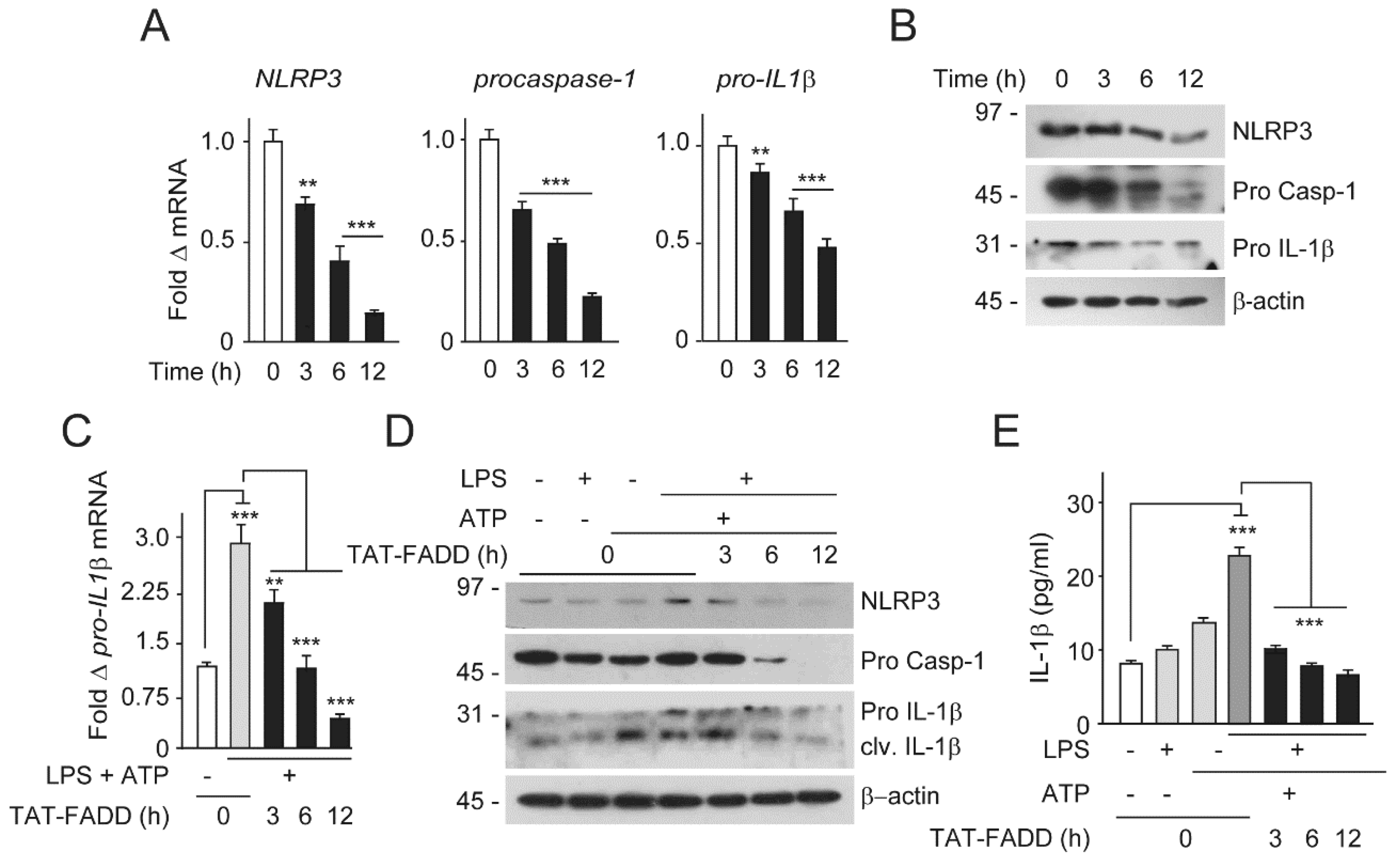
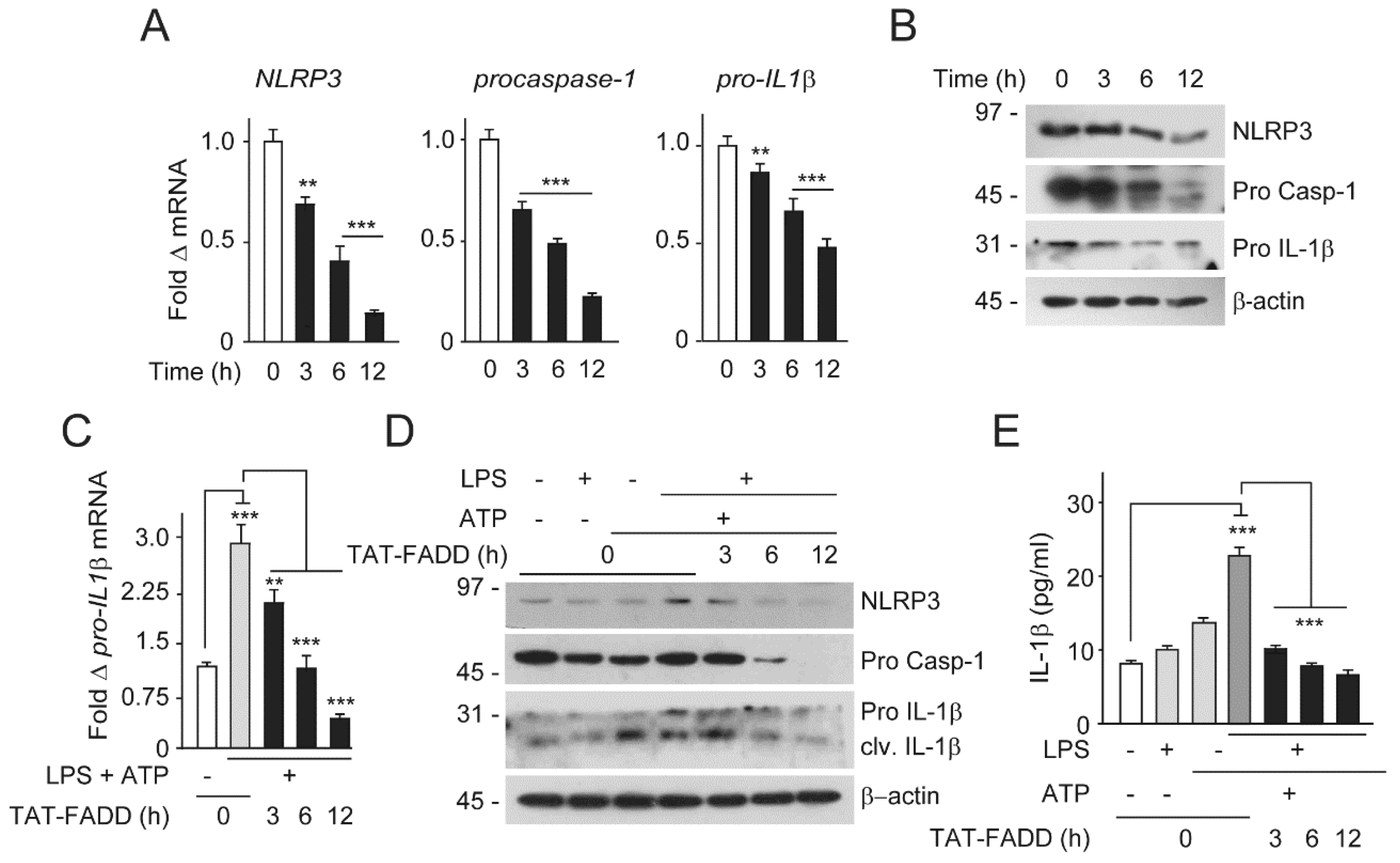
2.6. TAT-FADD Restricts Inflammasome Components and Secretion of Proinflammatory Cytokine IL-1β
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines and In Vitro Cell Culture
4.3. Cloning and Purification of Human FADD (hFADD)
4.4. Mass Spectrometry
4.5. Chemical Conjugation of hFADD with TAT Peptide (TAT-FADD)
4.6. Fourier-Transform Infrared Spectroscopy (FT-IR) Analysis
4.7. In Vitro Protein Interaction Assay
4.8. Intracellular Examination of TAT-FADD
4.9. Plasmid Constructs and Transfection
4.10. Cell Viability and Toxicity and Apoptotic Cell Death Analysis
4.11. Flow Cytometry for Apoptotic Death Analysis
4.12. Isolation of RNA and Real-Time-qPCR
4.13. Subcellular Fractions
4.14. Co-Immunoprecipitation Assay
4.15. Western Blotting
4.16. NF-κB Luciferase Reporter Assay
4.17. Measurement of Caspase 8 and Caspase 3 Activity
4.18. Measurement of Mitochondrial Membrane Potential (ΔΨm)
4.19. Enzyme-Linked Immunosorbent Assays (ELISA)
4.20. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mouasni, S.; Tourneur, L. Fadd at the crossroads between cancer and inflammation. Trends Immunol. 2018, 39, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.H.; Wu, C.; Walsh, C.M. Emerging roles for the death adaptor fadd in death receptor avidity and cell cycle regulation. Cell Cycle 2006, 5, 2332–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The fas-fadd death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavrik, I.; Krueger, A.; Schmitz, I.; Baumann, S.; Weyd, H.; Krammer, P.H.; Kirchhoff, S. The active caspase-8 heterotetramer is formed at the cd95 disc. Cell Death Differ. 2003, 10, 144–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.K.; Hawkins, C.J. Mammalian initiator apoptotic caspases. FEBS J. 2005, 272, 5436–5453. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Okano, H.; Shiraki, K.; Inoue, H.; Kawakita, T.; Yamanaka, T.; Deguchi, M.; Sugimoto, K.; Sakai, T.; Ohmori, S.; Fujikawa, K.; et al. Cellular flice/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab. Investig. 2003, 83, 1033–1043. [Google Scholar] [CrossRef] [Green Version]
- Ram, D.R.; Ilyukha, V.; Volkova, T.; Buzdin, A.; Tai, A.; Smirnova, I.; Poltorak, A. Balance between short and long isoforms of cflip regulates fas-mediated apoptosis in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1606–1611. [Google Scholar] [CrossRef] [Green Version]
- Alappat, E.C.; Feig, C.; Boyerinas, B.; Volkland, J.; Samuels, M.; Murmann, A.E.; Thorburn, A.; Kidd, V.J.; Slaughter, C.A.; Osborn, S.L.; et al. Phosphorylation of fadd at serine 194 by ckialpha regulates its nonapoptotic activities. Mol. Cell 2005, 19, 321–332. [Google Scholar] [CrossRef]
