Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses


Abstract
1. Introduction
2. Current Treatments for Neuropathic MPS
2.1. General and Specific Treatments
2.2. Current Enzyme Replacement Therapies and Their Limitations
3. Central Nervous System Pathology in Neuropathic MPS
3.1. Enzyme Deficiency and Substance Accumulation in CNS
3.2. Neuropathological Progression in Neuropathic MPS
3.3. GAG Derived Neuropathology and Its Transition to Clinical CNS Symptoms
4. Novel ERTs for Neuropathic MPS
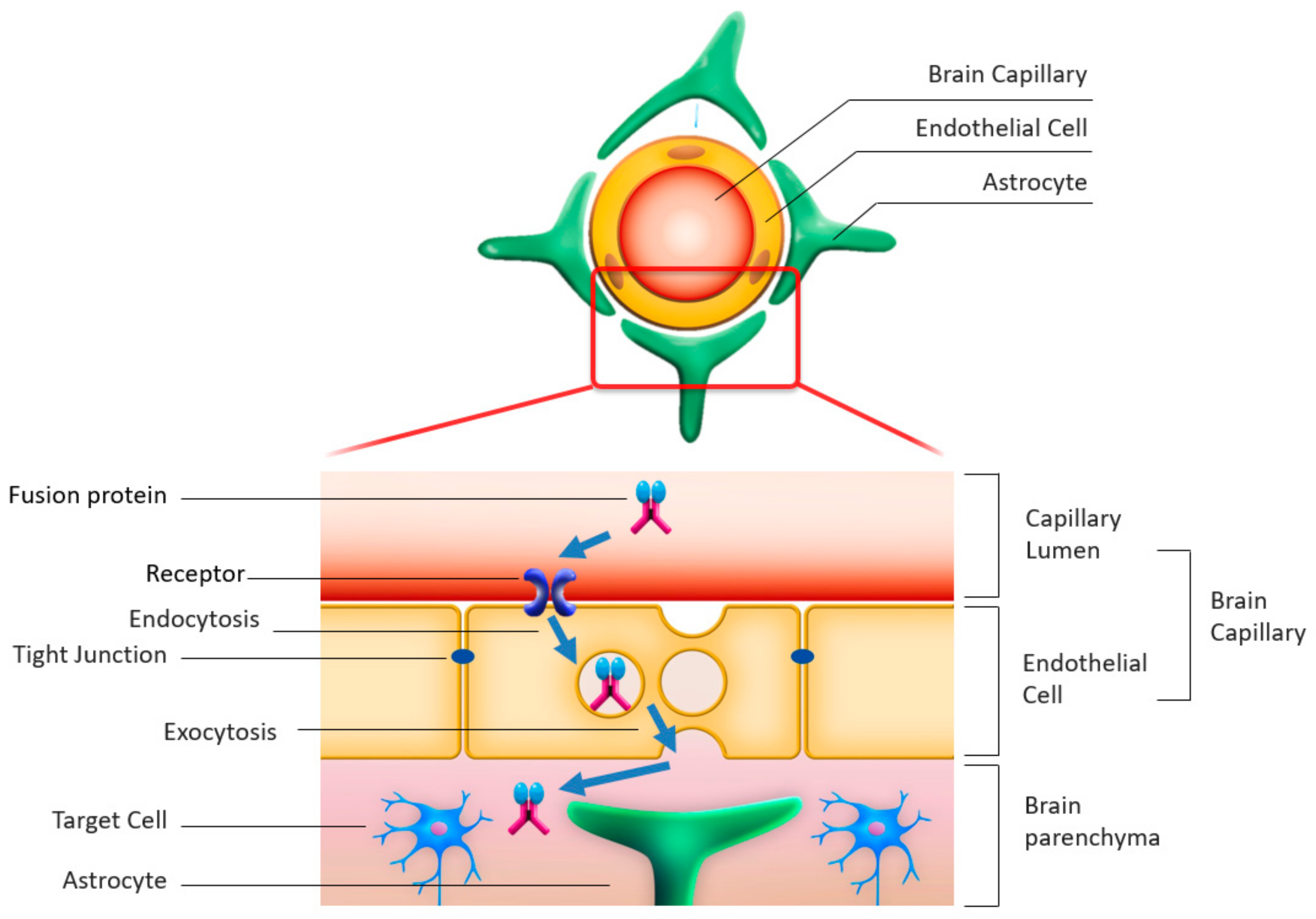
4.1. Rationale for Enzyme Delivery across the BBB
4.2. Enzyme Modulation by Fusion with Antibodies Enabling Transcytosis
4.3. Clinical Trials for Neuropathic MPS
4.4. Methodological Challenges in Clinical Development for Neuropathic MPS
5. Paths Forward
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cox, T.M. Current Treatments. In Lysosomal Storage Disorders—A Practical Guide; Mehta, A., Winchester, B., Eds.; Wiley-Blackwell: Chichester, UK, 2012; pp. 153–165. [Google Scholar]
- Giugliani, R.; Vairo, F.; Kubaski, F.; Poswar, F.; Riegel, M.; Baldo, G.; Saute, J.A.M. Neurological manifestations of lysosomal disorders and emerging therapies targeting the CNS. Lancet Child Adolesc. Health 2018, 2, 56–68. [Google Scholar] [CrossRef]
- Giuliani, R.; Giuliani, L.; de Oliveira Poswar, F.; Donis, K.C.; Corte, A.D.; Schmidt, M.; Boado, R.J.; Nestrasil, I.; Nguyen, C.; Chen, S.; et al. Neurocognitive and somatic stabilization in pediatric patients with severe Mucopolysaccharidosis Type I after 52 weeks of intravenous brain-penetrating insulin receptor antibody-iduronidase fusion protein (valanafusp alpha): An open label phase 1–2 trial. Orphanet J. Rare Dis. 2018, 13, 110. [Google Scholar] [CrossRef]
- Sonoda, H.; Morimoto, H.; Yoden, E.; Koshimura, Y.; Kinoshita, M.; Golovina, G.; Takagi, H.; Yamamoto, R.; Minami, K.; Mizoguchi, A.; et al. A Blood-Brain-Barrier-Penetrating Anti-human Transferrin Receptor Antibody Fusion Protein for Neuronopathic Mucopolysaccharidosis II. Mol. Ther. 2018, 26, 1366–1374. [Google Scholar] [CrossRef]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef]
- Eisengart, J.B.; Pierpont, E.I.; Kaizer, A.M.; Rudser, K.D.; King, K.E.; Pasquali, M.; Polgree, L.E.; Dickson, P.I.; Le, S.Q.; Miller, W.P.; et al. Intrathecal enzyme replacement for Hurler syndrome: Biomarker association with neurocognitive outcomes. Genet. Med. 2019, 21, 2552–2560. [Google Scholar] [CrossRef]
