Galectin-3 Modulates Macrophage Activation and Contributes Smooth Muscle Cells Apoptosis in Abdominal Aortic Aneurysm Pathogenesis


 , ,
, ,
Abstract
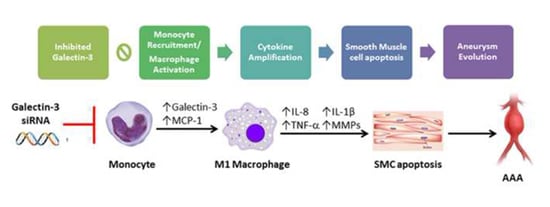
:
1. Introduction
2. Results
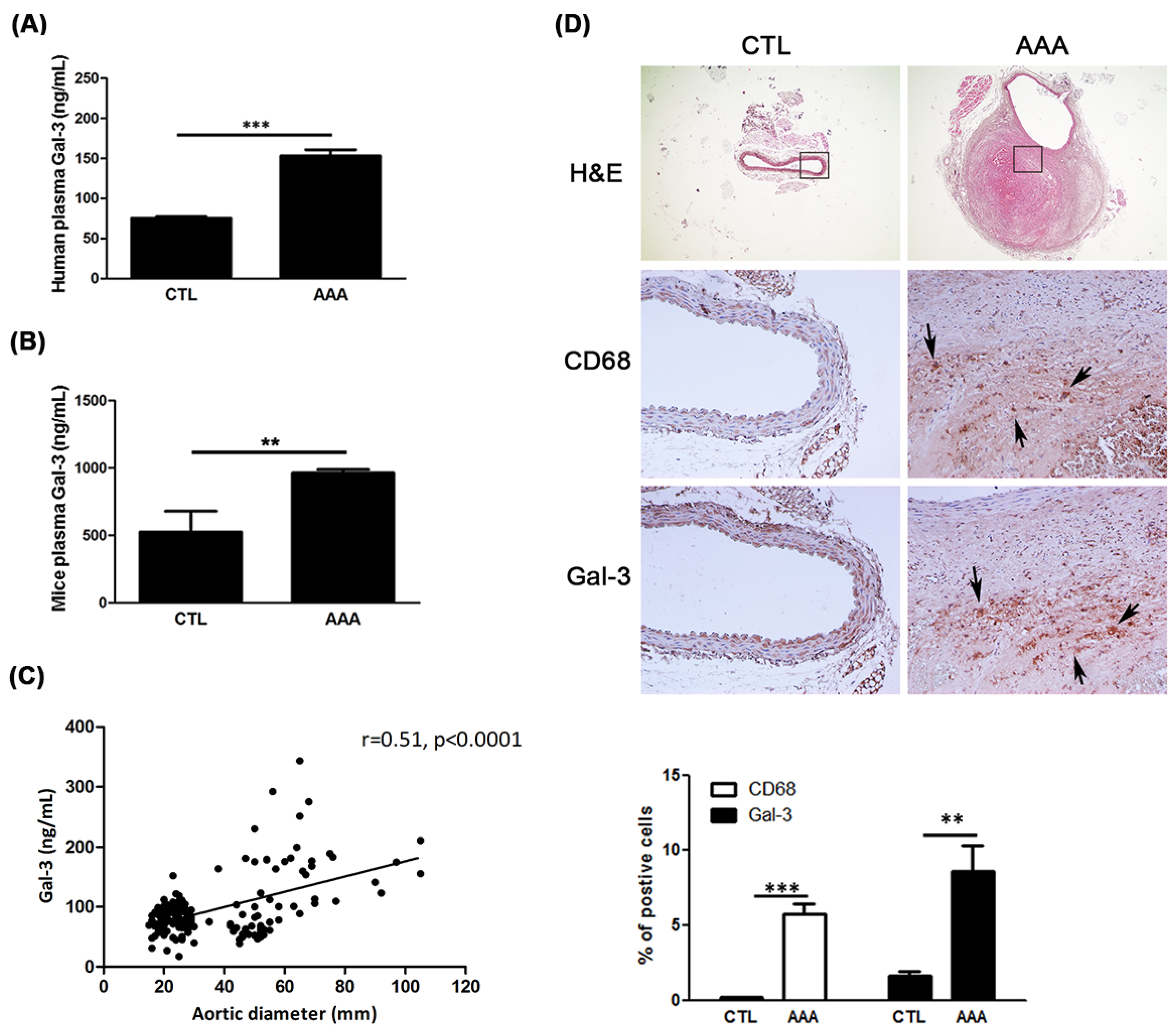
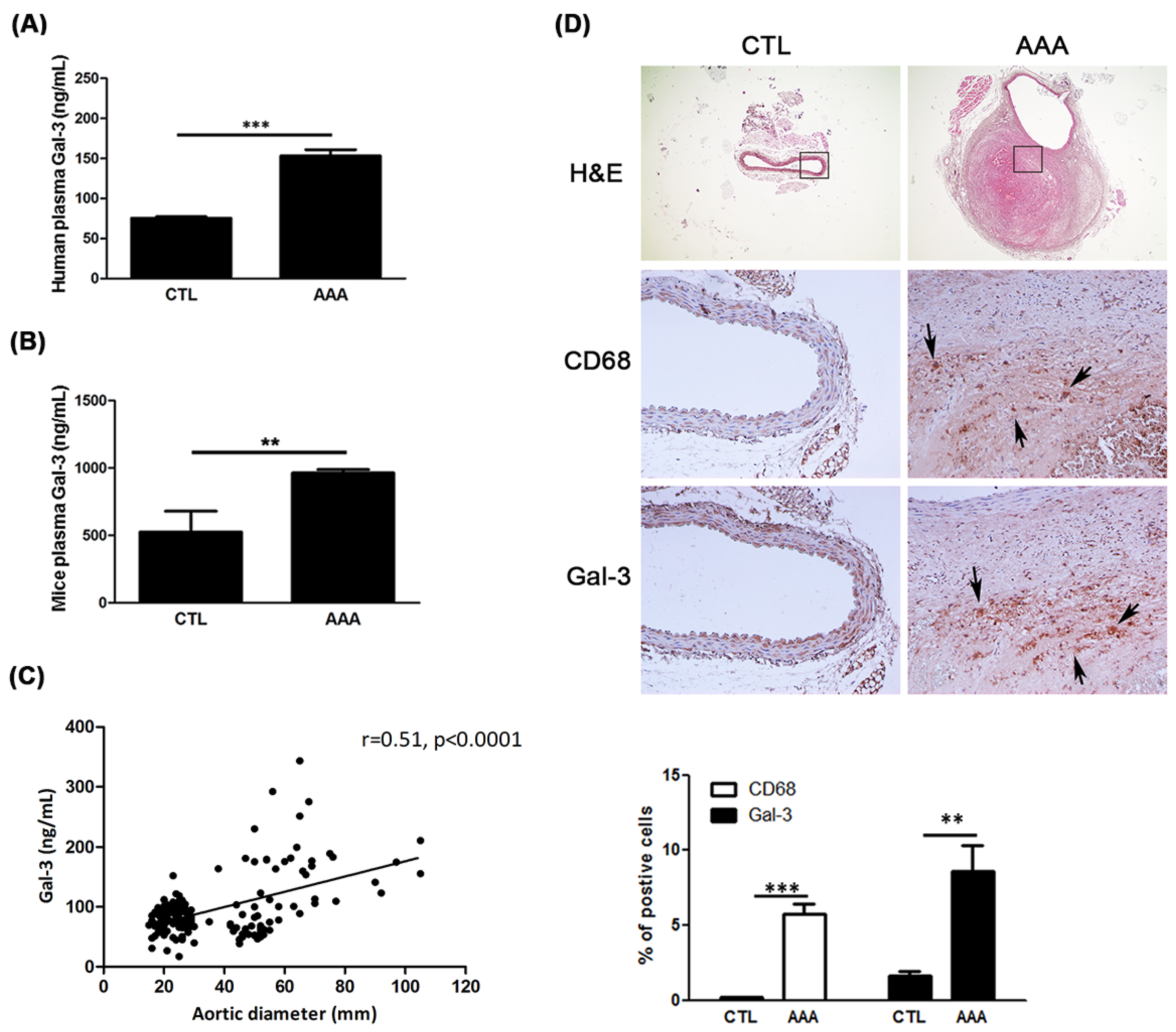
2.1. Increased Expression of Gal-3 in AAA
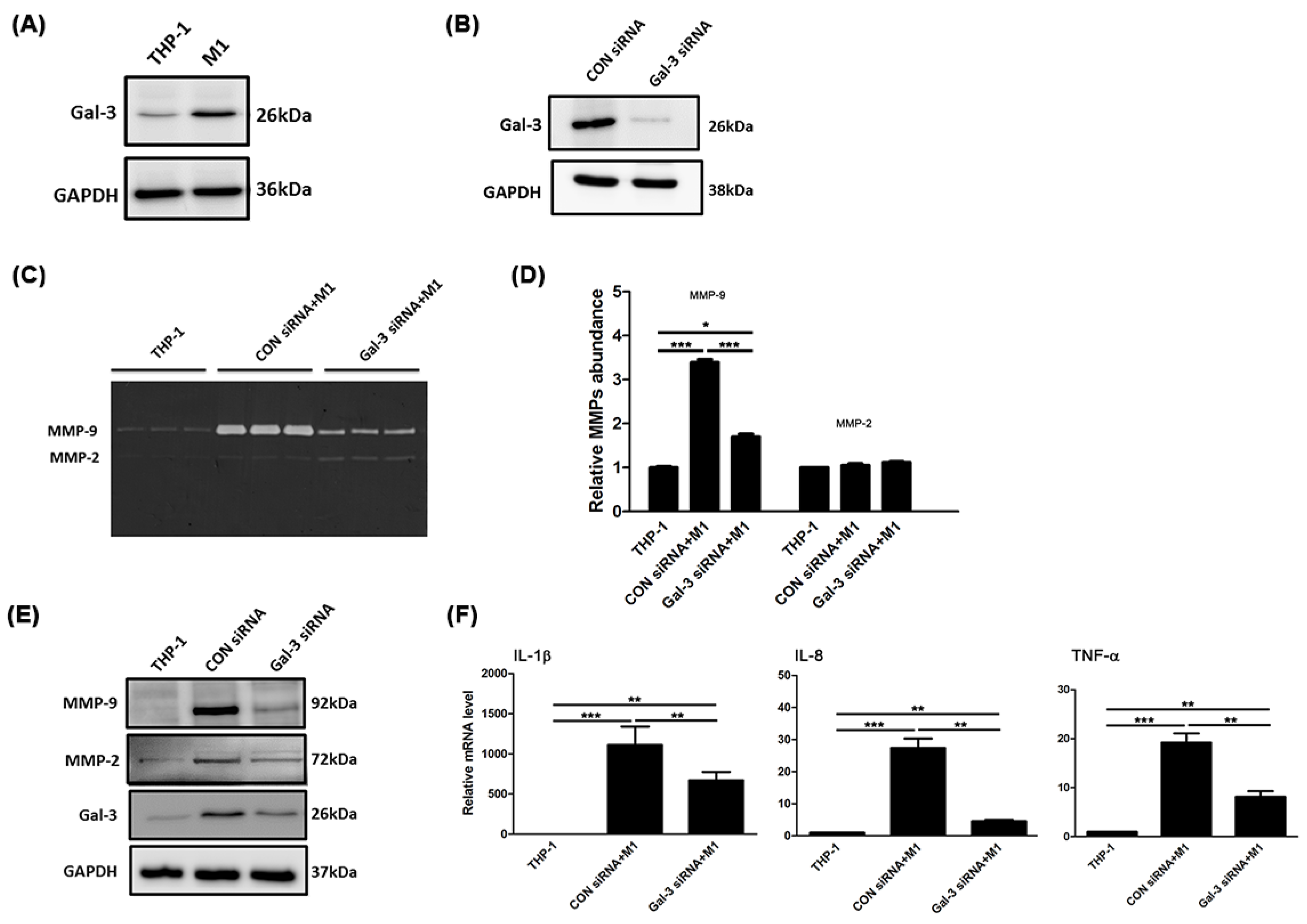
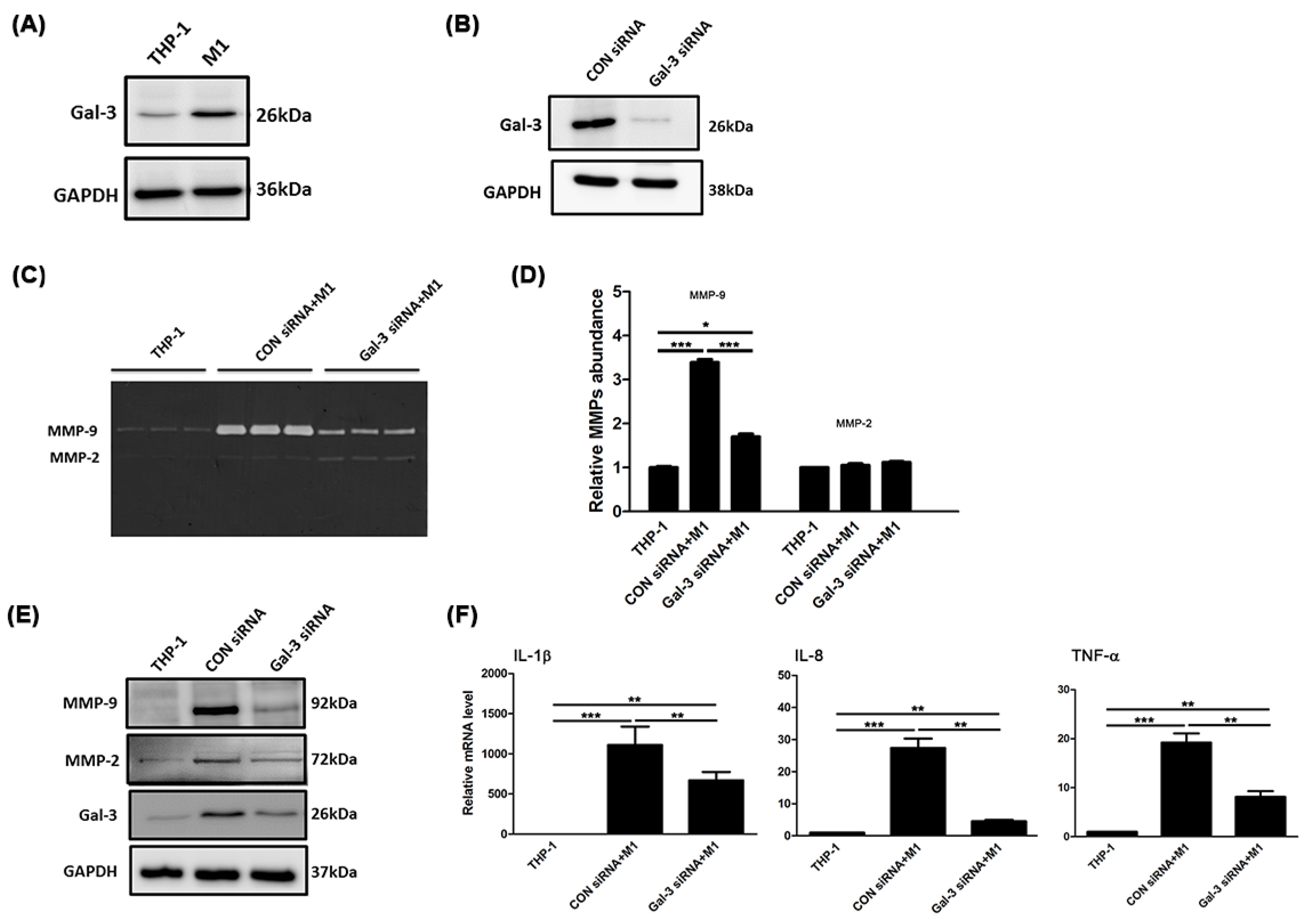
2.2. Gal-3 Modulated Inflammatory Regulators in Macrophages
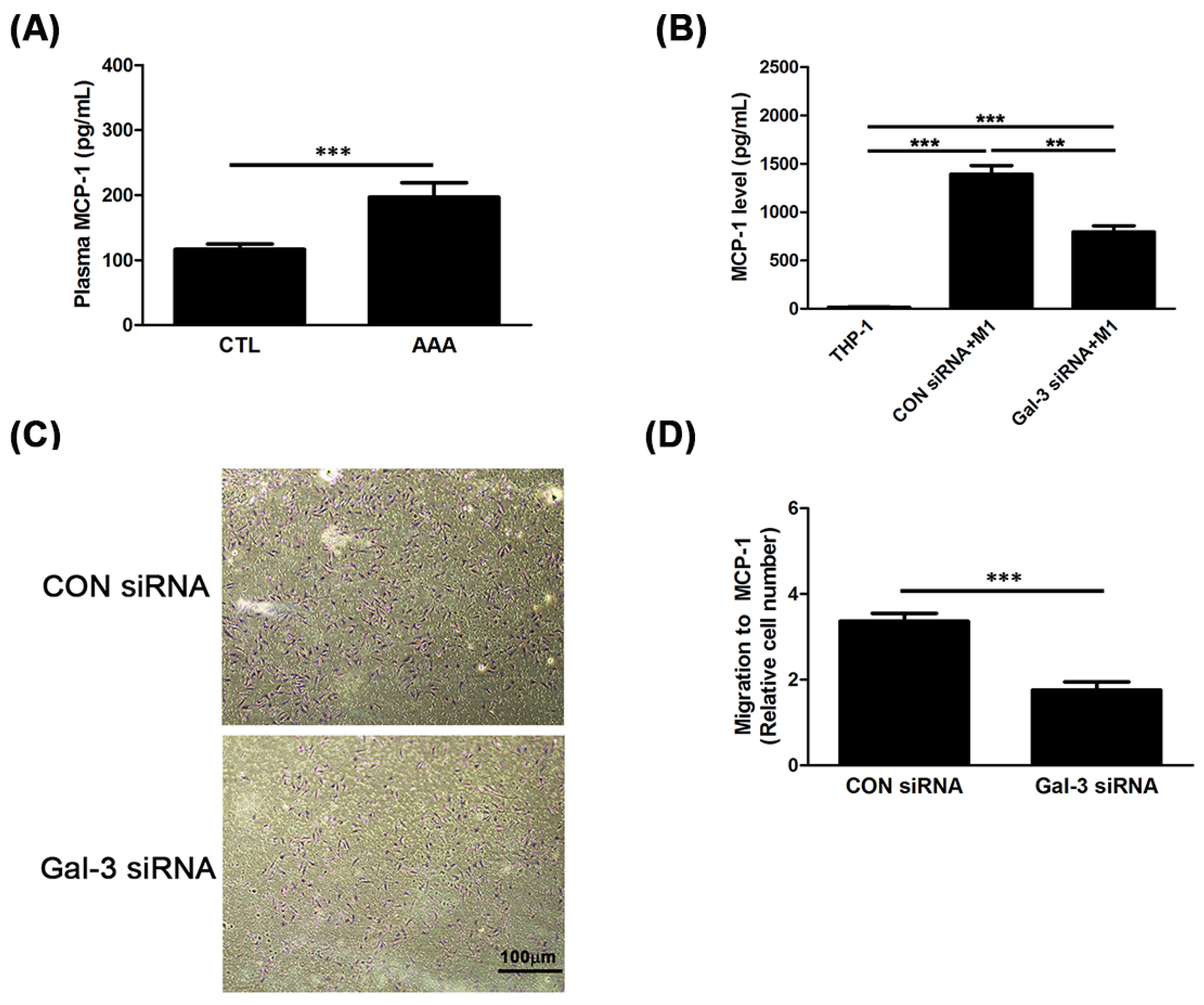
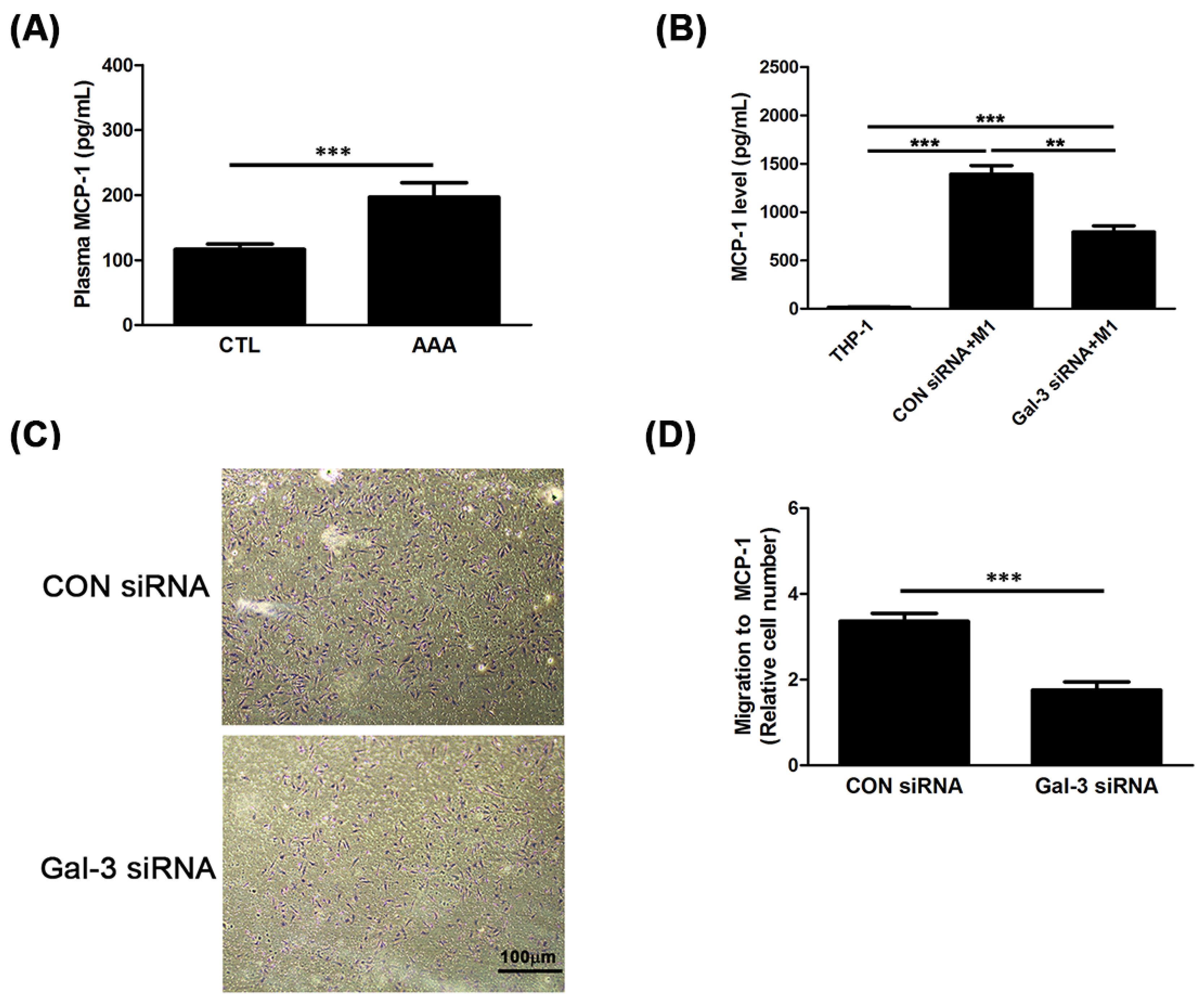
2.3. Gal-3 Promoted Up-Regulation of MCP-1 to Enhance the Chemotaxis of Monocytes
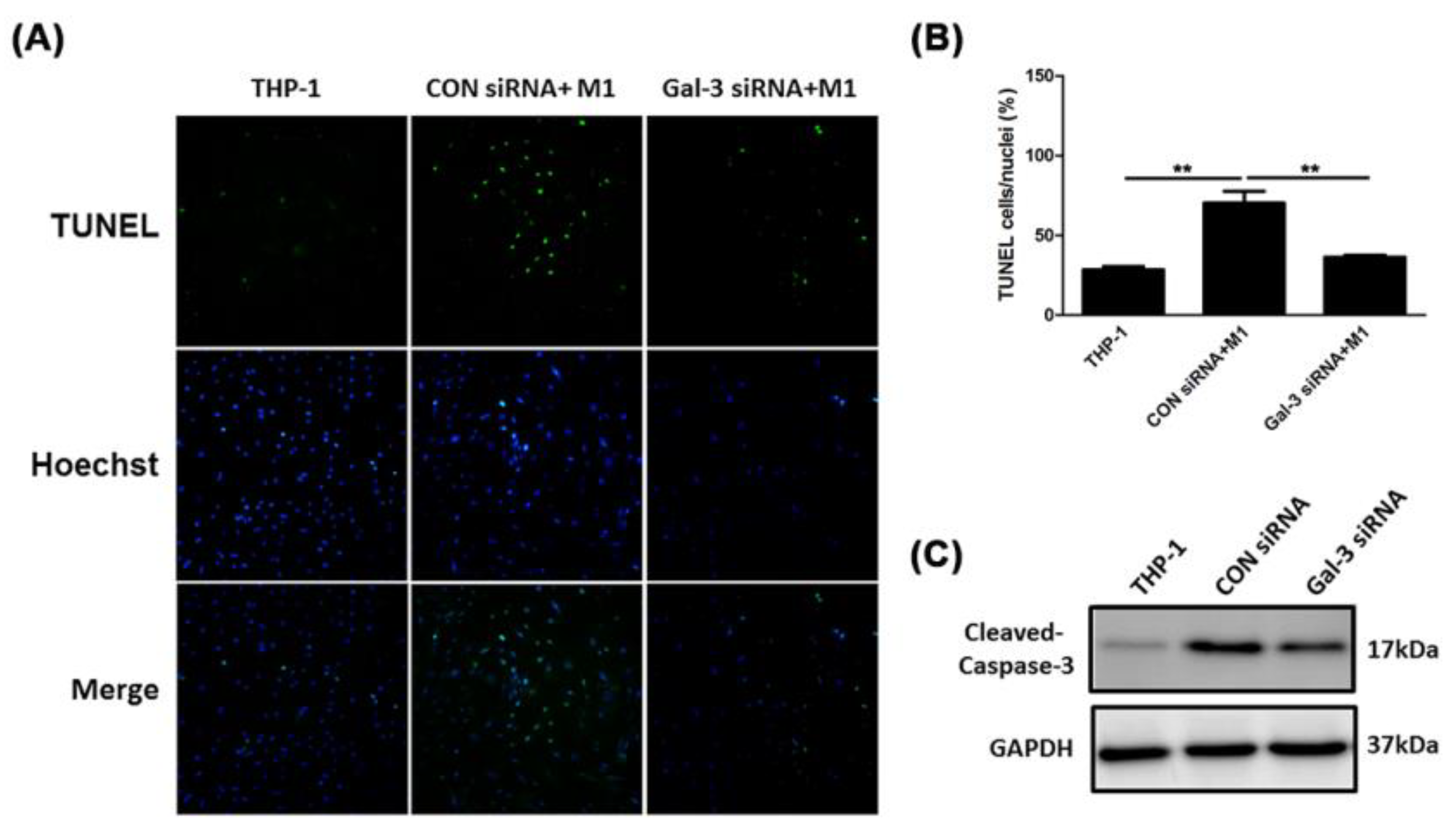
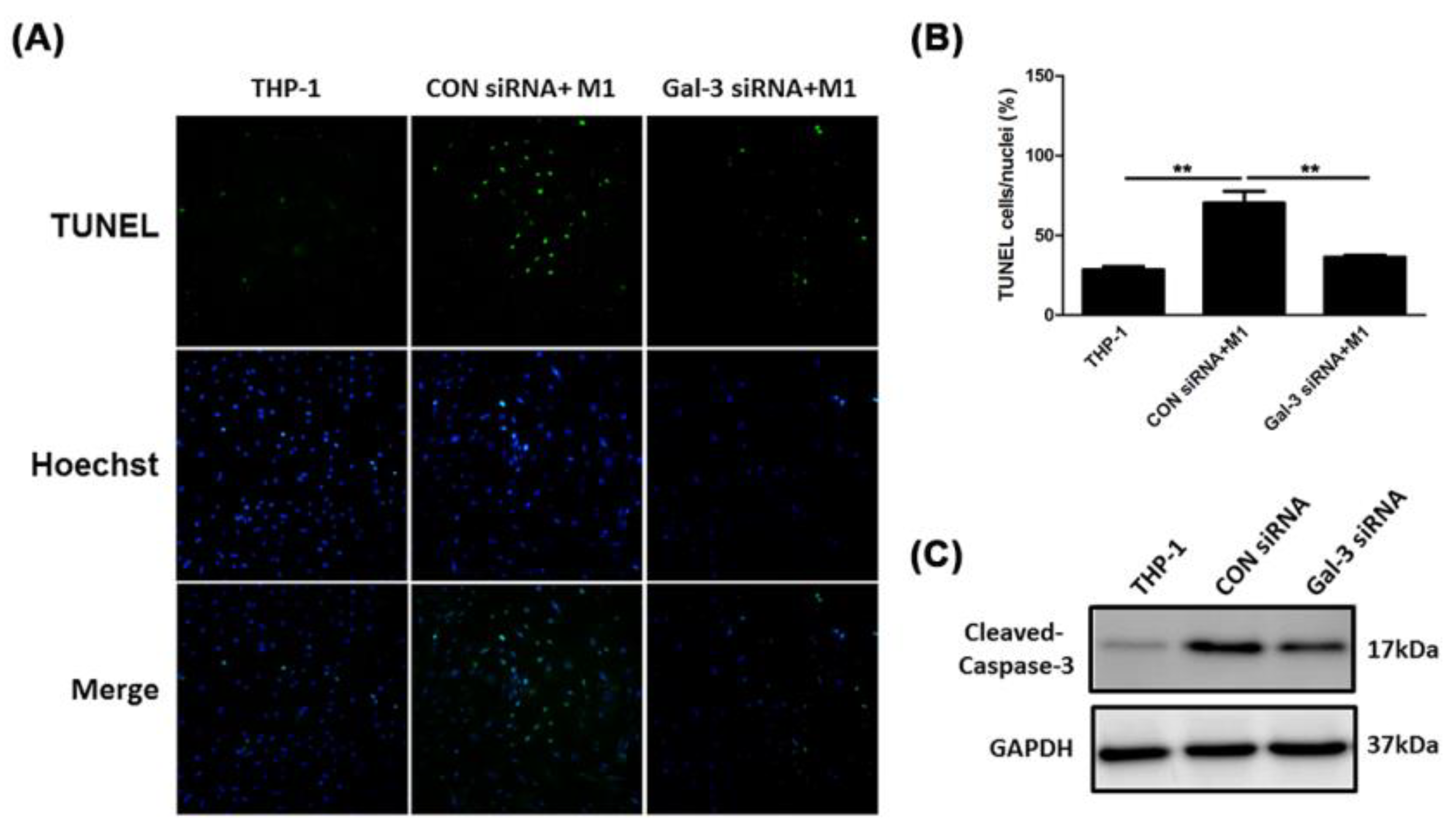
2.4. Inhibited Gal-3 Attenuated Macrophage-Induced Death of SMCs
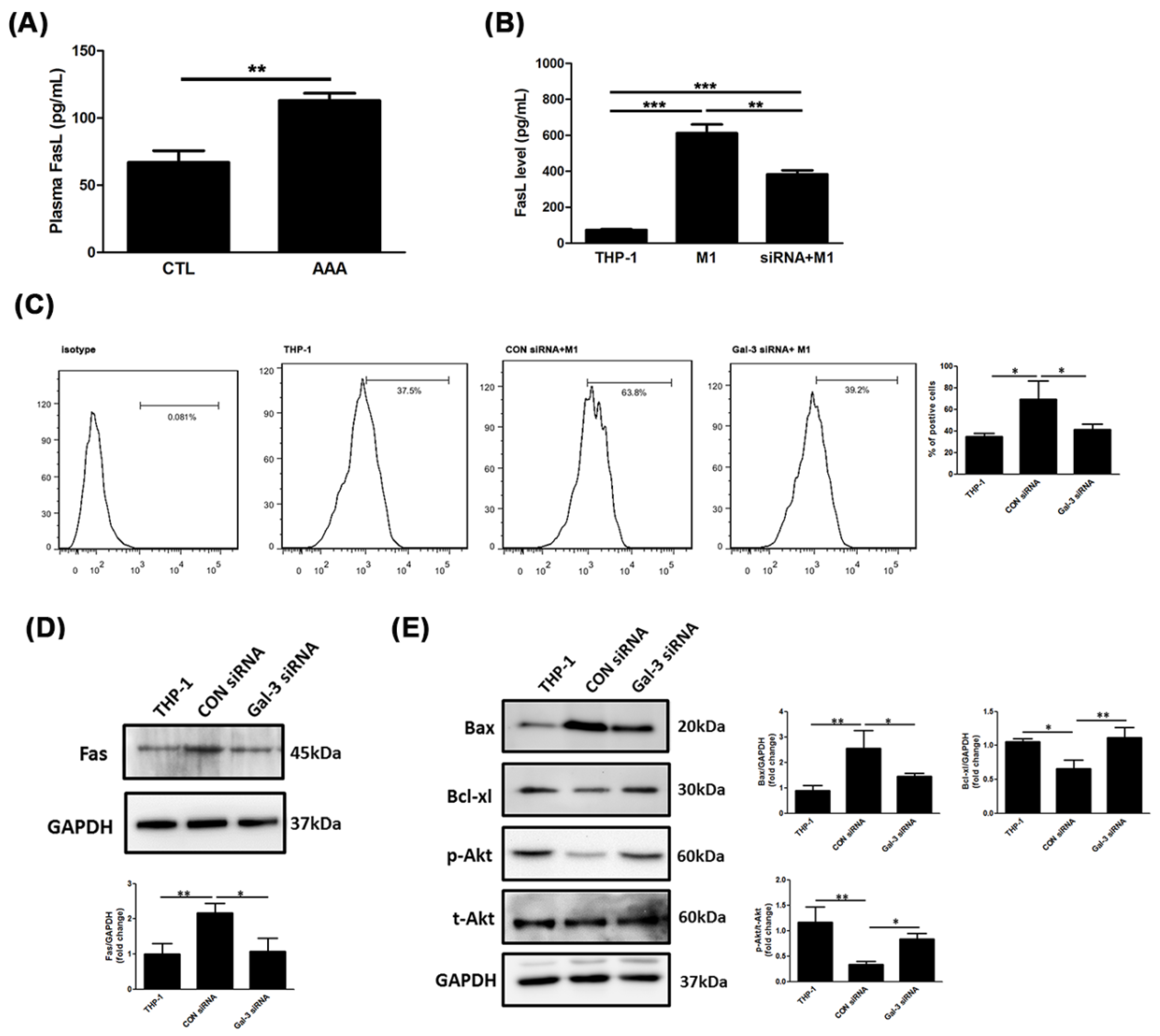
2.5. Resistance of Gal-3 to Macrophage-Induced SMC Apoptosis Associated With Fas Deactivation
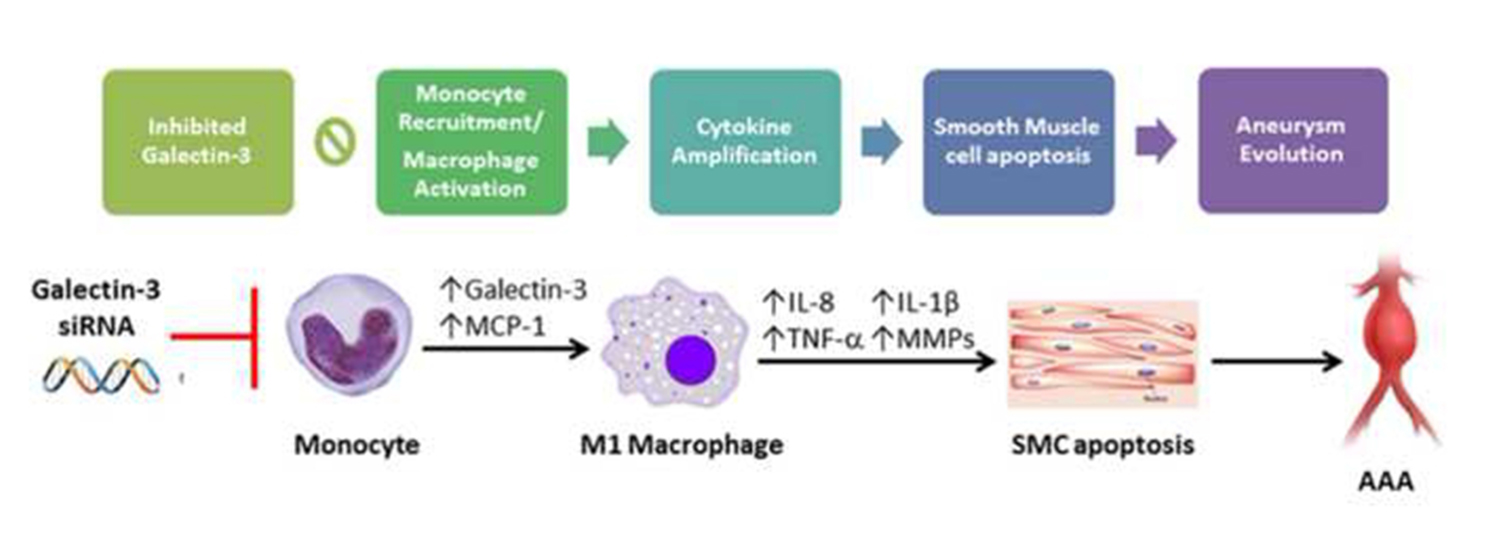
3. Discussion
4. Materials and Methods
4.1. Patient Recruitment and Blood Sample Collection
4.2. Enzyme-Linked Immunosorbent Assay
4.3. Animal Aortic Collection
4.4. Histological and Immunohistochemistical Staining
4.5. RNA Interference of Gal-3 Using Small-Interfering RNA
4.6. Cell Lines and Cell Culture
4.7. Chemotaxis Assay
4.8. Gelatin Zymography
4.9. Quantitative Real-Time Polymerase Chain Reaction
4.10. Western Blotting
4.11. Terminal Deoxynucleotidyl Transferase dUTP Nick end Labeling Staining
4.12. Flow Cytometry