- Gomez-Angelats, M.; Cidlowski, J.A. Molecular evidence for the nuclear localization of fadd. Cell Death Differ. 2003, 10, 791–797. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.W.; Kim, J.H.; Ahn, Y.H.; Seo, J.; Ko, A.; Jeong, M.; Kim, S.J.; Ro, J.Y.; Park, K.M.; Lee, H.W.; et al. Ubiquitination and degradation of the fadd adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat. Commun. 2012, 3, 978. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Bhojani, M.S.; Heaford, A.C.; Chang, D.C.; Laxman, B.; Thomas, D.G.; Griffin, L.B.; Yu, J.; Coppola, J.M.; Giordano, T.J.; et al. Phosphorylated fadd induces nf-kappab, perturbs cell cycle, and is associated with poor outcome in lung adenocarcinomas. Proc. Natl. Acad. Sci. USA 2005, 102, 12507–12512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Rubio, J.L.; Vela-Martin, L.; Fernandez-Piqueras, J.; Villa-Morales, M. Fadd in cancer: Mechanisms of altered expression and function, and clinical implications. Cancers 2019, 11, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourneur, L.; Delluc, S.; Levy, V.; Valensi, F.; Radford-Weiss, I.; Legrand, O.; Vargaftig, J.; Boix, C.; Macintyre, E.A.; Varet, B.; et al. Absence or low expression of fas-associated protein with death domain in acute myeloid leukemia cells predicts resistance to chemotherapy and poor outcome. Cancer Res. 2004, 64, 8101–8108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourneur, L.; Mistou, S.; Michiels, F.M.; Devauchelle, V.; Renia, L.; Feunteun, J.; Chiocchia, G. Loss of fadd protein expression results in a biased fas-signaling pathway and correlates with the development of tumoral status in thyroid follicular cells. Oncogene 2003, 22, 2795–2804. [Google Scholar] [CrossRef] [Green Version]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Nam, S.W.; Park, W.S.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Mutation of fadd gene is rare in human colon and stomach cancers. APMIS 2004, 112, 595–597. [Google Scholar] [CrossRef]
- Korkolopoulou, P.; Saetta, A.A.; Levidou, G.; Gigelou, F.; Lazaris, A.; Thymara, I.; Scliri, M.; Bousboukea, K.; Michalopoulos, N.V.; Apostolikas, N.; et al. C-flip expression in colorectal carcinomas: Association with fas/fasl expression and prognostic implications. Histopathology 2007, 51, 150–156. [Google Scholar] [CrossRef]
- Ranjan, K.; Pathak, C. Fadd regulates nf-kappab activation and promotes ubiquitination of cflipl to induce apoptosis. Sci. Rep. 2016, 6, 22787. [Google Scholar] [CrossRef]