- Muenzer, J.; Hendriksz, C.J.; Fan, Z.; Vijayaraghavan, S.; Perry, V.; Santra, S.; Solanki, G.A.; Mascelli, M.A.; Pan, L.; Wang, N.; et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet. Med. 2016, 18, 73–81. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Ko, A.R.; Seong, M.R.; Lee, S.; Kim, M.R.; Cho, S.Y.; Kim, J.S.; Sakaguchi, M.; Nakazawa, T.; Kosuga, M.; et al. The efficacy of intracerebroventricular idursulfase-beta enzyme replacement therapy in mucopolysaccharidosis II murine model: Heparan sulfate in cerebrospinal fluid as a clinical biomarker of neuropathology. J. Inherit. Metab. Dis. 2018, 41, 1235–1246. [Google Scholar] [CrossRef]
- Beck, M. Treatment strategies for lysosomal storage disorders. Med. Child Neurol. 2018, 60, 13–18. [Google Scholar] [CrossRef]
- Escolar, M.L.; Jones, S.A.; Shapiro, E.G.; Horovitz, D.D.G.; Lampe, C.; Amartino, H. Practical management of behavioral problems in mucopolysaccharidoses disorders. Mol. Genet. Metab. 2017, 122, 35–40. [Google Scholar] [CrossRef]
- Scarpa, M.; Lourenço, C.M.; Amartino, H. Epilepsy in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2017, 122, 55–61. [Google Scholar] [CrossRef]
- Almeciga-Diaz, C.; Barrera, L.A. Enyme Relacement Therapy for MPS: The Effects and Limitations. In Mucopolysaccharidoses Update (2 Volume Set); Tomtasu, S., Lavery, C., Giugliani, R., Harmatz, P.R., Scarpa, M., Wegrzyn, G., Orii, T., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2018; pp. 535–558. [Google Scholar]
- Giuliani, R. The Mucopolysaccharidoses. In Lysosomal Storage Disorders—A Practical Guide; Mehta, A., Winchester, B., Eds.; Wiley-Blackwell: Chichester, UK, 2012; pp. 94–100. [Google Scholar]
- Scarpa, M.; Orchard, P.J.; Schulz, A.; Dickson, P.I.; Haskins, M.E.; Escolar, M.L.; Giugliani, R. Treatment of brain disease in the mucopolysaccharidoses. Mol. Genet. Metab. 2017, 122, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Jakobkiewicz-Banecka, J.; Gabig-Ciminska, M.; Kloska, A.; Malinowska, M.; Piotrowska, E.; Banecka-Majkutewicz, Z.; Banecki, B.; Wegrzyn, A.; Wegrzyn, G. Glycosaminoglycans and mucopolysaccharidosis type III. Front. Biosci. 2016, 21, 1393–1409. [Google Scholar]
- Tomatsu, S.; Shimada, T.; Mason, R.W.; Montaño, A.M.; Kelly, J.; LaMarr, W.A.; Kubaski, F.; Giugliani, R.; Guha, A.; Yasuda, E.; et al. Establishment of glycosaminoglycan assays for mucopolysaccharidoses. Metabolites 2014, 11, 655–679. [Google Scholar] [CrossRef] [PubMed]