4.13. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAA | Abdominal Aortic Aneurysm |
| SMC | Smooth Muscle Cell |
| MMP | Matrix Metalloproteinase |
| CREB | cAMP-response element binding protein |
| NF-κB | Nuclear factor-κB |
| CCL5 | C-C motif chemokine 5 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| EDTA | Ethylene-Diamine-Tetra-Acetic Acid |
| LPS | Lipopolysaccharide |
| IFN-γ | Interferon-gamma |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| CM | Conditioned Media |
| IL | Interleukin |
| TNF-α | Tumor Necrosis Factor-alpha |
| GAPDH | Glyceraldehyde 3-phosphate Dehydrogenase |
| TUNEL | Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling |
| RT-PCR | Reverse Transcription Polymerase Chain Reaction |
References
- Sakalihasan, N.; Limet, R.; Defawe, O.D. Abdominal aortic aneurysm. Lancet 2005, 365, 1577–1589. [Google Scholar] [CrossRef]
- Lopez-Candales, A.; Holmes, D.R.; Liao, S.; Scott, M.J.; Wickline, S.A.; Thompson, R.W. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am. J. Pathol. 1997, 150, 993–1007. [Google Scholar] [PubMed]
- McCormick, M.L.; Gavrila, D.; Weintraub, N.L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Barondes, S.H.; Castronovo, V.; Cooper, D.N.; Cummings, R.D.; Drickamer, K.; Feizi, T.; Gitt, M.A.; Hirabayashi, J.; Hughes, C.; Kasai, K.; et al. Galectins: A family of animal beta-galactoside-binding lectins. Cell 1994, 76, 597–598. [Google Scholar] [CrossRef]