- Ranjan, K.; Surolia, A.; Pathak, C. Apoptotic potential of fas-associated death domain on regulation of cell death regulatory protein cflip and death receptor mediated apoptosis in hek 293t cells. J. Cell Commun. Signal. 2012, 6, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, K.; Pathak, C. Expression of fadd and cflipl balances mitochondrial integrity and redox signaling to substantiate apoptotic cell death. Mol. Cell. Biochem. 2016, 422, 135–150. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. Nf-kappab regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. Nf-kappab, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Eluard, B.; Thieblemont, C.; Baud, V. Nf-kappab in the new era of cancer therapy. Trends Cancer 2020, 6, 677–687. [Google Scholar] [CrossRef]
- Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Krappmann, D. Mechanisms and consequences of constitutive nf-kappab activation in b-cell lymphoid malignancies. Oncogene 2014, 33, 5655–5665. [Google Scholar] [CrossRef] [Green Version]
- Sau, A.; Lau, R.; Cabrita, M.A.; Nolan, E.; Crooks, P.A.; Visvader, J.E.; Pratt, M.A. Persistent activation of nf-kappab in brca1-deficient mammary progenitors drives aberrant proliferation and accumulation of DNA damage. Cell Stem Cell 2016, 19, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Karin, M. Nf-kappab, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of nlrp3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Bannerman, D.D.; Tupper, J.C.; Kelly, J.D.; Winn, R.K.; Harlan, J.M. The fas-associated death domain protein suppresses activation of nf-kappa b by lps and il-1 beta. J. Clin. Investig. 2002, 109, 419–425. [Google Scholar] [CrossRef]
- Mouasni, S.; Gonzalez, V.; Schmitt, A.; Bennana, E.; Guillonneau, F.; Mistou, S.; Avouac, J.; Ea, H.K.; Devauchelle, V.; Gottenberg, J.E.; et al. The classical nlrp3 inflammasome controls fadd unconventional secretion through microvesicle shedding. Cell Death Dis. 2019, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Ishizaka, Y.; Okada, T.; Kondo, Y.; Hitomi, M.; Tanaka, Y.; Haqqi, T.; Barnett, G.H.; Barna, B.P. Fadd gene therapy for malignant gliomas in vitro and in vivo. Hum. Gene Ther. 1998, 9, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Komata, T.; Koga, S.; Hirohata, S.; Takakura, M.; Germano, I.M.; Inoue, M.; Kyo, S.; Kondo, S.; Kondo, Y. A novel treatment of human malignant gliomas in vitro and in vivo: Fadd gene transfer under the control of the human telomerase reverse transcriptase gene promoter. Int. J. Oncol. 2001, 19, 1015–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.W.; Zhang, C.M.; Huang, X.J.; Zhang, X.X.; Zhang, L.K.; Li, J.H.; Hua, Z.C. Tumor-targeted delivery of a c-terminally truncated fadd (n-fadd) significantly suppresses the b16f10 melanoma via enhancing apoptosis. Sci. Rep. 2016, 6, 34178. