Targeted Stimuli-Responsive Mesoporous Silica Nanoparticles for Bacterial Infection Treatment



Abstract
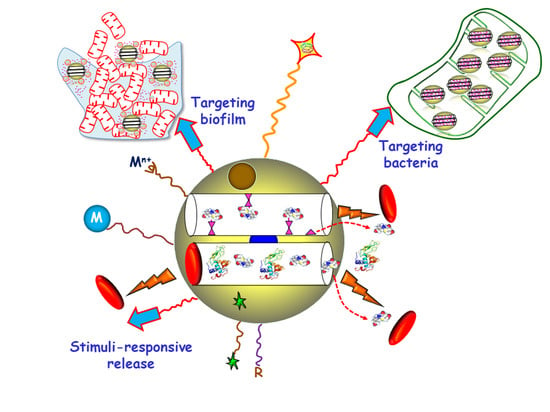
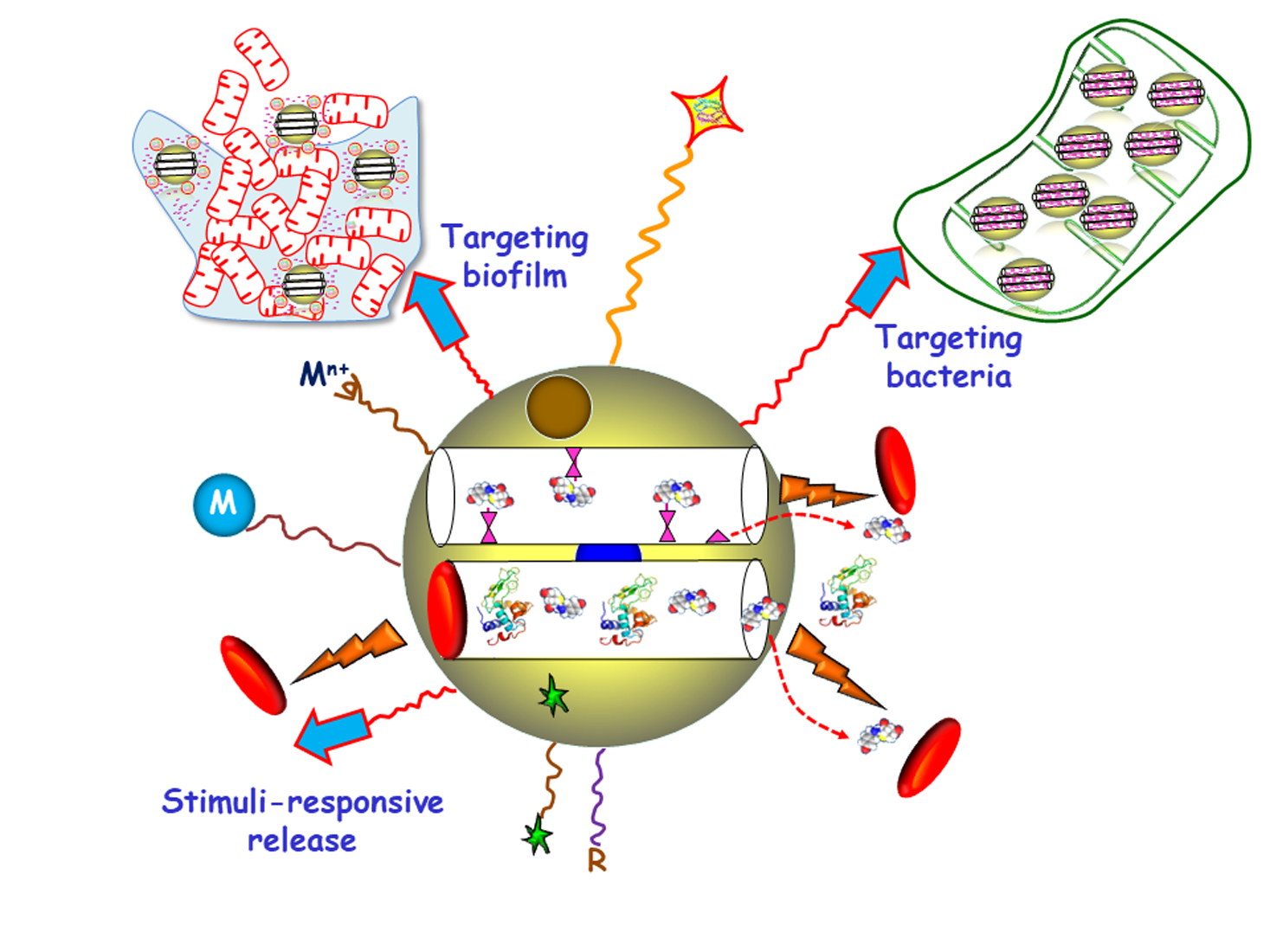
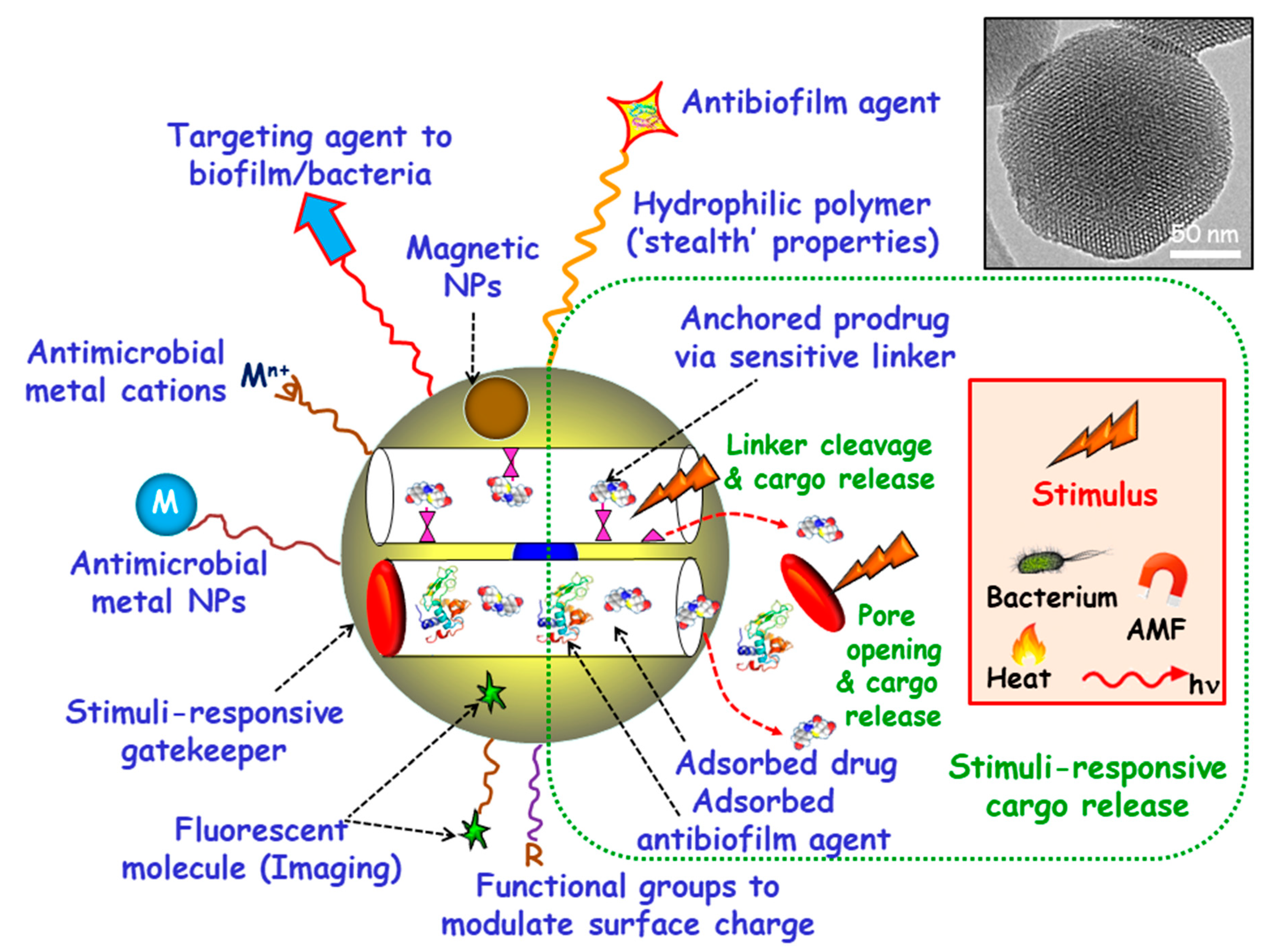
1. Introduction
2. Targeted Delivery of Antimicrobials
2.1. Targeting Bacteria
2.1.1. Antibodies
2.1.2. Aptamers
2.1.3. Peptides
2.1.4. Carbohydrates
2.1.5. Small Molecules
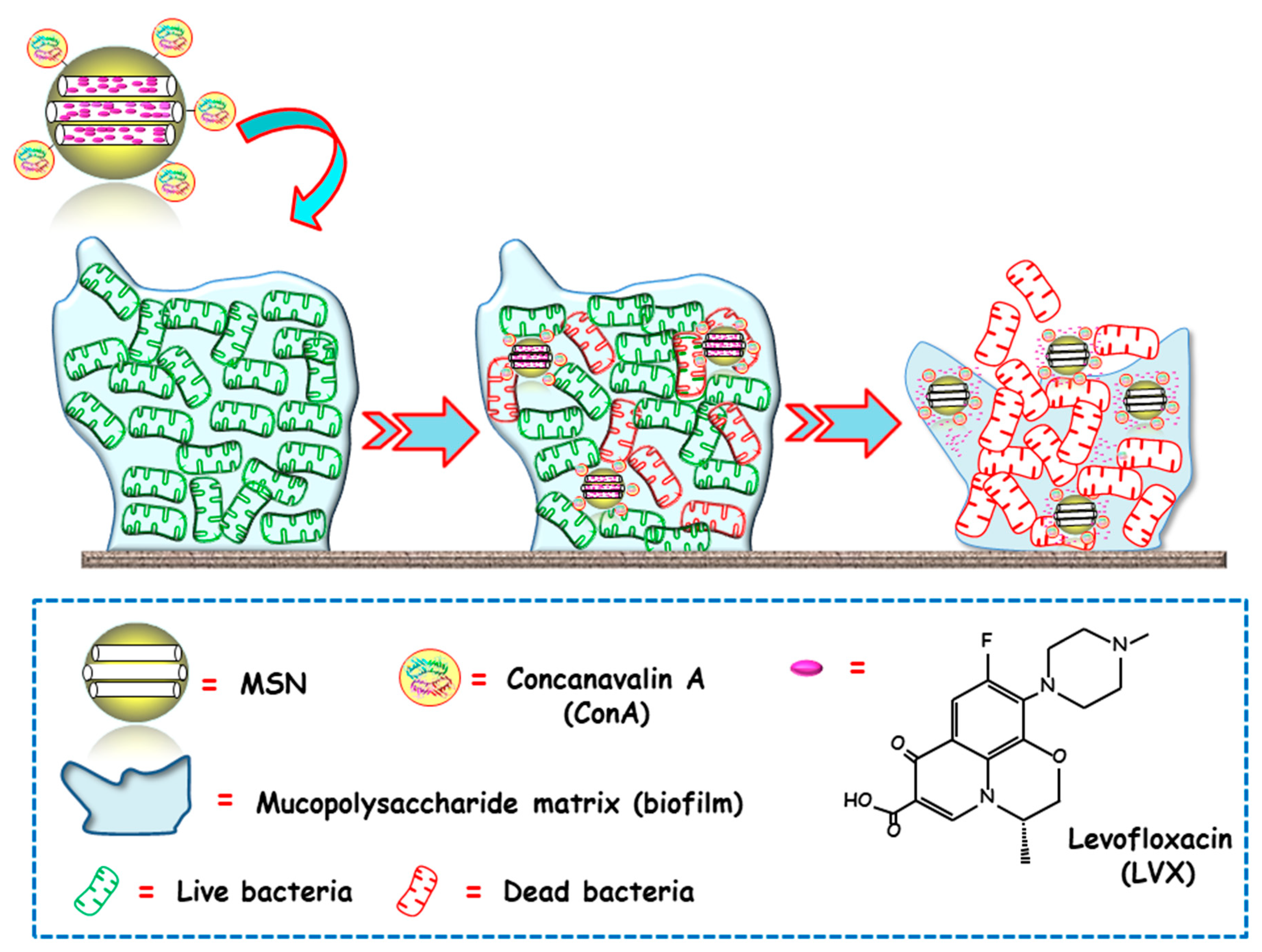
2.2. Targeting Biofilm
3. Stimuli-Responsive Antimicrobials Delivery
3.1. Internal Stimuli-Responsive MSNs
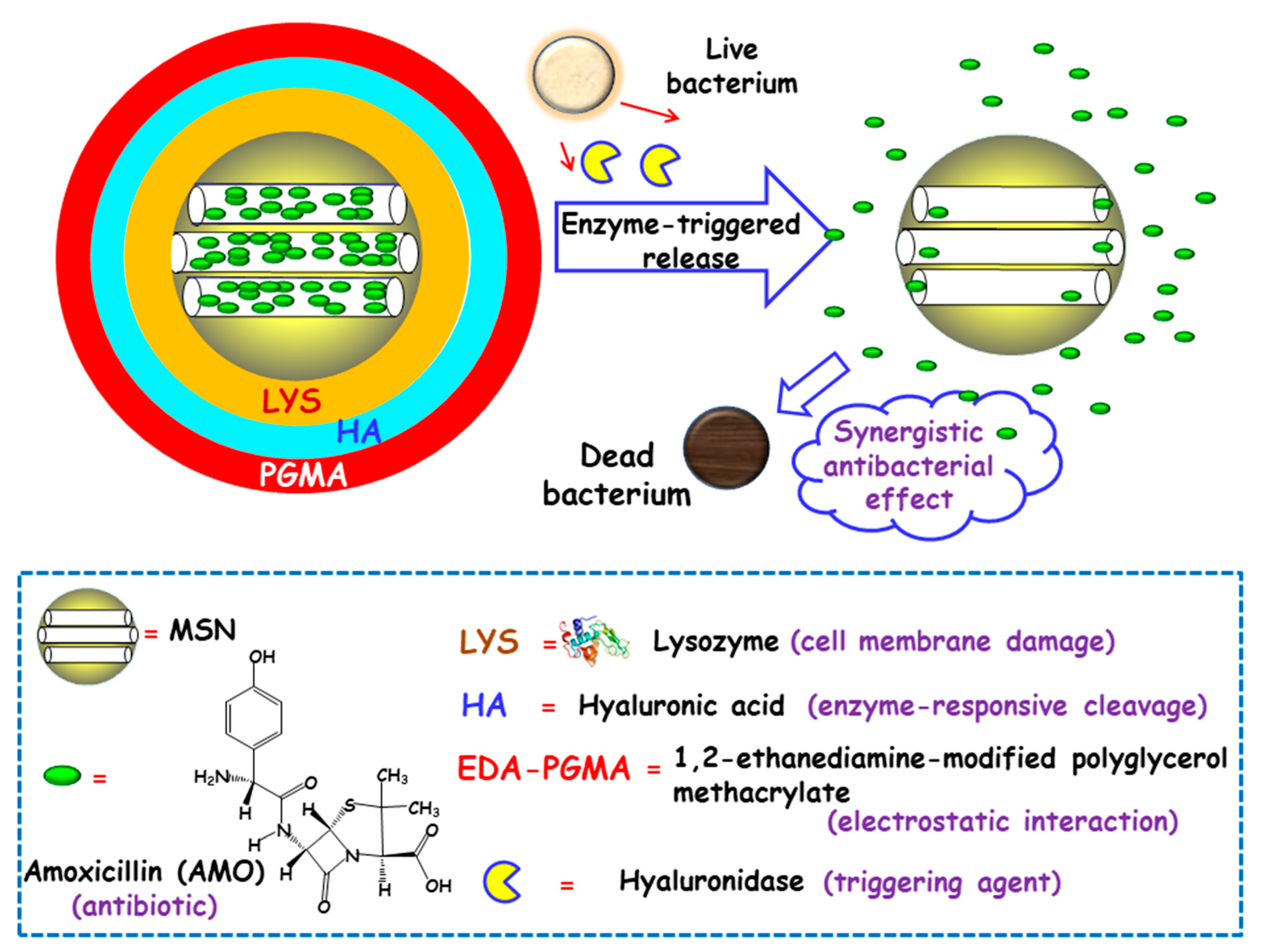
3.1.1. Presence of Bacteria
3.1.2. Bacterial Toxins
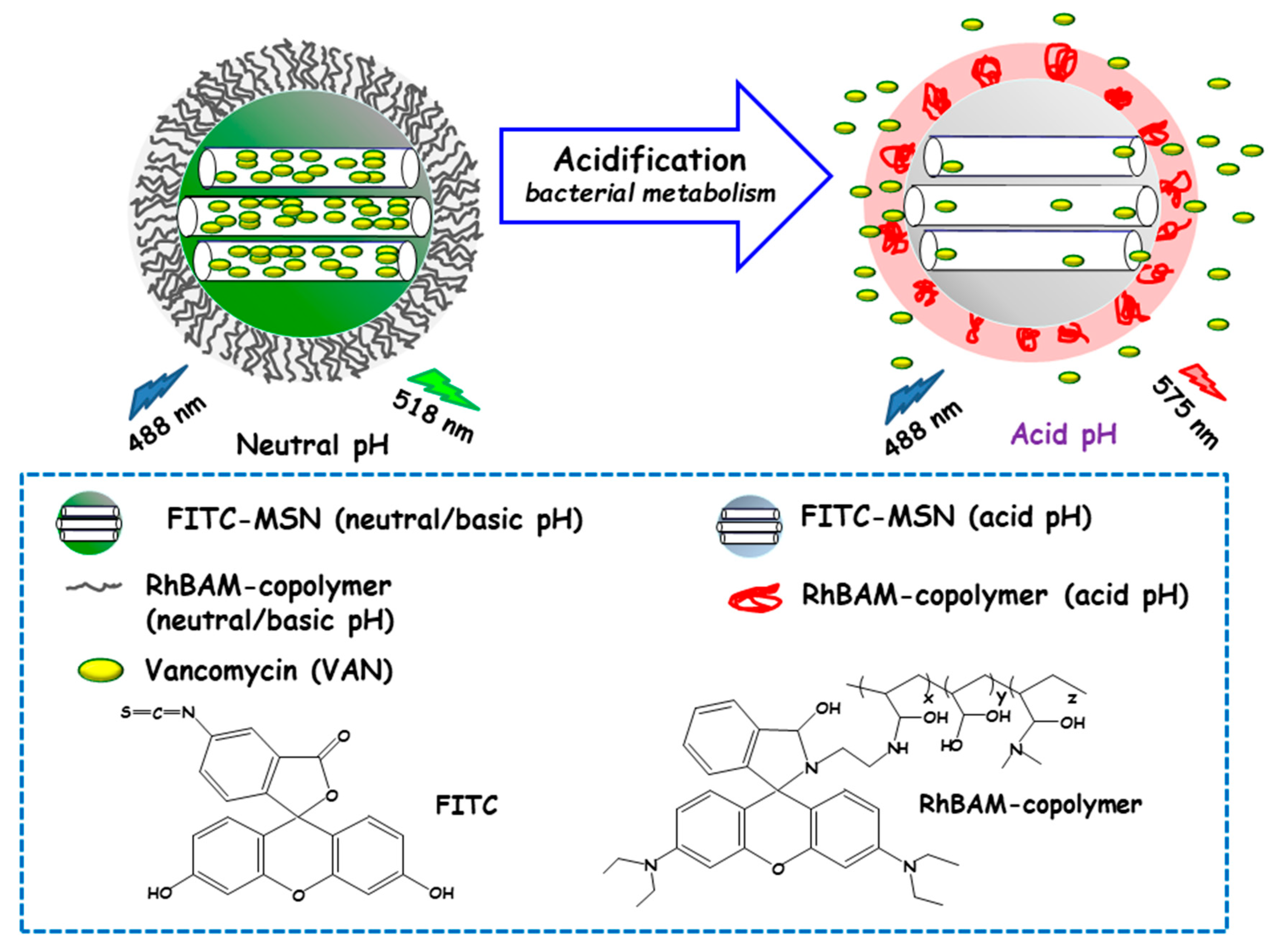
3.1.3. pH
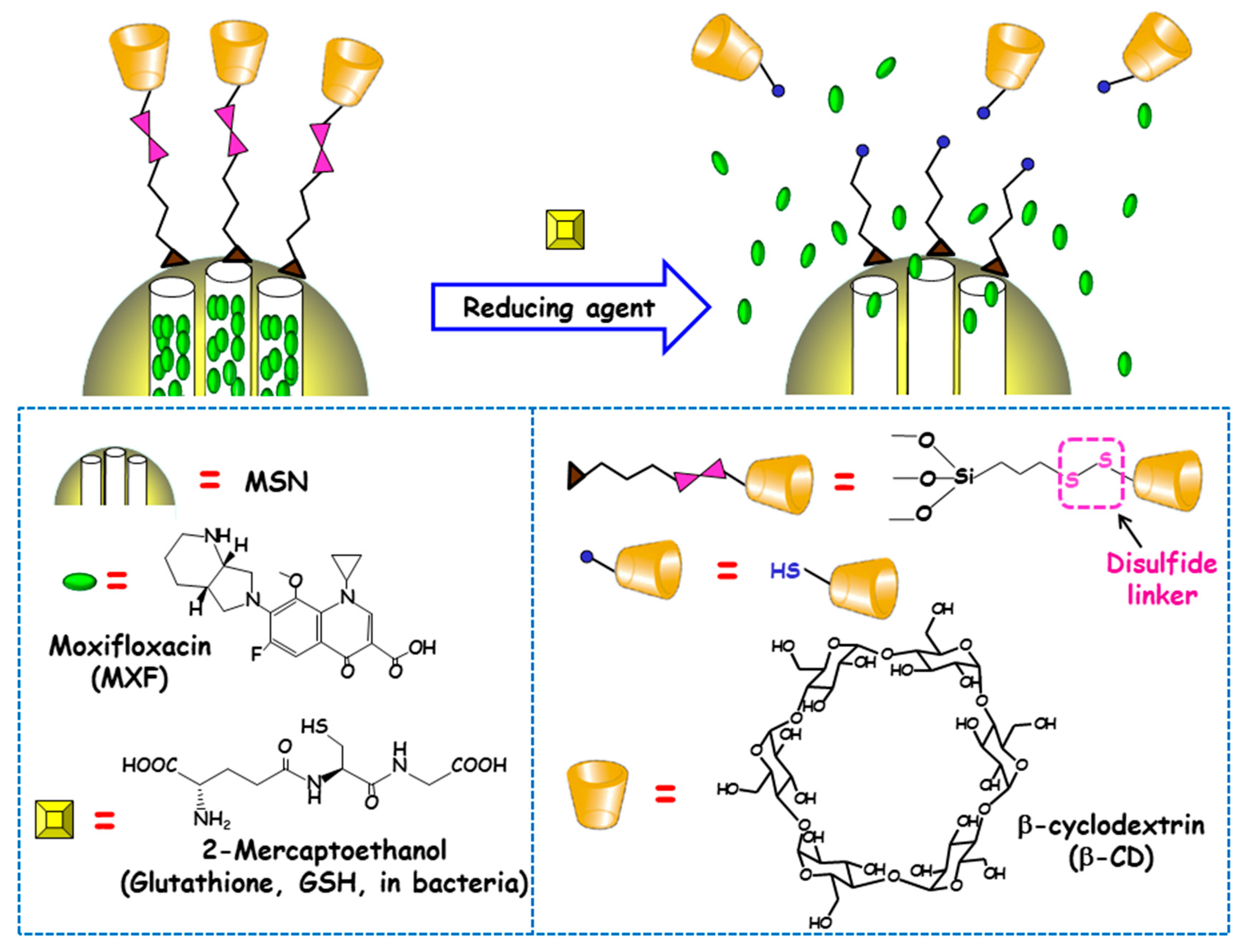
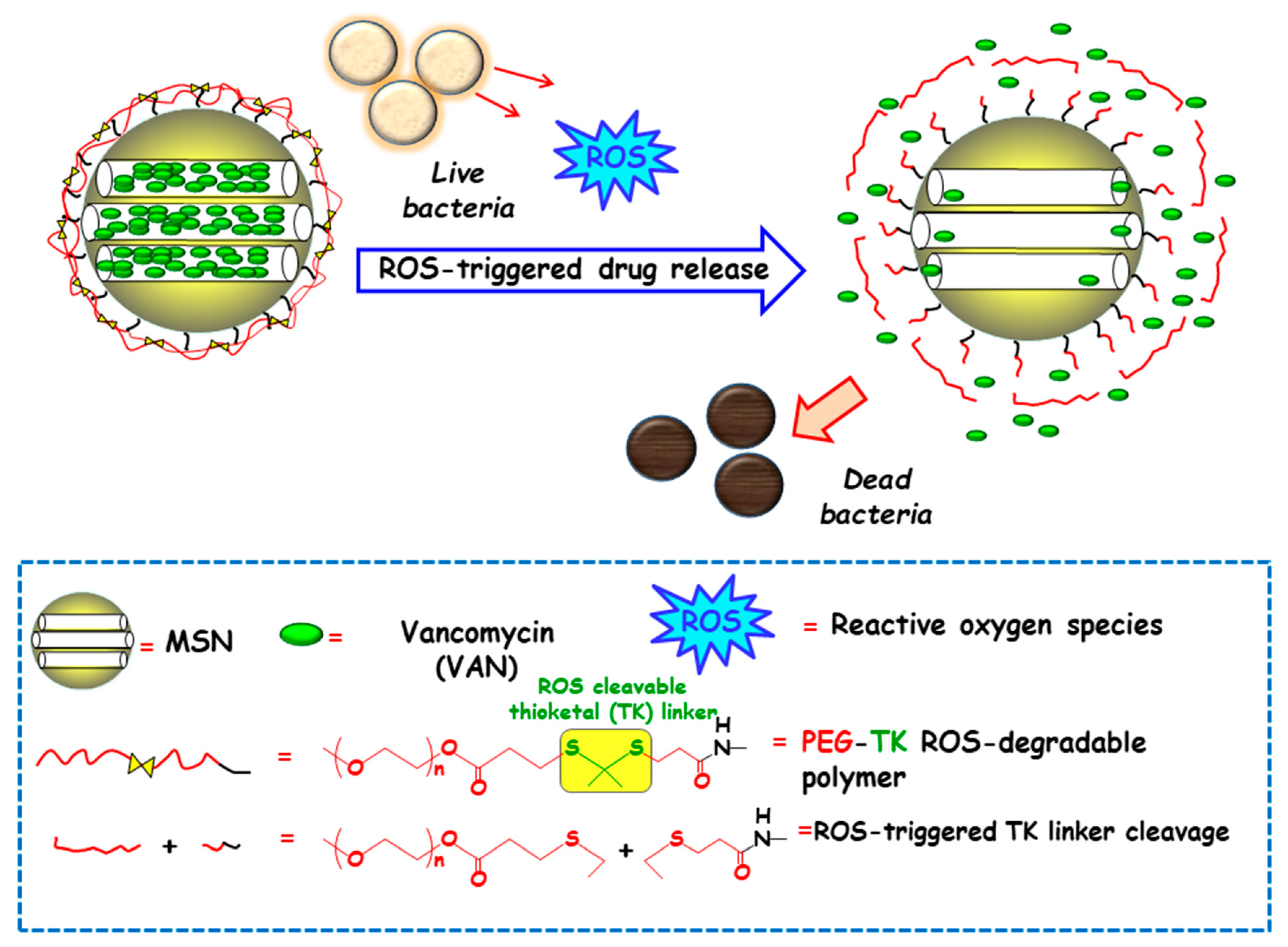
3.1.4. Redox Potential
3.1.5. Dual Stimuli
3.2. External Stimuli-Responsive MSNs
3.2.1. Chemical Species
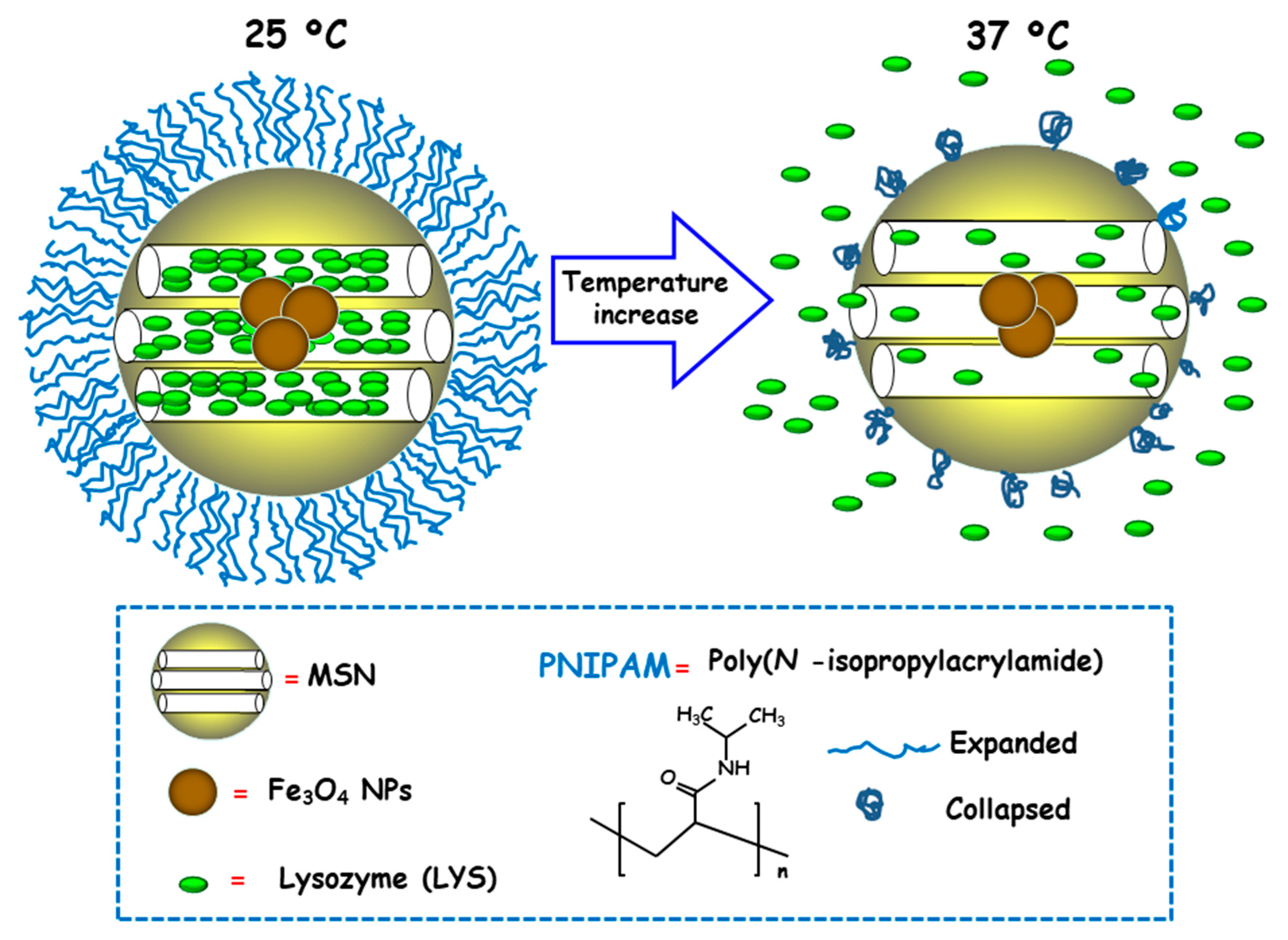
3.2.2. Temperature
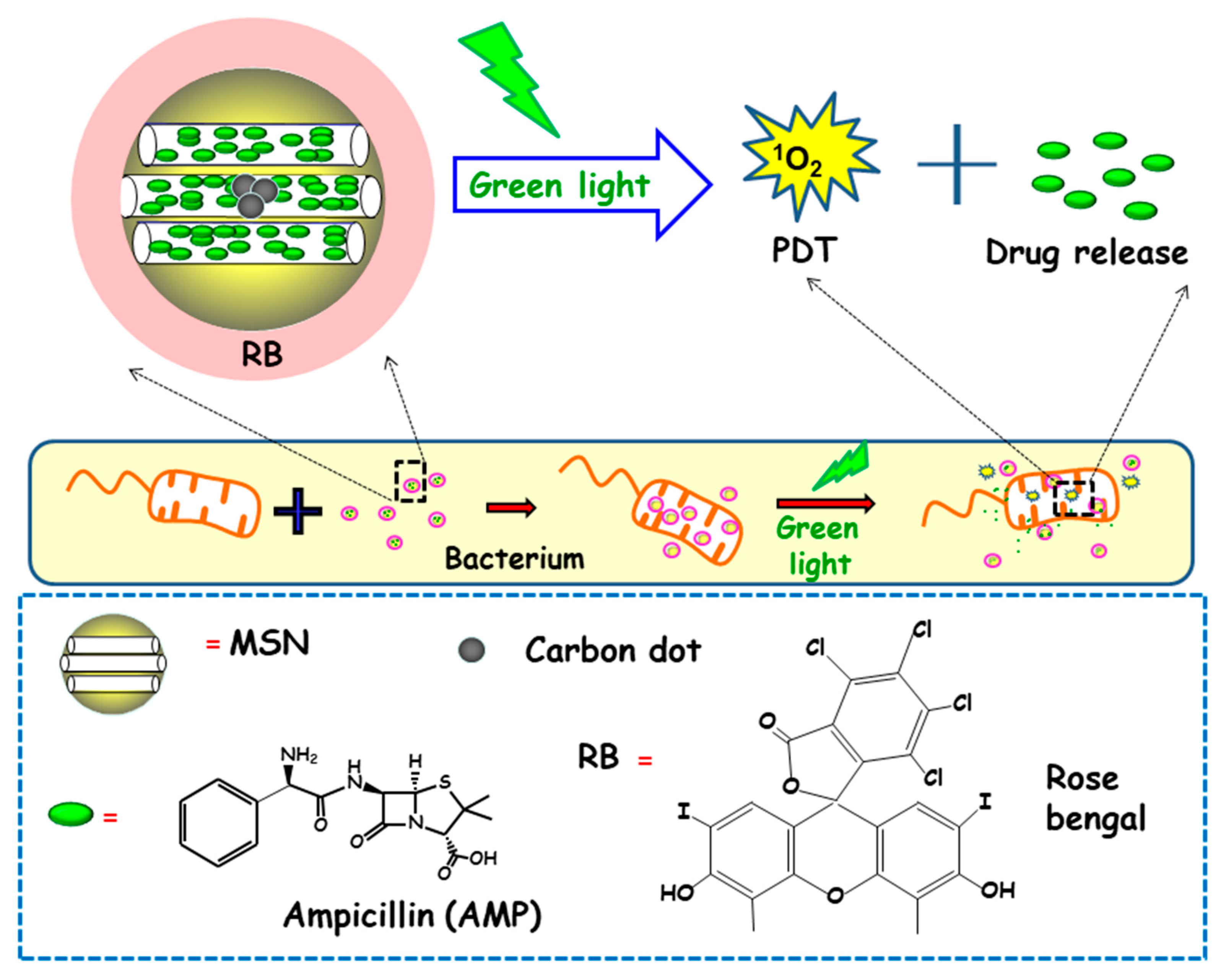
3.2.3. Light
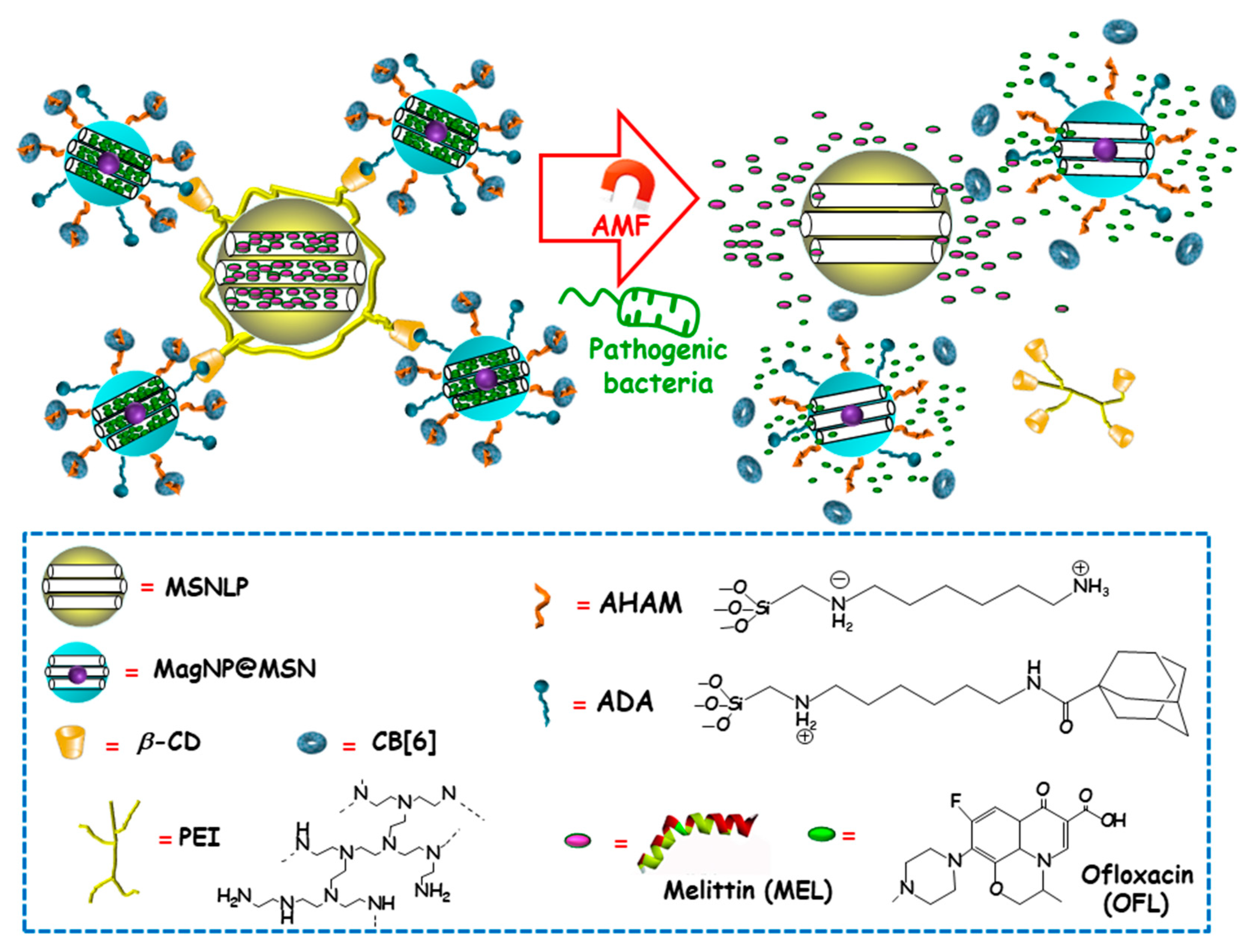
3.2.4. Alternating Magnetic Field
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ab@S-HA@MMSNs | Sulfonated-hyaluronic acid (S-HA) terminated magnetic MSNs with Anti-S. aureus antibody (Ab) |
| AD | Adamantaneamine |
| ADA | Adamantine |
| AD⊂CB[7] | Inclusion complex between adamantaneamine and curcubit[7]uril, |
| Ag NPs | Silver nanoparticles |
| Ag-SS-MSNs | Disulfide-bridged MSNs decorated with Ag NPs |
| Ag-SS-MSNs-CHX | Disulfide-bridged MSNs decorated with Ag NPs and loaded with chlorhexidine |
| AHAM | N-(6-N-aminohexyl)aminomethyl triethoxysilane |
| AIE | aggregation-induced emission |