- Moutsatsos, I.K.; Wade, M.; Schindler, M.; Wang, J.L. Endogenous lectins from cultured cells: Nuclear localization of carbohydrate-binding protein 35 in proliferating 3T3 fibroblasts. Proc. Natl. Acad. Sci. USA 1987, 84, 6452–6456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, H.; AlSadek, D.M. Galectin-3 as a Potential Target to Prevent Cancer Metastasis. Clin. Med. Insights Oncol. 2015, 9, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Lavine, K.J.; Epelman, S.; Evans, S.A.; Weinheimer, C.J.; Barger, P.M.; Mann, D.L. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J. Am. Heart Assoc. 2015, 4, e001993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, C.T.; Lood, C.; Ostergaard, O.; Iversen, L.V.; Voss, A.; Bengtsson, A.; Jacobsen, S.; Heegaard, N.H. Plasma levels of galectin-3-binding protein reflect type I interferon activity and are increased in patients with systemic lupus erythematosus. Lupus Sci. Med. 2014, 1, e000026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, U.C.; Pokharel, S.; van Brakel, T.J.; van Berlo, J.H.; Cleutjens, J.P.; Schroen, B.; Andre, S.; Crijns, H.J.; Gabius, H.J.; Maessen, J.; et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004, 110, 3121–3128. [Google Scholar] [CrossRef]
- De Boer, R.A.; Daniels, L.B.; Maisel, A.S.; Januzzi, J.L., Jr. State of the Art: Newer biomarkers in heart failure. Eur. J. Heart Fail. 2015, 17, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Djousse, L.; Matsumoto, C.; Petrone, A.; Weir, N.L.; Tsai, M.Y.; Gaziano, J.M. Plasma galectin 3 and heart failure risk in the Physicians’ Health Study. Eur. J. Heart Fail. 