- Węgrzyn, G.; Jakóbkiewicz-Banecka, J.; Narajczyk, M.; Wiśniewski, A.; Piotrowska, E.; Gabig-Cimińska, M.; Kloska, A.; Słomińska-Wojewódzka, M.; Korzon-Burakowska, A.; Węgrzyn, A. Why are behaviors of children suffering from various neuronopathic types of mucopolysaccharidoses different? Med. Hypotheses 2010, 75, 605–609. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Jones, S.A.; Escolar, M.L. Developmental and behavioral aspects of mucopolysaccharidoses with brain manifestations—Neurological signs and symptoms. Mol. Genet. Metab. 2017, 122, 1–7. [Google Scholar] [CrossRef]
- Węgrzyn, G.; Jakóbkiewicz-Banecka, J.; Gabig-Cimińska, M.; Kloska, A.; Malinowska, M.; Moskot, M.; Piotrowska, E.; Narajczyk, M.; Banecka-Majkutewicz, Z.; Banecki, B.; et al. Mechanisms of neurodegeneration in mucopolysaccharidoses. In Mucopolysaccharidoses Update; Tomatsu, S., Lavery, C., Giugliani, R., Harmatz, P.R., Scarpa, M., Wegrzyn, G., Orii, T., Eds.; Nova Science Publishers: New York, NY, USA, 2018; Volume 1, pp. 87–113. [Google Scholar]
- Sharma, J.; di Ronza, A.; Lotfi, P.; Sardiello, M. Lysosomes and Brain Health. Annu. Rev. Neurosci. 2018, 41, 255–276. [Google Scholar] [CrossRef]
- Mohammed, E.E.; Snella, E.M.; Rutz-Mendicino, M.M.; Echevarria, F.D.; Awedikian, R.; Whitley, E.M.; Ellinwood, N.M. Accelerated clinical disease and pathology in mucopolysaccharidosis type IIIB and GalNAc transferase double knockout mice. Mol. Genet. Metab. 2012, 107, 129–135. [Google Scholar] [CrossRef]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 7, 33–150. [Google Scholar] [CrossRef]
- Jardim, L.B.; Villanueva, M.M.; de Souza, C.F.; Netto, C.B. Clinical aspects of neuropathic lysosomal storage disorders. J. Inherit. Metab. Dis. 2010, 33, 315–329. [Google Scholar] [CrossRef]
- Guidolin, D.; Marcoli, M.; Maura, G.; Agnati, L.F. New dimensions of connectomics and network plasticity in the central nervous system. Rev. Neurosci. 2017, 28, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.J.; Quarta, E.; Bravi, R.; Granato, A.; Minciacchi, D. Neural plasticity and network remodeling: From concepts to pathology. Neuroscience 2017, 344, 326–345. [Google Scholar] [CrossRef] [PubMed]
- Agnati, L.F.; Zoli, M.; Biagini, G.; Fuxe, K. Neuronal plasticity and ageing processes in the frame of the ‘Red Queen Theory’. Acta Physiol. Scand. 1992, 145, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Parkinson’s disease and Parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef] [PubMed]
- Rüb, U.; Seidel, K.; Heinsen, H.; Vonsattel, J.P.; den Dunnen, W.F.; Korf, H.W. Huntington’s disease (HD): The neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. 2016, 26, 726–740. [Google Scholar] [CrossRef]