| AMF | Alternating magnetic field |
| AMO | Amoxicillin |
| AMP | Ampicillin |
| AMR | Antimicrobial resistance |
| Anti-S. aureus Ab | Anti Staphylococcus aureus antibody |
| Arg | Arginine |
| Arg-MSN | MCM-41 type MSNs functionalized with L-Arg |
| AuNC | Gold nanoclusters |
| AuNC@LYS | Gold nanoclusters functionalized with lysozime |
| B. safensis | Bacillus safensis |
| B. subtilis | Bacillus subtilis |
| C. albicans | Candida albicans |
| CaP | Calcium phosphate |
| Car | Carbenicillin, |
| CarMOF | Fe3+-Carbenicillin Metal Organic Framwork (MOF) |
| CB[6] | Cucurbit[6]uril |
| CB[7] | Cucurbit[7]uril |
| C-dots | carbon dots |
| β-CD | β−Cyclodextrin |
| CFU | Colony Forming Units |
| CHX | Chlorhexidine |
| CIP | Ciprofloxacin |
| CIP-Arg-MSNs | MSNs conjugated to L-arginine and loaded with ciprofloxacin |
| COL | Colistin |
| COL-MSN@LL-(LL-37) | MSNs loaded with colistin, capped with a liposomal shell and conjugated to P. aeruginosa peptide LL-37 |
| ConA | Concanavalin A |
| CUR | Curcumin |
| Cu-MSN-AgNPs-CUR | MSNs impregnated with Cu(II) and externally decorated with Ag NPs and loaded with curcumin |
| DAMO | N-(2-aminoethyl)-3-aminopropyltrimethoxy-silane |
| DNA | Deoxyribonucleic acid |
| DOX | Doxorubicin |
| E. coli | Escherichia coli |
| EDA-PGMA | 1,2-ethanediamine-modified polyglycerol metacrylate |
| EPR | Enhanced permeability and retention |
| EPS | Extracellular polymer substances |
| F. novocida | Francisella novocida (Fn) |
| F. tularensis | Francisella tularensis (Ft) |
| FA | Folic acid |
| FB11 | Antibody for lipopolysaccharide (LPS) present in Francisella tularensis (Ft) |
| FITC | Fluorescein isothiocyanate |