2014, 16, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Kusaka, H.; Yamamoto, E.; Hirata, Y.; Fujisue, K.; Tokitsu, T.; Sugamura, K.; Sakamoto, K.; Tsujita, K.; Kaikita, K.; Hokimoto, S.; et al. Clinical significance of plasma galectin-3 in patients with coronary artery disease. Int. J. Cardiol. 2015, 201, 532–534. [Google Scholar] [CrossRef]
- Fernandez-Garcia, C.E.; Tarin, C.; Roldan-Montero, R.; Martinez-Lopez, D.; Torres-Fonseca, M.; Lindhot, J.S.; Vega de Ceniga, M.; Egido, J.; Lopez-Andres, N.; Blanco-Colio, L.M.; et al. Increased galectin-3 levels are associated with abdominal aortic aneurysm progression and inhibition of galectin-3 decreases elastase-induced AAA development. Clin. Sci. (Lond.) 2017, 131, 2707–2719. [Google Scholar] [CrossRef]
- Holmes, D.R.; Lopez-Candales, A.; Liao, S.; Thompson, R.W. Smooth muscle cell apoptosis and p53 expression in human abdominal aortic aneurysms. Ann. N. Y. Acad. Sci. 1996, 800, 286–287. [Google Scholar] [CrossRef]
- Thompson, R.W.; Liao, S.; Curci, J.A. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron. Artery Dis. 1997, 8, 623–631. [Google Scholar] [CrossRef]
- Boyle, J.J.; Bowyer, D.E.; Weissberg, P.L.; Bennett, M.R. Human blood-derived macrophages induce apoptosis in human plaque-derived vascular smooth muscle cells by Fas-ligand/Fas interactions. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1402–1407. [Google Scholar] [CrossRef] [Green Version]