- Kobayashi, H.; Ariga, M.; Sato, Y.; Fujiwara, M.; Fukasawa, N.; Fukuda, T.; Takahashi, H.; Ikegami, M.; Kosuga, M.; Okuyama, T.; et al. P-Tau and Subunit c Mitochondrial ATP Synthase Accumulation in the Central Nervous System of a Woman with Hurler–Scheie Syndrome Treated with Enzyme Replacement Therapy for 12 Years. In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer: Berlin, Germany, 2018; Volume 41, pp. 101–107. [Google Scholar]
- David, A.; Fleminger, S.; Kopelman, M.; Lovestone, S.; Mellers, J. Lishman’s Organic Psychiatry: A Textbook of Neuropsychiatry, 4th ed.; Wiley-Blackwell: Chichester, UK, 2009; pp. 845–905. [Google Scholar]
- Barichello, T.; Collodel, A.; Hasbun, R.; Morales, R. An Overview of the Blood-Brain Barrier. In Blood-Brain Barrier; Barichello, T., Ed.; Neuromethods, Humana Press: New York, NY, USA, 2019; Volume 142, pp. 1–8. [Google Scholar]
- Jolly, R.D.; Marshall, N.R.; Marshall, J.; Hartman, A.; Hemsley, K.M.; Winner, L.K. Intracisternal enzyme replacement therapy in lysosomal storage diseases: Dispersal pathways, regional enzyme concentrations and the effect of posttreatment posture. Neuropathol. Appl. Neurobiol. 2013, 39, 681–692. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human blood-brain barrier insulin receptor. J. Neurochem. 1985, 44, 1771–1778. [Google Scholar] [CrossRef]
- Golden, P.L.; Maccagnan, T.J.; Pardridge, W.M. Human blood-brain barrier leptin receptor. Binding and endocytosis in isolated human brain microvessels. J. Clin. Investig. 1997, 99, 14–18. [Google Scholar] [CrossRef]
- Fishman, J.B.; Rubin, J.B.; Handrahan, J.V.; Connor, J.R.; Fine, R.E. Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J. Neurosci. Res. 1987, 18, 299–304. [Google Scholar] [CrossRef]
- Jefferies, W.A.; Brandon, M.R.; Hunt, S.V.; Williams, A.F.; Gatter, K.C.; Mason, D.Y. Transferrin receptor on endothelium of brain capillaries. Nature 1984, 312, 162–163. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human blood-brain barrier transferrin receptor. Metabolism 1987, 36, 892–895. [Google Scholar] [CrossRef]
- Boado, R.J.; Ka-Wai Hui, E.; Zhiqiang Lu, J.; Pardridge, W.M. Insulin receptor antibody-iduronate 2-sulfatase fusion protein: Pharmacokinetics, anti-drug antibody, and safety pharmacology in Rhesus monkeys. Biotechnol. Bioeng. 2014, 111, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.H.; Boado, R.J.; Lu, J.Z.; Hui, E.K.; Pardridge, W.M. Brain-penetrating IgG-iduronate 2-sulfatase fusion protein for the mouse. Drug Metab. Dispos. 