| Fn | Francisella novocida |
| FRα | Folate receptor alpha |
| Ft | Francisella tularensis |
| G | Guest |
| GEN | Gentamicin |
| GEN@MSU-LU | MSNs loaded with GEN, capped with a lipid bilayer and functionalized with UBI29–41 |
| GSH/GSSH | Oxidized/reduced glutathione redox couple |
| G3 | Poly(propyleneimine) third-generation dendrimer |
| H | Host |
| HA | Hyaluronic acid |
| HKAIs | Histidine kinase authophosphorylation Inhibitors |
| HOMSNs | Hollow oblate mesoporous silica nanoparticles |
| HOMSNs-Tre | HOMSNs funcionalized with trehalose |
| HOMSNs-Tre-INH | HOMSNs funcionalized with trehalose and loaded with isoniazid |
| IAAH-Ag | Silver-indole-3 acetic acid hydrazide |
| INH | Isoniazid |
| KANA | Kanamycin |
| LBL | Layer-by-layer |
| LBL@MSN-Ag NPs | MSNs encapsulating Ag NPs coated with polymers by layer-by-layer assembly |
| LL-37 | Human cathelicidin peptide |
| LPS | Lipopolysaccharide |
| LVX | Levofloxacin |
| LYS | Lysozyme |
| M. smegmatis | Mycobacterium smegmatis |
| M. tuberculosis | Micobacterium tuberculosis |
| MagNP@MSN | Magnetic NPs coated with a mesoporous silica layer |
| MagNP@MSNA-CB[6] | Magnetic NPs coated with a mesoporous silica layer (MagNP@MSN) decorated with both adamantine (ADA) and N-(6-N-aminohexyl)aminomethyl triethoxysilane (AHAM) and capped by cucurbit[6]uril (CB[6]) |
| MEL | Melittin |
| MCM-41 ConA-MSNs | MCM-41 type MSNs decorated with concanavalin A |
| MCM-41 DAMO-MSNs | MCM-41 type MSNs functionalized with DAMO |
| MCM-41 FB11mFt LPS-MSNs | MCM-41 type MSNs functionalized with FB11 antibody through a derivative of the O-antigen of Ft LPS |
| MCM-41 G3-MSNs | MCM-41 type MSNs functionalized with G3 |