- MacSweeney, S.T.; Ellis, M.; Worrell, P.C.; Greenhalgh, R.M.; Powell, J.T. Smoking and growth rate of small abdominal aortic aneurysms. Lancet 1994, 344, 651–652. [Google Scholar] [CrossRef]
- Vardulaki, K.A.; Walker, N.M.; Day, N.E.; Duffy, S.W.; Ashton, H.A.; Scott, R.A. Quantifying the risks of hypertension, age, sex and smoking in patients with abdominal aortic aneurysm. Br. J. Surg. 2000, 87, 195–200. [Google Scholar] [CrossRef]
- Blanchard, J.F.; Armenian, H.K.; Friesen, P.P. Risk factors for abdominal aortic aneurysm: Results of a case-control study. Am. J. Epidemiol. 2000, 151, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Brady, A.R.; Thompson, S.G.; Fowkes, F.G.; Greenhalgh, R.M.; Powell, J.T.; Participants, U.K.S.A.T. Abdominal aortic aneurysm expansion: Risk factors and time intervals for surveillance. Circulation 2004, 110, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Fashanu, O.E.; Folsom, A.R.; Oyenuga, A.; Ballantyne, C.M.; Lutsey, P.L.; Tang, W. Galectin-3 and the incidence of abdominal aortic aneurysm: The atherosclerosis risk in communities (ARIC) study. Am. J. Cardiovasc. Dis. 2017, 7, 114–121. [Google Scholar]
- Dumic, J.; Dabelic, S.; Flogel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635. [Google Scholar] [CrossRef]
- Maiolino, G.; Rossitto, G.; Pedon, L.; Cesari, M.; Frigo, A.C.; Azzolini, M.; Plebani, M.; Rossi, G.P. Galectin-3 predicts long-term cardiovascular death in high-risk patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Nachtigal, M.; Ghaffar, A.; Mayer, E.P. Galectin-3 gene inactivation reduces atherosclerotic lesions and adventitial inflammation in ApoE-deficient mice. Am. J. Pathol. 2008, 172, 247–255. [Google Scholar] [CrossRef] [Green Version]