2012, 40, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Jones, S.A.; Tylki-Szymańska, A.; Harmatz, P.; Mendelsohn, N.J.; Guffon, N.; Giugliani, R.; Burton, B.K.; Scarpa, M.; Beck, M.; et al. Ten years of the Hunter Outcome Survey (HOS): Insights, achievements, and lessons learned from a global patient registry. Orphanet J. Rare Dis. 2017, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://denalitherapeutics.com/investors/press-release/denali-therapeutics-receives-orphan-drug-and-rare-pediatric-disease-designation-for-dnl310-and-expands-its-portfolio-of-brain-penetrant-enzyme-replacement-programs (accessed on 3 October 2019).
- Available online: http://www.globalgreencross.com/newsroom_detail?b_idx=536 (accessed on 3 October 2019).
- Available online: https://clinicaltrials.gov/ct2/show/NCT02262338 (accessed on 3 October 2019).
- Available online: https://www.clinicaltrials.gov/ct2/show/NCT03811028 (accessed on 3 October 2019).
- Available online: https://www.clinicaltrials.gov/ct2/show/NCT02754076 (accessed on 3 October 2019).
- Whitley, C.B.; Vijay, S.; Yao, B.; Pineda, M.; Parker, G.J.M.; Rojas-Caro, S.; Zhang, X.; Dai, Y.; Cinar, A.; Bubb, G.; et al. Final results of the phase 1/2, open-label clinical study of intravenous recombinant human N-acetyl-α-d-glucosaminidase (SBC-103) in children with mucopolysaccharidosis IIIB. Mol. Genet. Metab. 2019, 126, 131–138. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Escolar, M.L.; Delaney, K.A.; Mitchell, J.J. Assessments of neurocognitive and behavioral function in the mucopolysaccharidoses. Mol. Genet. Metab. 2017, 122, 8–16. [Google Scholar] [CrossRef]
- Whiteman, D.A.; Kimura, A. Development of idursulfase therapy for mucopolysaccharidosis type II (Hunter syndrome): The past, the present and the future. Drug Des. Dev. Ther. 2017, 11, 2467–2480. [Google Scholar] [CrossRef]
- Van der Lee, J.H.; Morton, J.; Adams, H.R.; Clarke, L.; Ebbink, B.J.; Escolar, M.L.; Giugliani, R.; Harmatz, P.; Hogan, M.; Jones, S.; et al. Cognitive endpoints for therapy development for neuronopathic mucopolysaccharidoses: Results of a consensus procedure. Mol. Genet. Metab. 2017, 121, 70–79. [Google Scholar] [CrossRef]
- Holt, J.B.; Poe, M.D.; Escolar, M.L. Natural progression of neurological disease in mucopolysaccharidosis type II. Pediatrics 2011, 27, e1258–e1265. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Rare Diseases: Common Issues in Drug Development Guidance for Industry. February 2019. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/rare-diseases-common-issues-drug-development-guidance-industry (accessed on 3 October 2019).