| MCM-41 MSNs⊂VAN | MCM-41 type MSNs functionalized with vancomycin |
| MCM-41 SA20hp-MSNs | MCM-41 type MSNs functionalized with SA20hp |
| MCM-41 ε−pLys-MSNs | MCM-41 type MSNs functionalized with ε-pLys |
| MBC | Minimum bactericidal concentration |
| MBIC | Minimal biofilm inhibitory concentration |
| MGCE | Magnetic glassy carbon electrode |
| MGCE/Ab@S-HA@MMSNs | Ab@S-HA@MMSNs immobilized Magnetic glassy carbon electrode |
| MIC | Minimum Inhibitory Concentrations |
| MMSNs | Magnetic MSNs |
| MSNs | Mesoporous Silica Nanoparticles |
| MSN@C-dots/RB | Core-shell MSNs embedding C-dots and rose-bengal |
| MSN@C-dots/RB/AMP | Core-shell MSNs embedding C-dots and rose-bengal and loaded with ampicillin |
| MSN@C-dots/RB/DOX | Core-shell MSNs embedding C-dots and rose-bengal and loaded with doxorubicin |
| MSN@FA@CaP@FA | MSNs covered by double folic acid (FA) and calcium phosphate (CaP) |
| MSN@LL-(LL-37) | MCM-41 type MSNs coated with a lipidic layer and conjugated with LL-37 |
| MSNLP@PEICD | Large pore MSNs capped by β-cyclodextrin-modified polyethylenimine (PEICD) |
| mPEG-TK | Methoxy poly(ethyleneglycol) fuctionalized with thioketal |
| MRSA | Methicillin-resistant S. aureus |
| MSN-Ag | MSNs with Ag NPs encapsulates |
| MSN-LU | MSNs modified with a lipidic bilayer surface shell and conjugated with UBI29–41 |
| MSN-NH2 | MSNs functionalized with aminopropyl groups |
| MXF | Moxifloxacin |
| M-PFPA-Tre | MSNs functionalized with PFPA-Si and decorated with Tre via azide-mediated surface photoligation |
| M-PFPA-Tre-INH | MSNs loaded with isoniazid, functionalized with perfluorophenylazide and decorated with trehalose |