- MacKinnon, A.C.; Liu, X.; Hadoke, P.W.; Miller, M.R.; Newby, D.E.; Sethi, T. Inhibition of galectin-3 reduces atherosclerosis in apolipoprotein E-deficient mice. Glycobiology 2013, 23, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Hsu, D.K.; Yu, L.; Apgar, J.R.; Kuwabara, I.; Yamanaka, T.; Hirashima, M.; Liu, F.T. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J. Immunol. 2000, 165, 2156–2164. [Google Scholar] [CrossRef] [Green Version]
- Kahari, V.M.; Saarialho-Kere, U. Matrix metalloproteinases and their inhibitors in tumour growth and invasion. Ann. Med. 1999, 31, 34–45. [Google Scholar] [CrossRef]
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Investig. 2002, 110, 625–632. [Google Scholar] [CrossRef]
- Gong, Y.; Hart, E.; Shchurin, A.; Hoover-Plow, J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J. Clin. Investig. 2008, 118, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.; Peng, Y.; Bardeesi, A.S.; Bardisi, E.S.; Liao, X.; Luo, C. Vessel heterogeneity of TIMI frame count and its relation to P-wave dispersion in patients with coronary slow flow. J. Thorac. Dis. 2016, 8, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, I.; Liu, F.T. Galectin-3 promotes adhesion of human neutrophils to laminin. J. Immunol. 1996, 156, 3939–3944. [Google Scholar]