{kind=link}
{kind=link}
{kind=link}
| Neuropathic MPS | Deficient Enzyme | Main GAG Stored | Neuropsychiatric Symptoms | Systemic Symptoms |
|---|---|---|---|---|
| MPS-I (Hurler syndrome; Hurler–Scheie syndrome; Scheie syndrome) | α-L-iduronidase (IDUA) | HS, DS | Hurler: severe Hurler–Scheie and Scheie: mild to absent | Wide spectrum of severity |
| MPS-II (Hunter syndrome) | Iduronate-2-sulfatase (IDS) | HS, DS | Severe (rapidly progressing phenotypes) or mild to absent (slowly progressing phenotypes) | Wide spectrum of severity |
| MPS-IIIA (Sanfilippo A syndrome) | Heparan-N-sulfatase (SGSH) | HS | Severe | Mild to absent |
| MPS-IIIB (Sanfilippo B syndrome) | α-N-acetylglucosaminidase (NAGLU) | HS | Severe | Mild to absent |
| MPS-IIIC (Sanfilippo C syndrome) | α-glucosaminidase acetyltransferase (HGSNAT) | HS | Severe | Mild to absent |
| MPS-IIID (Sanfilippo D syndrome) | N-acetylglucosamine 6-sulfatase (GNS) | HS | Severe | Mild to absent |
| MPS-VII | β-D-glucuronidase (GUSB) | HS, DS | Severe (rapidly progressing phenotypes) or mild to absent (slowly progressing phenotypes) | Wide spectrum of severity |
| MPS | Compound | Targeted Receptor to Cross BBB | Route of Administration | Developmental Status | Manufacturer/Sponsor | Source |
|---|---|---|---|---|---|---|
| MPS-I (Hurler syndrome) | Valanafusp alpha | Insulin receptor | Intravenous | Ph I/II trial completed | ArmaGen | Giugliani et al. [3] |
| Iduronidase | N/A | Intrathecal (iv ERT + HCT + intrathecal ERT) | An open label Ph I trial | Sanofi Genzyme/NIH | Eisengart et al. [6] | |
| MPS-II (Hunter syndrome) | Idursulfase-IT | N/A | Intrathecal | Ph I/II trial completed | Shire | Muenzer et al. [41] |
| DNL 310 | Transferrin receptor | Intravenous | Ph I/II trial planned | Denali therapeutics | Press release [42] | |
| Idursulfase- beta ICV | N/A | Intracerebroventricular | Ph I/II trial completed | Greencross | Press release [43] | |
| AGT-182 | Insulin receptor | Intravenous | Ph I trial completed | ArmaGen | NCT02262338 [44] | |
| JR-141 | Transferrin receptor | Intravenous | Ph I/II completed Ph III ongoing | JCR pharmaceuticals | Okuyama et al. [5] | |
| MPS-IIIA (Sanfilippo A syndrome) | SOBI003 | N/A | Intravenous | Ph I/II ongoing | Swedish Orphan Biovitrum (Sobi) | NCT03811028 [45] |
| MPS-IIIB (Sanfilippo B syndrome) | Tralesinidase alfa (BMN 250) | N/A | Intracerebroventricular | Ph I/II ongoing | BioMarin | NCT02754076 [46] |
| SBC-103 | N/A | Intravenous | Ph I/II completed | Alexion | Whitley et al. [47] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, Y.; Okuyama, T. Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 400. https://doi.org/10.3390/ijms21020400
Sato Y, Okuyama T. Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses. International Journal of Molecular Sciences. 2020; 21(2):400. https://doi.org/10.3390/ijms21020400
Chicago/Turabian StyleSato, Yuji, and Torayuki Okuyama. 2020. "Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses" International Journal of Molecular Sciences 21, no. 2: 400. https://doi.org/10.3390/ijms21020400
APA StyleSato, Y., & Okuyama, T. (2020). Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses. International Journal of Molecular Sciences, 21(2), 400. https://doi.org/10.3390/ijms21020400