| NPs | Nanoparticles |
| OFL | Ofloxacin |
| P. aeruginosa | Pseudomonas aeruginosa |
| PAG | Polyallylamine hydrochloride |
| PDI | Photodynamic inactivation |
| PEI-PEG | poly(ethylene imine)-poly(ethylene glycol) |
| PEG | Polyethylene glycol |
| PEICD | Polyethylenimine modified with b-cyclodextrin (β-CD) |
| PFPA-Si | Perfluorophenilazide silane |
| PG | poly-L-glutamic acid |
| PLA | Polylactic acid |
| PLA-NF | Polylactic acid nanoflowers |
| PNIPAM | poly(N-isopropylacrylamide) |
| PNIPAAM | poly(N-isopropyacrylamide-co-acrylic acid) |
| RhBAM | Rhodamine B derivative |
| ROS | Reactive oxygen species |
| RB | Rose bengal |
| S. aureus | Staphylococcus aureus |
| S. marcescens | Serratia marcescens |
| S. mutans | Streptococcus mutans |
| S. typhimuruim | Salmonella typhimuruim. |
| SA20hp | S. aureus 20 aptamer with hairpin structure |
| Sul | Sulbactam |
| S-HA | Sulfonated-hyaluronic acid |
| TB | Tuberculosis |
| TK | Thioketal |
| TMS-EDTA | N-[(3-trimethoxysilyl)propyl] ethylendiamine triacetic acid trisodium salt |
| TPE-(COOH)4 | Negatively charged tetraphenylethylene tetracarboxylate |
| Tre-HOMSNs | Trehalose-functionalized hollow oblate mesoporous silica nanoparticles |
| UBI29–41 | Ubiquicin |
| UV | Ultraviolet |
| VAN | Vancomycin |
| β-CD | Beta-cyclodextrin (β-CD) |
| ε-pLys | ε-poly-L-lysine |
References
- WHO. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- WHO. New Report Calls for Urgent Action to Avert Antimicrobial Resistance Crisis. Available online: https://www.who.int/news/item/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis (accessed on 6 November 2020).