- Frigeri, L.G.; Zuberi, R.I.; Liu, F.T.; Epsilon, B.P. A beta-galactoside-binding animal lectin, recognizes IgE receptor (Fc epsilon RI) and activates mast cells. Biochemistry 1993, 32, 7644–7649. [Google Scholar] [CrossRef]
- Matter, C.M.; Chadjichristos, C.E.; Meier, P.; von Lukowicz, T.; Lohmann, C.; Schuler, P.K.; Zhang, D.; Odermatt, B.; Hofmann, E.; Brunner, T.; et al. Role of endogenous Fas (CD95/Apo-1) ligand in balloon-induced apoptosis, inflammation, and neointima formation. Circulation 2006, 113, 1879–1887. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.Y.; Hsu, D.K.; Liu, F.T. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 6737–6742. [Google Scholar] [CrossRef] [Green Version]
- Fukumori, T.; Takenaka, Y.; Yoshii, T.; Kim, H.R.; Hogan, V.; Inohara, H.; Kagawa, S.; Raz, A. CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res. 2003, 63, 8302–8311. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total No. | CTL | AAA | p Value |
|---|---|---|---|
| 97 | 76 | ||
| Aortic diameter (mm) | 20.1 ± 1.7 | 61.4 ± 10.8 | <0.0001 |
| Age, yr | 70.5 ± 10.3 | 78.8 ± 8.6 | 0.007 |
| Sex (male/female) | 46/51 | 66/10 | <0.001 |
| Body weight, kg | 66.3 ± 11.8 | 64.4 ± 12.6 | 0.659 |
| Height, cm | 160.4 ± 9.0 | 163.3 ± 7.9 | 0.226 |
| Hypertension (%yes) | 86.6 | 75.0 | 0.075 |
| Smoke (%yes) | 15.5 | 61.8 | <0.001 |
| Hypercholesterolemia (%yes) | 50.5 | 27.6 | 0.003 |
| DM (%yes) | 31.9 | 17.1 | 0.034 |
| Peripheral vascular disease(%yes) | 1.0 | 4.0 | 0.321 |
| COPD (%yes) | 4.1 | 19.7 | 0.001 |
| Medications: | |||
| ACEi/ARB (%yes) | 65.0 | 48.7 | 0.043 |
| Statin (%yes) | 42.3 | 25 | 0.024 |