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Okamoto, K.; Kobata, T.; Hasunuma, T.; Kato, T.; Hamada, H.; Nishioka, K. Novel gene therapy for rheumatoid arthritis by fadd gene transfer: Induction of apoptosis of rheumatoid synoviocytes but not chondrocytes. Gene Ther. 2000, 7, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.W.; Luther, D.C.; Kretzmann, J.A.; Burden, A.; Jeon, T.; Zhai, S.; Rotello, V.M. Protein delivery into the cell cytosol using non-viral nanocarriers. Theranostics 2019, 9, 3280–3292. [Google Scholar] [CrossRef]
- Liu, X.; Wu, F.; Ji, Y.; Yin, L. Recent advances in anti-cancer protein/peptide delivery. Bioconjug. Chem. 2019, 30, 305–324. [Google Scholar] [CrossRef]
- Zhang, Y.; Roise, J.J.; Lee, K.; Li, J.; Murthy, N. Recent developments in intracellular protein delivery. Curr. Opin. Biotechnol. 2018, 52, 25–31. [Google Scholar] [CrossRef]
- Jafari, B.; Pourseif, M.M.; Barar, J.; Rafi, M.A.; Omidi, Y. Peptide-mediated drug delivery across the blood-brain barrier for targeting brain tumors. Expert Opin. Drug Deliv. 2019, 16, 583–605. [Google Scholar] [CrossRef]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharm. 2005, 145, 1093–1102. [Google Scholar] [CrossRef]
- Ferrari, A.; Pellegrini, V.; Arcangeli, C.; Fittipaldi, A.; Giacca, M.; Beltram, F. Caveolae-mediated internalization of extracellular hiv-1 tat fusion proteins visualized in real time. Mol. Ther. 2003, 8, 284–294. [Google Scholar] [CrossRef]
- Fittipaldi, A.; Ferrari, A.; Zoppe, M.; Arcangeli, C.; Pellegrini, V.; Beltram, F.; Giacca, M. Cell membrane lipid rafts mediate caveolar endocytosis of hiv-1 tat fusion proteins. J. Biol. Chem. 2003, 278, 34141–34149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudt, L.M. Oncogenic activation of nf-kappab. Cold Spring Harb. Perspect. Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Golks, A.; Brenner, D.; Fritsch, C.; Krammer, P.H.; Lavrik, I.N. C-flipr, a new regulator of death receptor-induced apoptosis. J. Biol. Chem. 2005, 280, 14507–14513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micheau, O.; Tschopp, J. Induction of tnf receptor i-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J. Ubiquitin signalling in the nf-kappab pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef] [Green Version]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of ikk by tnfalpha requires site-specific ubiquitination of rip1 and polyubiquitin binding by nemo. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Gaynor, R.B. Ikappab kinases: Key regulators of the nf-kappab pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef]