- O’Neill, J. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist. 2014, 1, 1–16. [Google Scholar]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Geesey, G.G.; Cheng, K.J. How bacteria stick. Sci. Am. 1978, 238, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Alam, A.; Rani, M.; Ehtesham, N.Z.; Hasnain, S.E. Biofilms: Survival and defense strategy for pathogens. Int. J. Med. Microbiol. 2017, 307, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M. Role of biofilms in antimicrobial resistance. Asaio J. 2000, 46, S47–S52. [Google Scholar] [CrossRef]
- Watnick, P.; Kolter, R. Biofilm, City of Microbes. J. Bacteriol. 2000, 182, 2675–2679. [Google Scholar] [CrossRef]
- Årdal, C.; Balasegaram, M.; Laxminarayan, R.; McAdams, D.; Outterson, K.; Rex, J.H.; Sumpradit, N. Antibiotic development—Economic, regulatory and societal challenges. Nat. Rev. Microbiol. 2020, 18, 267–274. [Google Scholar] [CrossRef]
- Gupta, S.K.; Nayak, R.P. Dry antibiotic pipeline: Regulatory bottlenecks and regulatory reforms. J. Pharmacol. Pharmacother. 2014, 5, 4–7. [Google Scholar] [CrossRef]
- Canaparo, R.; Foglietta, F.; Giuntini, F.; Della Pepa, C.; Dosio, F.; Serpe, L. Recent Developments in Antibacterial Therapy: Focus on Stimuli-Responsive Drug-Delivery Systems and Therapeutic Nanoparticles. Molecules 2019, 24, 1991. [Google Scholar] [CrossRef]
- Gao, W.; Chen, Y.; Zhang, Y.; Zhang, Q.; Zhang, L. Nanoparticle-based local antimicrobial drug delivery. Adv. Drug Deliv. Rev. 2018, 127, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Vallet-Regí, M.; González, B.; Izquierdo-Barba, I. Nanomaterials as Promising Alternative in the Infection Treatment. Int. J. Mol. Sci. 2019, 20, 3806. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, L.; Su, L.; van der Mei, H.C.; Jutte, P.C.; Ren, Y.; Busscher, H.J. Nanotechnology-based antimicrobials and delivery systems for biofilm-infection control. Chem. Soc. Rev. 2019, 48, 428–446. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Baptista, P.V.; McCusker, M.P.; Carvalho, A.; Ferreira, D.A.; Mohan, N.M.; Martins, M.; Fernandes, A.R. Nano-Strategies to Fight Multidrug Resistant Bacteria—”A Battle of the Titans”. Front. Microbiol. 2018, 9, 1441. [Google Scholar] [CrossRef]
- Lu, J.; Liong, M.; Li, Z.; Zink, J.I.; Tamanoi, F. Biocompatibility, biodistribution, and drug-delivery efficiency of mesoporous silica nanoparticles for cancer therapy in animals. Small 2010, 6, 1794–1805. [Google Scholar] [CrossRef]
- Paris, J.L.; Colilla, M.; Izquierdo-Barba, I.; Manzano, M.; Vallet-Regí, M. Tuning mesoporous silica dissolution in physiological environments: A review. J. Mater. Sci. 2017, 52, 8761–8771. [Google Scholar] [CrossRef]
- Vallet-Regi, M.; Rámila, A.; del Real, R.P.; Pérez-Pariente, J. A New Property of MCM-41: Drug Delivery System. Chem. Mater. 2001, 13, 308–311. [Google Scholar] [CrossRef]
- Yang, P.; Gai, S.; Lin, J. Functionalized mesoporous silica materials for controlled drug delivery. Chem. Soc. Rev. 2012, 41, 3679–3698. [Google Scholar] [CrossRef]
- Argyo, C.; Weiss, V.; Bräuchle, C.; Bein, T. Multifunctional Mesoporous Silica Nanoparticles as a Universal Platform for Drug Delivery. Chem. Mater. 2014, 26, 435–451. [Google Scholar] [CrossRef]
- Colilla, M.; González, B.; Vallet-Regí, M. Mesoporous silica nanoparticles for the design of smart delivery nanodevices. Biomater. Sci. 2013, 1, 114–134. [Google Scholar] [CrossRef] [PubMed]
- Baeza, A.; Colilla, M.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery. Expert Opin. Drug Deliv. 2015, 12, 319–337. [Google Scholar] [CrossRef] [PubMed]
- Castillo, R.R.; Colilla, M.; Vallet-Regí, M. Advances in mesoporous silica-based nanocarriers for co-delivery and combination therapy against cancer. Expert Opin. Drug Deliv. 2017, 14, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Rosenholm, J.M.; Sahlgren, C.; Lindén, M. Towards multifunctional, targeted drug delivery systems using mesoporous silica nanoparticles--Opportunities & challenges. Nanoscale 2010, 2, 1870–1883. [Google Scholar] [CrossRef] [PubMed]
- Castillo, R.R.; de la Torre, L.; García-Ochoa, F.; Ladero, M.; Vallet-Regí, M. Production of MCM-41 Nanoparticles with Control of Particle Size and Structural Properties: Optimizing Operational Conditions during Scale-Up. Int. J. Mol. Sci. 2020, 21, 7899. [Google Scholar] [CrossRef]
- Hoffmann, F.; Cornelius, M.; Morell, J.; Fröba, M. Silica-based mesoporous organic-inorganic hybrid materials. Angew. Chem. Int. Ed. Engl. 2006, 45, 3216–3251. [Google Scholar] [CrossRef]
- Trewyn, B.G.; Slowing, I.I.; Giri, S.; Chen, H.T.; Lin, V.S. Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol-gel process and applications in controlled release. Acc. Chem. Res. 2007, 40, 846–853. [Google Scholar] [CrossRef]
- Vallet-Regí, M.; Colilla, M.; González, B. Medical applications of organic–inorganic hybrid materials within the field of silica-based bioceramics. Chem. Soc. Rev. 2011, 40, 596–607. [Google Scholar] [CrossRef]
- Martínez-Carmona, M.; Gun’ko, Y.K.; Vallet-Regí, M. Mesoporous Silica Materials as Drug Delivery: “The Nightmare” of Bacterial Infection. Pharmaceutics 2018, 10, 279. [Google Scholar] [CrossRef]
- Bernardos, A.; Piacenza, E.; Sancenón, F.; Hamidi, M.; Maleki, A.; Turner, R.J.; Martínez-Máñez, R. Mesoporous Silica-Based Materials with Bactericidal Properties. Small 2019, 15, 1900669. [Google Scholar] [CrossRef]
- Gisbert-Garzarán, M.; Manzano, M.; Vallet-Regí, M. Mesoporous Silica Nanoparticles for the Treatment of Complex Bone Diseases: Bone Cancer, Bone Infection and Osteoporosis. Pharmaceutics 2020, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Selvarajan, V.; Obuobi, S.; Ee, P.L.R. Silica Nanoparticles—A Versatile Tool for the Treatment of Bacterial Infections. Front. Chem. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, G.C.; Sábio, R.M.; de Cássia Ribeiro, T.; Monteiro, A.S.; Pereira, D.V.; Ribeiro, S.J.L.; Chorilli, M. Highlights in Mesoporous Silica Nanoparticles as a Multifunctional Controlled Drug Delivery Nanoplatform for Infectious Diseases Treatment. Pharm. Res. 2020, 37, 191. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Colomer, A.; Colilla, M.; Izquierdo-Barba, I.; Jiménez-Jiménez, C.; Mahillo, I.; Esteban, J.; Vallet-Regí, M. Impact of the antibiotic-cargo from MSNs on gram-positive and gram-negative bacterial biofilms. Microporous Mesoporous Mater. 2021, 311, 110681. [Google Scholar] [CrossRef]
- D’Agostino, A.; Taglietti, A.; Grisoli, P.; Dacarro, G.; Cucca, L.; Patrini, M.; Pallavicini, P. Seed mediated growth of silver nanoplates on glass: Exploiting the bimodal antibacterial effect by near IR photo-thermal action and Ag+ release. RSC Adv. 2016, 6, 70414–70423. [Google Scholar] [CrossRef]
- Xu, J.-W.; Yao, K.; Xu, Z.-K. Nanomaterials with a photothermal effect for antibacterial activities: An overview. Nanoscale 2019, 11, 8680–8691. [Google Scholar] [CrossRef]
- Pallavicini, P.; Dacarro, G.; Diaz-Fernandez, Y.A.; Taglietti, A. Coordination chemistry of surface-grafted ligands for antibacterial materials. Coord. Chem. Rev. 2014, 275, 37–53. [Google Scholar] [CrossRef]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef]
- Hao, N.; Chen, X.; Jeon, S.; Yan, M. Carbohydrate-Conjugated Hollow Oblate Mesoporous Silica Nanoparticles as Nanoantibiotics to Target Mycobacteria. Adv. Healthc. Mater. 2015, 4, 2797–2801. [Google Scholar] [CrossRef]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef]
- Maeda, H. Vascular permeability in cancer and infection as related to macromolecular drug delivery, with emphasis on the EPR effect for tumor-selective drug targeting. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Abdelbar, M.F.; Shams, R.S.; Morsy, O.M.; Hady, M.A.; Shoueir, K.; Abdelmonem, R. Highly ordered functionalized mesoporous silicate nanoparticles reinforced poly (lactic acid) gatekeeper surface for infection treatment. Int. J. Biol. Macromol. 2020, 156, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Schneewind, O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 1999, 63, 174–229. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, T.J. Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol. 1999, 181, 4725–4733. [Google Scholar] [CrossRef] [PubMed]