| β-blocker (%yes) | 30.9 | 39.5 | 0.263 |
| calcium channel blocker (%yes) | 44.3 | 55.3 | 0.170 |
| Aspirin (%yes) | 8.2 | 17.1 | 0.101 |
| anti-coagulants (%yes) | 5.2 | 2.6 | 0.468 |
| Antiplatelets (%yes) | 6.2 | 31.6 | <0.001 |
| Variables | OR (95% CI) | p Value |
|---|---|---|
| Model 0 | 1.106 (1.062−1.151) | 0.000 |
| Model 1 | 1.026 (1.015−1.037) | 0.000 |
| Model 2 | 1.104 (1.057−1.152) | 0.000 |
| Model 3 | 1.018 (1.009−1.027) | 0.000 |
| Model 4 | 1.017 (1.009−1.025) | 0.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.-Y.; Shih, C.-M.; Huang, C.-Y.; Wu, A.T.H.; Cheng, T.-M.; Mi, F.-L.; Shih, C.-C. Galectin-3 Modulates Macrophage Activation and Contributes Smooth Muscle Cells Apoptosis in Abdominal Aortic Aneurysm Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8257. https://doi.org/10.3390/ijms21218257
Lu H-Y, Shih C-M, Huang C-Y, Wu ATH, Cheng T-M, Mi F-L, Shih C-C. Galectin-3 Modulates Macrophage Activation and Contributes Smooth Muscle Cells Apoptosis in Abdominal Aortic Aneurysm Pathogenesis. International Journal of Molecular Sciences. 2020; 21(21):8257. https://doi.org/10.3390/ijms21218257
Chicago/Turabian StyleLu, Hsin-Ying, Chun-Ming Shih, Chun-Yang Huang, Alexander T. H. Wu, Tsai-Mu Cheng, Fwu-Long Mi, and Chun-Che Shih. 2020. "Galectin-3 Modulates Macrophage Activation and Contributes Smooth Muscle Cells Apoptosis in Abdominal Aortic Aneurysm Pathogenesis" International Journal of Molecular Sciences 21, no. 21: 8257. https://doi.org/10.3390/ijms21218257