- Baratchian, M.; Davis, C.A.; Shimizu, A.; Escors, D.; Bagneris, C.; Barrett, T.; Collins, M.K. Distinct activation mechanisms of nf-kappab regulator inhibitor of nf-kappab kinase (ikk) by isoforms of the cell death regulator cellular flice-like inhibitory protein (cflip). J. Biol. Chem. 2016, 291, 7608–7620. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, T.; Tschopp, J. N-terminal fragment of c-flip(l) processed by caspase 8 specifically interacts with traf2 and induces activation of the nf-kappab signaling pathway. Mol. Cell. Biol. 2004, 24, 2627–2636. [Google Scholar] [CrossRef] [Green Version]
- Golks, A.; Brenner, D.; Krammer, P.H.; Lavrik, I.N. The c-flip-nh2 terminus (p22-flip) induces nf-kappab activation. J. Exp. Med. 2006, 203, 1295–1305. [Google Scholar] [CrossRef]
- Tourneur, L.; Chiocchia, G. Fadd: A regulator of life and death. Trends Immunol. 2010, 31, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Cimino, Y.; Costes, A.; Damotte, D.; Validire, P.; Mistou, S.; Cagnard, N.; Alifano, M.; Regnard, J.F.; Chiocchia, G.; Sautes-Fridman, C.; et al. Fadd protein release mirrors the development and aggressiveness of human non-small cell lung cancer. Br. J. Cancer 2012, 106, 1989–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Rubio, J.L.; de Arriba, M.C.; Cobos-Fernandez, M.A.; Gonzalez-Sanchez, L.; Ors, I.; Sastre, I.; Fernandez-Piqueras, J.; Villa-Morales, M. Deregulated fadd expression and phosphorylation in t-cell lymphoblastic lymphoma. Oncotarget 2016, 7, 61485–61499. [Google Scholar] [CrossRef]
- Marin-Rubio, J.L.; Perez-Gomez, E.; Fernandez-Piqueras, J.; Villa-Morales, M. S194-p-fadd as a marker of aggressiveness and poor prognosis in human t-cell lymphoblastic lymphoma. Carcinogenesis 2019, 40, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Foger, N.; Bulfone-Paus, S.; Chan, A.C.; Lee, K.H. Subcellular compartmentalization of fadd as a new level of regulation in death receptor signaling. FEBS J. 2009, 276, 4256–4265. [Google Scholar] [CrossRef]
- Stewart, M.P.; Sharei, A.; Ding, X.; Sahay, G.; Langer, R.; Jensen, K.F. In vitro and ex vivo strategies for intracellular delivery. Nature 2016, 538, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Goverdhana, S.; Puntel, M.; Xiong, W.; Zirger, J.M.; Barcia, C.; Curtin, J.F.; Soffer, E.B.; Mondkar, S.; King, G.D.; Hu, J.; et al. Regulatable gene expression systems for gene therapy applications: Progress and future challenges. Mol. Ther. 2005, 12, 189–211. [Google Scholar] [CrossRef]