- González, B.; Colilla, M.; Díez, J.; Pedraza, D.; Guembe, M.; Izquierdo-Barba, I.; Vallet-Regí, M. Mesoporous silica nanoparticles decorated with polycationic dendrimers for infection treatment. Acta Biomater. 2018, 68, 261–271. [Google Scholar] [CrossRef]
- Mas, N.; Galiana, I.; Mondragón, L.; Aznar, E.; Climent, E.; Cabedo, N.; Sancenón, F.; Murguía, J.R.; Martínez-Máñez, R.; Marcos, M.D.; et al. Enhanced Efficacy and Broadening of Antibacterial Action of Drugs via the Use of Capped Mesoporous Nanoparticles. Chem. Eur. J. 2013, 19, 11167–11171. [Google Scholar] [CrossRef]
- Velikova, N.; Mas, N.; Miguel-Romero, L.; Polo, L.; Stolte, E.; Zaccaria, E.; Cao, R.; Taverne, N.; Murguía, J.R.; Martinez-Manez, R.; et al. Broadening the antibacterial spectrum of histidine kinase autophosphorylation inhibitors via the use of ε-poly-L-lysine capped mesoporous silica-based nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 569–581. [Google Scholar] [CrossRef]
- Ruehle, B.; Clemens, D.L.; Lee, B.-Y.; Horwitz, M.A.; Zink, J.I. A Pathogen-Specific Cargo Delivery Platform Based on Mesoporous Silica Nanoparticles. J. Am. Chem. Soc. 2017, 139, 6663–6668. [Google Scholar] [CrossRef]
- Xu, T.; Li, J.; Zhang, S.; Jin, Y.; Wang, R. Integration of diagnosis and treatment in the detection and kill of S. aureus in the whole blood. Biosens. Bioelectron. 2019, 142, 111507. [Google Scholar] [CrossRef]
- Kavruk, M.; Celikbicak, O.; Ozalp, V.C.; Borsa, B.A.; Hernandez, F.J.; Bayramoglu, G.; Salih, B.; Arica, M.Y. Antibiotic loaded nanocapsules functionalized with aptamer gates for targeted destruction of pathogens. Chem. Commun. 2015, 51, 8492–8495. [Google Scholar] [CrossRef]
- Yang, S.; Han, X.; Yang, Y.; Qiao, H.; Yu, Z.; Liu, Y.; Wang, J.; Tang, T. Bacteria-Targeting Nanoparticles with Microenvironment-Responsive Antibiotic Release To Eliminate Intracellular Staphylococcus aureus and Associated Infection. ACS Appl. Mater. Interfaces 2018, 10, 14299–14311. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, K.; Patel, U.; Pham, C.; McAlpin, A.; Budisalich, T.; Jayawardena, S.N. Targeted Delivery of Antibiotic Therapy to Inhibit Pseudomonas aeruginosa Using Lipid-Coated Mesoporous Silica Core–Shell Nanoassembly. ACS Appl. Biomater. 2020, 3, 6708–6721. [Google Scholar] [CrossRef]
- Zhou, J.; Jayawardana, K.W.; Kong, N.; Ren, Y.; Hao, N.; Yan, M.; Ramström, O. Trehalose-Conjugated, Photofunctionalized Mesoporous Silica Nanoparticles for Efficient Delivery of Isoniazid into Mycobacteria. ACS Biomater. Sci. Eng. 2015, 1, 1250–1255. [Google Scholar] [CrossRef]
- Mudakavi, R.J.; Vanamali, S.; Chakravortty, D.; Raichur, A.M. Development of arginine based nanocarriers for targeting and treatment of intracellular Salmonella. RSC Adv. 2017, 7, 7022–7032. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Lin, A.; Huang, N.; Long, L.; Gang, Y.; Liu, J. Folic acid-modified mesoporous silica nanoparticles with pH-responsiveness loaded with Amp for an enhanced effect against anti-drug-resistant bacteria by overcoming efflux pump systems. Biomater. Sci. 2018, 6, 1923–1935. [Google Scholar] [CrossRef]
- Qi, G.; Li, L.; Yu, F.; Wang, H. Vancomycin-Modified Mesoporous Silica Nanoparticles for Selective Recognition and Killing of Pathogenic Gram-Positive Bacteria Over Macrophage-Like Cells. ACS Appl. Mater. Inter. 2013, 5, 10874–10881. [Google Scholar] [CrossRef]
- Radovic-Moreno, A.F.; Lu, T.K.; Puscasu, V.A.; Yoon, C.J.; Langer, R.; Farokhzad, O.C. Surface Charge-Switching Polymeric Nanoparticles for Bacterial Cell Wall-Targeted Delivery of Antibiotics. ACS Nano 2012, 6, 4279–4287. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; McCarthy, K.A.; Kelly, M.A.; Gao, J. Targeting bacteria via iminoboronate chemistry of amine-presenting lipids. Nat. Commun. 2015, 6, 6561. [Google Scholar] [CrossRef]
- Ruiz-Rico, M.; Pérez-Esteve, É.; de la Torre, C.; Jiménez-Belenguer, A.I.; Quiles, A.; Marcos, M.D.; Martínez-Máñez, R.; Barat, J.M. Improving the Antimicrobial Power of Low-Effective Antimicrobial Molecules Through Nanotechnology. J. Food Sci. 2018, 83, 2140–2147. [Google Scholar] [CrossRef]
- Bebbington, C.; Yarranton, G. Antibodies for the treatment of bacterial infections: Current experience and future prospects. Curr. Opin. Biotechnol. 2008, 19, 613–619. [Google Scholar] [CrossRef]
- Özalp, V.C.; Bilecen, K.; Kavruk, M.; Öktem, H.A. Antimicrobial aptamers for detection and inhibition of microbial pathogen growth. Fut. Microbiol. 2013, 8, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.D.; Claypool, S.E.; Liu, R. The smart targeting of nanoparticles. Curr. Pharm. Des. 2013, 19, 6315–6329. [Google Scholar] [CrossRef] [PubMed]
- Nobre, A.; Alarico, S.; Maranha, A.; Mendes, V.; Empadinhas, N. The molecular biology of mycobacterial trehalose in the quest for advanced tuberculosis therapies. Microbiology 2014, 160, 1547–1570. [Google Scholar] [CrossRef] [PubMed]
- Backus, K.M.; Boshoff, H.I.; Barry, C.S.; Boutureira, O.; Patel, M.K.; D’Hooge, F.; Lee, S.S.; Via, L.E.; Tahlan, K.; Barry, C.E.; et al. Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat. Chem. Biol. 2011, 7, 228–235. [Google Scholar] [CrossRef]
- Bermingham, A.; Derrick, J.P. The folic acid biosynthesis pathway in bacteria: Evaluation of potential for antibacterial drug discovery. BioEssays 2002, 24, 637–648. [Google Scholar] [CrossRef]
- Pedraza, D.; Díez, J.; Colilla, M.; Vallet-Regí, M. Amine-Functionalized Mesoporous Silica Nanoparticles: A New Nanoantibiotic for Bone Infection Treatment. Biomed. Glas. 2018, 4, 1. [Google Scholar] [CrossRef]
- Martínez-Carmona, M.; Izquierdo-Barba, I.; Colilla, M.; Vallet-Regí, M. Concanavalin A-targeted mesoporous silica nanoparticles for infection treatment. Acta Biomater. 2019, 96, 547–556. [Google Scholar] [CrossRef]
- Xu, C.; He, Y.; Li, Z.; Ahmad Nor, Y.; Ye, Q. Nanoengineered hollow mesoporous silica nanoparticles for the delivery of antimicrobial proteins into biofilms. J. Mater. Chem. B 2018, 6, 1899–1902. [Google Scholar] [CrossRef]
- Tasia, W.; Lei, C.; Cao, Y.; Ye, Q.; He, Y.; Xu, C. Enhanced eradication of bacterial biofilms with DNase I-loaded silver-doped mesoporous silica nanoparticles. Nanoscale 2020, 12, 2328–2332. [Google Scholar] [CrossRef]
- Fulaz, S.; Devlin, H.; Vitale, S.; Quinn, L.; O’Gara, J.P.; Casey, E. Tailoring Nanoparticle-Biofilm Interactions to Increase the Efficacy of Antimicrobial Agents Against Staphylococcus aureus. Int. J. Nanomed. 2020, 15, 4779–4791. [Google Scholar] [CrossRef]
- Moreira, A.F.; Dias, D.R.; Correia, I.J. Stimuli-responsive mesoporous silica nanoparticles for cancer therapy: A review. Microporous Mesoporous Mater. 2016, 236, 141–157. [Google Scholar] [CrossRef]
- Castillo, R.R.; Lozano, D.; González, B.; Manzano, M.; Izquierdo-Barba, I.; Vallet-Regí, M. Advances in mesoporous silica nanoparticles for targeted stimuli-responsive drug delivery: An update. Expert Opin. Drug Deliv. 2019, 16, 415–439. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carmona, M.; Colilla, M.; Vallet-Regí, M. Smart Mesoporous Nanomaterials for Antitumor Therapy. Nanomaterials 2015, 5, 1906–1937. [Google Scholar] [CrossRef] [PubMed]
- Alsaiari, S.K.; Hammami, M.A.; Croissant, J.G.; Omar, H.W.; Neelakanda, P.; Yapici, T.; Peinemann, K.-V.; Khashab, N.M. Colloidal Gold Nanoclusters Spiked Silica Fillers in Mixed Matrix Coatings: Simultaneous Detection and Inhibition of Healthcare-Associated Infections. Adv. Healthc. Mater. 2017, 6, 1601135. [Google Scholar] [CrossRef] [PubMed]
- Lemichez, E.; Barbieri, J.T. General Aspects and Recent Advances on Bacterial Protein Toxins. Cold Spring Harb. Perspect. Biol. 2013, 3. [Google Scholar] [CrossRef]
- Wu, Y.; Long, Y.; Li, Q.-L.; Han, S.; Ma, J.; Yang, Y.-W.; Gao, H. Layer-by-Layer (LBL) Self-Assembled Biohybrid Nanomaterials for Efficient Antibacterial Applications. ACS Appl. Mater. Interfaces 2015, 7, 17255–17263. [Google Scholar] [CrossRef]
- Ding, Y.; Hao, Y.; Yuan, Z.; Tao, B.; Chen, M.; Lin, C.; Liu, P.; Cai, K. A dual-functional implant with an enzyme-responsive effect for bacterial infection therapy and tissue regeneration. Biomater. Sci. 2020, 8, 1840–1854. [Google Scholar] [CrossRef]
- Fuchs, S.; Pané-Farré, J.; Kohler, C.; Hecker, M.; Engelmann, S. Anaerobic Gene Expression in Staphylococcus aureus. J. Bacteriol. 2007, 189, 4275–4289. [Google Scholar] [CrossRef]
- Bistrian, B. Systemic response to inflammation. Nutr. Rev. 2007, 65, S170–S172. [Google Scholar] [CrossRef]
- Simmen, H.P.; Blaser, J. Analysis of pH and pO2 in abscesses, peritoneal fluid, and drainage fluid in the presence or absence of bacterial infection during and after abdominal surgery. Am. J. Surg. 1993, 166, 24–27. [Google Scholar] [CrossRef]
- Kuthati, Y.; Kankala, R.K.; Lin, S.-X.; Weng, C.-F.; Lee, C.-H. pH-Triggered Controllable Release of Silver–Indole-3 Acetic Acid Complexes from Mesoporous Silica Nanoparticles (IBN-4) for Effectively Killing Malignant Bacteria. Mol. Pharm. 2015, 12, 2289–2304. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Shi, P.; Ren, J.; Qu, X. A “Sense-and-Treat” Hydrogel Used for Treatment of Bacterial Infection on the Solid Matrix. Small 2015, 11, 5540–5544. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Feng, X.; Jin, Y.; Liu, D.; Yang, X.; Zhou, G.; Liu, D.; Li, Z.; Liang, X.-J.; Zhang, J. Metal–carbenicillin framework-based nanoantibiotics with enhanced penetration and highly efficient inhibition of MRSA. Biomaterials 2017, 144, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Clemens, D.L.; Lee, B.-Y.; Xue, M.; Thomas, C.R.; Meng, H.; Ferris, D.; Nel, A.E.; Zink, J.I.; Horwitz, M.A. Targeted intracellular delivery of antituberculosis drugs to Mycobacterium tuberculosis-infected macrophages via functionalized mesoporous silica nanoparticles. Antimicrob. Agents Chemother. 2012, 56, 2535–2545. [Google Scholar] [CrossRef]
- Hwang, A.A.; Lee, B.-Y.; Clemens, D.L.; Dillon, B.J.; Zink, J.I.; Horwitz, M.A. pH-Responsive Isoniazid-Loaded Nanoparticles Markedly Improve Tuberculosis Treatment in Mice. Small 2015, 11, 5066–5078. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Clemens, D.L.; Lee, B.-Y.; Dillon, B.J.; Horwitz, M.A.; Zink, J.I. Mesoporous Silica Nanoparticles with pH-Sensitive Nanovalves for Delivery of Moxifloxacin Provide Improved Treatment of Lethal Pneumonic Tularemia. ACS Nano 2015, 9, 10778–10789. [Google Scholar] [CrossRef] [PubMed]
- Pieters, J. Mycobacterium tuberculosis and the Macrophage: Maintaining a Balance. Cell Host Microbe 2008, 3, 399–407. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.; Lodish, H. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Lee, B.Y.; Li, Z.; Clemens, D.L.; Dillon, B.J.; Hwang, A.A.; Zink, J.I.; Horwitz, M.A. Redox-Triggered Release of Moxifloxacin from Mesoporous Silica Nanoparticles Functionalized with Disulfide Snap-Tops Enhances Efficacy Against Pneumonic Tularemia in Mice. Small 2016, 12, 3690–3702. [Google Scholar] [CrossRef]
- Li, J.; Ding, Z.; Li, Y.; Miao, J.; Wang, W.; Nundlall, K.; Chen, S. Reactive oxygen species-sensitive thioketal-linked mesoporous silica nanoparticles as drug carrier for effective antibacterial activity. Mater. Des. 2020, 195, 109021. [Google Scholar] [CrossRef]
- Spooner, R.; Yilmaz, O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.M.; Ge, Y.; Qiu, J.; Shao, D.; Zhang, Y.; Bai, J.; Zheng, X.; Chang, Z.M.; Wang, Z.; Dong, W.F.; et al. Redox/pH dual-controlled release of chlorhexidine and silver ions from biodegradable mesoporous silica nanoparticles against oral biofilms. Int. J. Nanomed. 2018, 13, 7697–7709. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, Y.; Lu, H.; Wu, X.; Chen, S.; Song, N.; Yang, Y.-W.; Gao, H. Construction of Supramolecular Nanoassembly for Responsive Bacterial Elimination and Effective Bacterial Detection. ACS Appl. Mater. Interfaces 2017, 9, 10180–10189. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Galiana, I.; Martínez-Máñez, R.; Stroeve, P.; Marcos, M.D.; Aznar, E.; Sancenón, F.; Murguía, J.R.; Amorós, P. Poly(N-isopropylacrylamide)-gated Fe3O4/SiO2 core shell nanoparticles with expanded mesoporous structures for the temperature triggered release of lysozyme. Colloid. Surf. B 2015, 135, 652–660. [Google Scholar] [CrossRef]
- Kuthati, Y.; Kankala, R.K.; Busa, P.; Lin, S.-X.; Deng, J.-P.; Mou, C.-Y.; Lee, C.-H. Phototherapeutic spectrum expansion through synergistic effect of mesoporous silica trio-nanohybrids against antibiotic-resistant gram-negative bacterium. J. Photochem. Photobiol. B 2017, 169, 124–133. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Xiao, Y.; Chen, F.; Xiao, F. A multifunctional nanoplatform based on mesoporous silica nanoparticles for imaging-guided chemo/photodynamic synergetic therapy. RSC Adv. 2017, 7, 31133–31141. [Google Scholar] [CrossRef]
- Zhang, C.; Du, C.; Liao, J.-Y.; Gu, Y.; Gong, Y.; Pei, J.; Gu, H.; Yin, D.; Gao, L.; Pan, Y. Synthesis of magnetite hybrid nanocomplexes to eliminate bacteria and enhance biofilm disruption. Biomater. Sci. 2019, 7, 2833–2840. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Li, P.; Wang, L.; Zhou, J.; Zhang, G.; Li, X.; Hu, B.; Xing, X. Microenvironment-Responsive Magnetic Nanocomposites Based on Silver Nanoparticles/Gentamicin for Enhanced Biofilm Disruption by Magnetic Field. ACS Appl. Mater. Interfaces 2018, 10, 34905–34915. [Google Scholar] [CrossRef]
- Yu, Q.; Deng, T.; Lin, F.-C.; Zhang, B.; Zink, J.I. Supramolecular Assemblies of Heterogeneous Mesoporous Silica Nanoparticles to Co-deliver Antimicrobial Peptides and Antibiotics for Synergistic Eradication of Pathogenic Biofilms. ACS Nano 2020, 14, 5926–5937. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeting Ligand 1 | Drug Loaded 2 | Nanocarrier 3 | Bacteria 4 | Assay | Ref. |
|---|---|---|---|---|---|
| G3 | Levofloxacin | MCM-41 G3-MSNs | E. coli | In vitro | [47] |
| ε-pLys | Vancomycin | MCM-41 ε-pLys-MSNs | E. coli | In vitro | [48] |
| ε-pLys | HKAIs | MCM-41 ε-pLys-MSNs | E. coli, S. marcescens | In vitro | [49] |
| FB11 | Model drugs (Fluorescein, Hoechst 33342) | MCM-41 FB11mFt LPS-MSNs | F. tularensis | In vitro | [50] |
| Anti-S. aureus Ab | Vancomycin | Ab@S-HA@MMSNs | S. aureus | In vitro | [51] |
| SA20hp | Vancomycin | MCM-41 SA20hp-MSNs | S. aureus | In vitro | [52] |
| UBI29–41 | Gentamicin | MSN-LU | S. aureus | In vitro & in vivo | [53] |
| LL-37 | Colistin | MSN@LL-(LL-37) | P. aeruginosa | In vitro | [54] |
| Trehalose | Isoniazid | M-PFPA-Tre | M. smegmatis | In vitro | [55] |
| Trehalose | Isoniazid | Tre-HOMSNs | M. smegmatis | In vitro | [41] |
| Arginine | Ciprofloxacin | Arg-MSNs | S. typhimuruim | In vitro & in vivo | [56] |
| Folic acid | Ampicillin | MSN@FA@CaP@FA | E. coli, S. aureus | In vitro & in vivo | [57] |
| Vancomycin | Vancomycin (grafted) | MCM-41 MSNs⊂VAN | S. aureus | In vitro | [58] |
| Targeting Ligand 1 | Drug Loaded | Nanocarrier 2 | Bacterial Biofilm 3 | Assay | Ref. |
|---|---|---|---|---|---|
| DAMO | Levofloxacin | MCM-41 DAMO-MSNs | S. aureus | In vitro | [47,68] |
| G3 | Levofloxacin | MCM-41 G3-MSNs | E. coli | In vitro | [47] |
| ConA | Levofloxacin | MCM-41 ConA-MSNs | E. coli | In vitro | [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colilla, M.; Vallet-Regí, M. Targeted Stimuli-Responsive Mesoporous Silica Nanoparticles for Bacterial Infection Treatment. Int. J. Mol. Sci. 2020, 21, 8605. https://doi.org/10.3390/ijms21228605
Colilla M, Vallet-Regí M. Targeted Stimuli-Responsive Mesoporous Silica Nanoparticles for Bacterial Infection Treatment. International Journal of Molecular Sciences. 2020; 21(22):8605. https://doi.org/10.3390/ijms21228605
Chicago/Turabian StyleColilla, Montserrat, and María Vallet-Regí. 2020. "Targeted Stimuli-Responsive Mesoporous Silica Nanoparticles for Bacterial Infection Treatment" International Journal of Molecular Sciences 21, no. 22: 8605. https://doi.org/10.3390/ijms21228605
APA StyleColilla, M., & Vallet-Regí, M. (2020). Targeted Stimuli-Responsive Mesoporous Silica Nanoparticles for Bacterial Infection Treatment. International Journal of Molecular Sciences, 21(22), 8605. https://doi.org/10.3390/ijms21228605