- Essafi, M.; Baudot, A.D.; Mouska, X.; Cassuto, J.P.; Ticchioni, M.; Deckert, M. Cell-penetrating tat-foxo3 fusion proteins induce apoptotic cell death in leukemic cells. Mol. Cancer Ther. 2011, 10, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Orzechowska, E.J.; Kozlowska, E.; Czubaty, A.; Kozlowski, P.; Staron, K.; Trzcinska-Danielewicz, J. Controlled delivery of bid protein fused with tat peptide sensitizes cancer cells to apoptosis. BMC Cancer 2014, 14, 771. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.M.; Frederiksen, J.K.; Zacharias, D.A.; Chan, F.K.; Johnson, M.; Lynch, D.; Tsien, R.Y.; Lenardo, M.J. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 2000, 288, 2354–2357. [Google Scholar] [CrossRef] [Green Version]
- Orzechowska, E.J.; Girstun, A.; Staron, K.; Trzcinska-Danielewicz, J. Synergy of bid with doxorubicin in the killing of cancer cells. Oncol. Rep. 2015, 33, 2143–2150. [Google Scholar] [CrossRef]
- Karin, M.; Lin, A. Nf-kappab at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.T.; Jasmin, A.; Kumar, A.; Liu, L.; Hood, L. Activation of the nf-kappab pathway by caspase 8 and its homologs. Oncogene 2000, 19, 4451–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.H.; Johnson, H.; Shu, H.B. Activation of nf-kappab by fadd, casper, and caspase-8. J. Biol. Chem. 2000, 275, 10838–10844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smale, S.T. Hierarchies of nf-kappab target-gene regulation. Nat. Immunol. 2011, 12, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Bossaller, L.; Chiang, P.I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting edge: Fas (cd95) mediates noncanonical il-1beta and il-18 maturation via caspase-8 in an rip3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. Fadd and caspase-8 mediate priming and activation of the canonical and noncanonical nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, K.; Sharma, A.; Surolia, A.; Pathak, C. Regulation of ha14-1 mediated oxidative stress, toxic response, and autophagy by curcumin to enhance apoptotic activity in human embryonic kidney cells. BioFactors 2014, 40, 157–169. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjan, K.; Waghela, B.N.; Vaidya, F.U.; Pathak, C. Cell-Penetrable Peptide-Conjugated FADD Induces Apoptosis and Regulates Inflammatory Signaling in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 6890. https://doi.org/10.3390/ijms21186890
Ranjan K, Waghela BN, Vaidya FU, Pathak C. Cell-Penetrable Peptide-Conjugated FADD Induces Apoptosis and Regulates Inflammatory Signaling in Cancer Cells. International Journal of Molecular Sciences. 2020; 21(18):6890. https://doi.org/10.3390/ijms21186890
Chicago/Turabian StyleRanjan, Kishu, Bhargav N Waghela, Foram U Vaidya, and Chandramani Pathak. 2020. "Cell-Penetrable Peptide-Conjugated FADD Induces Apoptosis and Regulates Inflammatory Signaling in Cancer Cells" International Journal of Molecular Sciences 21, no. 18: 6890. https://doi.org/10.3390/ijms21186890